
Abstract
As a reinforcing phase, Graphene (Gr) can effectively enhance the strength of the aluminum substrate (Al). Yet most of the previous research focuses primarily on the interfacial adhesion, the mechanical performances of the graphene/aluminum interface structure and the corresponding strengthening mechanism remain insufficiently understood. In this study, employing density functional theory (DFT), the electronic and mechanical properties of different graphene/aluminum interface structures (Gr/Al) are investigated. By tensile simulations of the Gr/Al interface structures, the enhancement mechanism of Gr on aluminum substrate is revealed at a microscopic scale. The ideal strength of the Gr/Al interface structure increases with the increasing number of graphene layers, where the three-layer graphene structure (AAA-Gr/Al) exhibits the highest ideal strength of 5.02 N/m. For the tensile of the Gr/Al interface structure, the distortion of the graphene lattice enhances the interfacial binding ability and resists tensile deformation. Eventually, the breaking of the C-C bonds in graphene occurs, preventing the reformation of in-plane strong σ covalent bonds. For the AAA-Gr/Al interface structure under biaxial tension, the resistance to deformation primarily arises from the pz orbital of graphene and the px orbital of graphene hybridizes with the s orbital of Al, which synergistically increase the ideal strength of the interface structure. A comprehensive framework on evaluating the mechanical performance of Gr/Al interface structures has been developed by combining adhesion energy analysis, stress-strain calculations, and electronic structure analysis, which will advance the understanding of the structure-property relationship in Gr/Al composites.
Similar content being viewed by others


Introduction
Graphene1,2, known for its exceptional mechanical properties, is frequently utilized as a nano-particle reinforcement3,4 in the production of metal matrix composites. Aluminum-based composites, commonly used as structural materials in aerospace engineering, are currently undergoing development trends that prioritize lightweight, high strength, and low cost. It has been demonstrated that the incorporation of about 2 wt% of graphene into aluminum can significantly improve the mechanical performance of aluminum-based composites5,6. However, during the experimental production of graphene/aluminum composites, the poor wettability between graphene and aluminum substrate will result in defects in the interface structures7, hence affecting the mechanical properties of the composites.
In the previous researches on the fabrication and performance of graphene/aluminum composites, the enhancement of macroscopic mechanical strength has been achieved through microscopic modulation of the interface structure8. G. Guo et al.9 successfully fabricated biomimetic sheet-like graphene/aluminum composites with layered nanostructure and the in-situ tensile tests under a transmission electron microscope revealed an ultimate tensile strength of 302 MPa, 50% higher than that of pure aluminum. S.E. Shin et al.10 prepared few-layer graphene/aluminum composites with excellent dispersibility, conducted microscopic atomic-scale investigations into the interface structure and properties, and experimentally revealed an impressive tensile strength of 440 MPa, approximately double that of monolithic aluminum. J. Cao et al.11 fabricated a composite material with a multi-scale Al2O3/GNPs/Al nanointerface structure with the Young’s modulus of 76.9 GPa, higher than the value of pure aluminum (69.1GPa), and tensile strength of 497 MPa, 317% improvement compared to 1060 pure aluminum, with only 27% loss in plasticity. Nevertheless, there is still a lack of investigating about the mechanical properties and the reinforcement mechanisms of graphene/aluminum composite interface structures at the microscopic scale.
Based on the first-principles method, the mechanical properties of graphene/aluminum interface structures can be investigated at the atomic scale, allowing for detailed analysis of their interfacial bonding strength and electronic properties to optimize the interface structure and enhance its mechanical performance. For examples, H.O. Frota et al.12 investigated the electronic properties and transport properties of a stable Gr(0001)/Al2O3(0001) interface structure; J. Xie et al.13 conducted a study on the valence bond properties and doping effects of the Gr(0001)/Al(111) interface structure; Yue Qi et al.14 examined the interfacial binding properties of the Gr(0001)/Al(111) interface structure; H. Gao et al.15 combined experimental observations with first principles methods to study the crystal plane orientation laws of the graphene/aluminum composite’s interface structure. Currently, various properties of interface structures have been successfully investigated with first principles methods, including the microscopic mechanical properties16, the interfacial binding properties17, and the ideal strength18, etc. However, there’re relatively scarce researches focusing on the ideal tensile strength of graphene/aluminum interface structures at the microscopic scale considering the graphene’s anisotropy and the in-plane characteristics.
In this study, based on first principles methods, we construct Gr(0001) and Al(111) surface structures, as well as four graphene/aluminum interface structures including single-layer graphene (A-Gr), double-layer graphene (AA-Gr), triple-layer graphene (AAA-Gr), and vacancy-containing graphene (V-Gr). The ideal tensile strengths of the Gr/Al interface structures in three different directions x, y, and biaxial are investigated. The variations in the interface structure and electronic properties of the Gr/Al interface structures during tensile deformation are analyzed and the reinforcement mechanisms are explored. By integrating multilayer interface modeling, orbital-level analysis, and loading-mode-dependent mechanical evaluation, the present research can provide a unique perspective for deeply understanding the enhancement of mechanical performances of Gr/Al composites.
Structure modeling and calculation method
Surface and interface structure modeling
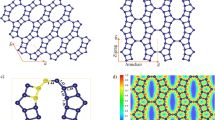
Figure 1a and b presents the surface structure of Gr(0001) and Al(111) surface with an ABC stacking sequence respectively. Generally, for constructing interface structures, the lattice constant difference between the two surface structures, namely interface mismatch, is required to be less than 5%17. Figure 1c presents the Gr/Al interface consisting of a 2 × 2 Gr(0001) surface and a \(\:\sqrt{3}\times\:\sqrt{3}\) supercell of the Al(111) surface, following the top-fcc configuration. The interlayer spacing of the Gr/Al interface is 3.5 Å. The lattice mismatch of the Gr/Al interface model is only 0.28%. Considering interfacial reactions in metal substrates typically occur between 1 ~ 2 layers at the interface17,19, a 6-layer Al(111) structure is enough to model the Al substrate and the interface. Figure 1d ~ f are the Gr/Al interface structures containing single-layer Gr(0001), the AA-Gr/Al interface structure containing double-layer Gr(0001), and the AAA-Gr/Al interface structure containing three-layer Gr(0001) and the V-Gr/Al interface structure of single-layer Gr(0001) containing single vacancy, respectively. Although practical factors that enhance interface strength, such as oxide layers, chemical bonding, surface functionalization, alloying and microstructural features, will be considered, it should be noted that although the real Gr/Al interfaces may include more complex defect types, the single vacancy is one of the most common and well-studied intrinsic defects in graphene, and the simple structure and high computational efficiency make it widely used in first-principles studies. The current study aims to establish a basic theoretical understanding based on the monovacancy model.

Atomic configurations diagrams of the surface/interface structures: (a) Gr(0001), (b) Al(111), (c)~(d) A-Gr/Al, (e) AA-Gr/Al, (f) AA-Gr/Al and (g) V-Gr/Al.
Stress-strain curve and ideal strength calculation
Stress tensor can be obtained through the derivative of the total energy with respect to the strain tensor, and strain \(\:\epsilon\:\) is determined by the varying of the lattice constant, i.e20,21. :
where \(\:\epsilon\:\) is the strain, \(\:a\) the lattice constant under strain, and \(\:{a}_{0}\) the equilibrium lattice constant.
In this study, the x-direction corresponds to the armchair direction of graphene, while the y-direction aligns with its zigzag direction. To determine the tensile stress-strain relationship along the x-direction, a series of strain increments are added to the cell of the interface structure along the x direction. The strain step of 0.02 and the strain range of 0–40% adopted in this study are chosen to balance computational cost and numerical stability, which will not introduce time-dependent or strain-rate effects like molecular dynamics simulations and experimental tests. Similar treatments are applied along the y-direction to calculate the tensile stress-strain response along y-direction. For biaxial (corresponding to the a- and b-axes of graphene) stress-strain analysis, equal strain increments are applied to the a and b axes of the interfacial structure cells, respectively. To ensure that the interface structure is strictly under uniaxial/biaxial stress state, it is necessary to fix the corresponding tensile direction (without optimization) when optimizing the atomic coordinates of the cell.
The ideal strength of the interface structure can be quantified using the in-plane stress σ, a two-dimensional stress per unit length measured in N/m. Taking into account the variations in the thickness of the interface structure, normalization is required22,23,24. In first-principles calculations, any atomic configuration under periodic boundary conditions—whether defective or not—can be regarded as an ideal system, with its ideal strength characterizing the intrinsic mechanical limit of the specific configuration. Comparing of the ideal strengths of materials helps illustrate the strengthening mechanisms. The normalized ideal strength is calculated by:
where σ is the in-plane stress (N/m), F the Cauchy stress extracted from the VASP output file, L the total thickness of the unit cell, and D the effective atomic thickness of the interface structure excluding the vacuum layer. Specifically, FXX is uniaxial tension in the x-direction, and Fyy is uniaxial tension in the y-direction. For biaxial tension, the stress is (Fxx+Fyy)/2. Following the above scheme, from the stress-strain curve, the ideal tensile strengths can be obtained for different Gr(0001) surface structures and Gr/Al interface structures.
Introduction of calculation method
Based on density functional theory (DFT) with periodic boundary conditions, first-principles calculations are implemented by using the Vienna Ab Initio Simulation Package (VASP) to investigate the structural response of the Gr/Al interface under tensile deformation. The projector augmented wave (PAW) method with a plane-wave basis set is utilized to model interactions between valence electrons and atomic cores. The exchange-correlation effects are described by the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)25,26.
Pseudopotentials for C (2s22p2) and Al (3s23p1) are constructed based on their valence electron configurations. The plane-wave energy cutoff is set to 550 eV, and Brillouin zone sampling is performed using a Monkhorst-Pack (MP) 9 × 9 × 3 k-point grid. Gaussian smearing with a width of 0.01 eV is applied to smooth partial electronic occupancies. Convergence criteria are atomic forces below 1.0 × 10−3 eV/Å and total energy variation below 1.0 × 10−6 eV/atom. Full atomic relaxation is carried out. By using the modified ADAIS code based on VASP, at each strain step the lattice constant along the tensile direction is fixed to produce the prescribed deformation, whereas in other directions all atoms are fully relaxed. To prevent interactions between neighboring periodic images along the c-axis, a 15 Å vacuum layer is introduced to the model. To correct the system energy, the Van der Waals interactions on the performance of the interface structure is considered and the DFT-D3 method with zero damping form27 is used.
The VESTA software (Visualization for Electronic and Structural Analysis) and the ADAIS program are used to visualize the interface structure and electronic distribution and to simulate tensile deformation of the interface structure, respectively.
Results and discussions
Surface and interface stability
Surface stability
The stability of surface structure can be assessed by two fundamental physical parameters: surface energy and work function. Surface energy reflects the energy required for surface formation and indicates the thermodynamic stability, while the work function represents the electronic properties and provides insights into the stability of surface atomic arrangements. The surface energy \(\:{E}_{surf}\) is calculated by28:
where \(\:{E}_{surf}\) is the surface energy of the structure, \(\:{E}_{slab}\) the total energy of the optimized surface structure, \(\:n\) the total number of atoms, \(\:{E}_{Al}\) the energy of an individual atom, and \(\:S\) the total surface area of the structure.
The work function \(\:{W}_{f}\) is calculated by29:
where \(\:{W}_{f}\) is the work function of the surface structure, \(\:\chi\:\) the electrostatic potential in vacuum, and \(\:{E}_{f}\) the Fermi energy level.
Table 1 presents the structural and electronic properties for both Al(111) and Gr(0001) surface configurations, including the lattice constant (a), surface energy (\(\:{E}_{surf}\)), and work function (\(\:{W}_{f}\)). For the Al(111) surface structure, the calculated total energy (\(\:{E}_{slab}\)) is −21.70 eV, the energy per atom (\(\:{E}_{Al}\)) is −3.72 eV/atom, and the surface area (S) is 14.17 Å2, yielding a surface energy of 0.70 J/m2. The electrostatic potential (χ) is 1.67 eV and the Fermi energy (\(\:{E}_{f}\)) is −2.35 eV, resulting in a work function of 4.02 eV. The optimized lattice constant is 2.86 Å. These computed values for Al(111) are consistent well with previous theoretical calculations and experiments30,31,32,34,35, demonstrating both the stability of our computational approach and the reliability of the 6-layer Al(111) surface model approximating the bulk Al structure.
For the Gr(0001) surface structure, the electrostatic potential (χ) is 2.00 eV, and the Fermi energy (\(\:{E}_{f}\)) is −2.26 eV, yielding a work function of 4.27 eV and the optimized lattice constant is 2.47 Å. These results agree well with both experimental measurements and theoretical predictions36,37,38, reflecting good consistency with calculation values37,38, further validating the reliability of the computational approach and modeling method.
Interface stability
Interfacial structural stability can effectively reflect the interfacial bond strength and reveal the potential failure mechanism of the material interface, playing an important role in advanced material design and performance enhancement. The interface binding energy is a core quantitative characterization parameter to characterize interface interaction strength and evaluate the stability of interface configurations. The interface binding energy \(\:{E}_{b}\) can be calculated by39:
where \(\:{E}_{b}\) is the binding energy of the interface structure, \(\:{E}_{Gr/Al}^{total}\) the total energy of the Gr/Al interface, \(\:{E}_{Al}^{slab}\) the total energy of the Al(111) surface, and \(\:{E}_{Gr}^{slab}\) the total energy of the Gr(0001) slab.
For the A-Gr/Al, AA-Gr/Al, AAA-Gr/Al, and V-Gr/Al interface structures, the optimized performance parameters are listed in Table 2, including the lattice constant (a), interlayer spacing (\(\:{d}_{0}\)), interface binding energy (\(\:{E}_{b}\)), work function (\(\:{W}_{f}\)), and Fermi level (\(\:{E}_{f}\)). For these interface structures, all the interlayer spacing (\(\:{d}_{0}\)) exceeds 3 Å and except for the V-Gr/Al configuration, the corresponding interface binding energies of A-Gr/Al, AA-Gr/Al and AAA-Gr/Al are below 0.1 eV. This result suggests these three interface structures have the physical adsorption characteristics39 with van der Waals interactions dominating the interlayer bonding, without significant chemical reactions at the interface.
Considering the influence of the graphene layer numbers, by comparing the three interfacial structures A-Gr/Al, AA-Gr/Al and AAA-Gr/Al, it can be seen that the lattice constant, layer spacing and interface binding energy of the interface structure do not change with the increasing of the graphene layers. This indicates that these interfaces are relatively stable and the Gr(0001) and Al(111) planes match well without lattice distortion.
When analyzing the influence of vacancy defect, comparing the A-Gr/Al and V-Gr/Al interface structures, the V-Gr/Al structure exhibits an increased lattice constant from 4.93 Å to 5.03 Å, indicating the lattice distortion. In V-Gr/Al structure, the single vacancy graphene induces lattice distortion when matching Al(111), resulting in the increase of lattice constant, the reduce of interlayer spacing (\(\:{d}_{0}\)) and the enhancement of interface binding energy (\(\:{E}_{b}\)). This suggests that vacancies in graphene can induce lattice distortion and the structural symmetry is broken, thereby enhancing its electronic coupling effects with the aluminum substrate. It improves the wetting properties40 and strengthens the interface binding performance41.
Although the V-Gr/Al interface structure has strong interface bonding performance, its work function increases and Fermi level decreases, which affects the electronic properties of the interface structure and changes the connection mode of the strong σ-bond in the graphene surface, so as to impact on the mechanical properties. Therefore, graphene structures containing vacancy defects should be avoided in the production and preparation process.
Stress-strain response and ideal strength of the Gr/Al interface structure
Stress-strain response of the interface structures
For material mechanical properties, stress-strain curve is the basis of performance assessment, which can be predicted by the first principles calculation based on density functional theory (DFT). It can relatively accurately provide the elastic deformation behavior, the theoretical strength limit and failure mechanism of materials.
The stress-strain curves and ideal strength values for the four Gr/Al interface structures under different tensile loading directions are shown in Fig. 2. Among the four interface structures, the V-Gr/Al exhibits the lowest ideal strength because the Gr structure is disrupted by the single vacancy, accompanying with its in-plane strong σ-bond turning into weaker multi-center bonds, which doesn’t present the enhancing effect. The AAA-Gr/Al interface structure demonstrates the highest ideal strength. Although the graphene layers primarily interact through van der Waals forces, stretching simulations along the in-plane direction, i.e., parallel to the graphene basal plane, show that each graphene layer exhibits a very high ideal strength, much higher than that of the aluminum substrate layer. When normalizing the interface thickness, the proportion of graphene layers with intrinsic high strength increases with the number of layers, effectively enhancing the overall strength of the composite interface structures. Therefore, the strengthening mechanism can be effectively illustrated by studying the mechanical properties of the AAA-Gr/Al interface structure. Simulated and experimental values are not directly compared due to fundamental differences in loading modes and failure mechanisms. In-plane tensile simulations are dominated by the intrinsic strength of graphene, whereas experiments typically show interface failure under out-of-plane stress caused by weak bonding. Consequently, this study focuses on atomic-scale mechanisms of in-plane reinforcement, providing theoretical insight into graphene’s contribution under idealized loading conditions.

Stress-strain curves of the four Gr/Al interface structures under (a) biaxial, (b) x-direction, and (c) y-direction stretching. (The dotted lines indicate the strains corresponding to the elastic stage, ultimate strength and failure stage).
The ideal strength of the surface/interface structures
As shown in Fig. 3, the ideal strengths of both Gr(0001) surface structures and Gr/Al interface structures exhibit significant anisotropy. From Fig. 3a, for the A-Gr, AA-G, and AAA-Gr surface structures, no matter how many layers the structure has, the ideal strengths are in the same order in descending, that is σx > σbiaxial > σy. The highest ideal strength occurs when stretching along the x-armchair direction. The results of the Gr(0001) surface structures along x- and y-directions are consistent well with Refs22,42,43. In contrast, for the V-Gr surface structure with a vacancy defect, the ideal strength follows the order: σy > σx > σbiaxial, indicating that the presence of vacancies influences the anisotropic mechanical response of graphene. When comparing ideal strengths in different directions, it can be found that the ideal strengths of different Gr(0001) surface structures decrease in the same order of σA−Gr > σV−Gr > σAA−Gr > σAAA−Gr. This indicates that, for pristine graphene structures, the ideal strength decreases with the increasing of layer numbers. Notably, the ideal strength of defective single-layer graphene exceeds that of multi-layer graphene, which can be attributed to the fact that interlayer interactions in graphene are primarily governed by weak van der Waals forces, whose effect diminishes with the increase of layers.
As shown in Fig. 3b, the anisotropy of ideal strength of Gr/Al interface structures is not as significant as Gr(0001) surface structures, especially for A-Gr/Al interface structure. It can be seen that for the ideal strength of all the same Gr/Al interface structures, the lowest strength is under the biaxial stretching. For Gr/Al interface structures except V-Gr/Al, the rank of ideal strength along different tensile directions from high to low is: σx > σy > σbiaxial. For different Gr/Al interface structures, under the same tensile direction, the ideal strengths of the A-Gr/Al, AA-Gr/Al and AAA-Gr/Al interface structures increase with the graphene layers, while the V-Gr/Al interface structure exhibits the lowest ideal strength. The existence of vacancy in the Gr/Al greatly reduces the strength of the Gr/Al interface structure.
The ideal tensile strengths of the Al(111) are 1.74 N/m, 1.68 N/m, 1.04 N/m along x, y, and biaxal directions, respectively. Compared with the strengths of the Al(111) matrix, the ideal strengths of all the perfect Gr/Al interface structures are much higher than those of the matrix Al, improved by 167.82% ~ 278.85%. However, for the V-Gr/Al interface structure, the ideal strength is weakened in all directions, slightly weakened in x and y directions and along biaxal direction and significantly weakened in biaxal direction. Among the four interface structures, the AAA-Gr/Al interface structure exhibits the highest ideal strength and the enhancing effect for the matrix Al is the most significant. Therefore, the AAA-Gr/Al interface structure is selected as the typical model for the strengthening mechanism investigation.

Ideal strength of (a) Gr(0001) and (b) Gr/Al structures under stretching in different directions.
Strengthening mechanism investigation of Gr/Al structures
Deformation evolution of the AAA-Gr/Al interface structure
To further understand the strengthening mechanism, the deformation evolution is explored at the microscopic level by analyzing the atomic position changes within the interface structure during the tensile process Fig. 4 shows the atomic structure diagram of the AAA-Gr/Al interface structure under different strains.
The parameters used for the discussions about the interlayer distance along the c-axis of the AAA-Gr/Al interface structure are defined in Fig. 4a. For example, the direct distance dC10−C12 between C10 and C12 atoms represents a measure of the distances between the second and third graphene layers, corresponding to the bond length. dAl13−Al17 is used to indirectly represent the interlayer distances within the aluminum matrix. For the discussions of interfacial interlayer distances between graphene and aluminum, dAl17−C1 and dAl17−C7 are utilized. The atomic distance within the ab plane is defined in Fig. 4b. To characterize the distance of carbon atoms in the Gr(0001) layer at the interface, dC1−C2, dC2−C7, and dC7−C8 are selected. Similarly, to describe the distance of aluminum atoms in the Al(111) layer at the interface, dAl16−Al17 and dAl17−Al18 are chosen.
Figure 4c ~ e presents the atomic structures at three specific strain points under biaxial, x-, and y-direction stretching, which corresponding to the representative elastic period, ultimate strength, fracture points in the stress-strain curves in Fig. 2. When stretched along the biaxal direction, Fig. 4c reveals that in the elastic deformation stage, the change of Al(111) structure is relatively uniform and the lattice constant and the interlayer distance increases. At the ultimate strength point, the lattice distortion of Al(111) structure occurs and the deformation increases with the distance of atoms from the interface. Meanwhile, both the in-layer and interlayer atomic distance of the Gr(0001) structure increase, the hexagonal honeycomb structure is still not destroyed. When fracture occurs, both Al and C atoms migrate toward the interface and correspondingly the interlayer distance of Al-Gr decreases. Severe lattice distortion appears in the Al(111) and Gr(0001) structures. Severe distortion of the Gr(0001) structure leads to the breaking of σ-bond. Similar deformation behaviors in x and y directions are observed, as shown in Fig. 4d ~ e. It can be drawn that, during the elastic deformation stage, the deformation resistance of the Gr/Al interface structure is primarily governed by the distortion of the Al(111) matrix, while in the plastic deformation stage, the resistance is mainly provided by the distortion of the Gr(0001) structure. During the fracture stage, the breaking of σ-bonds in graphene leads to a sharp decrease in the deformation resistance of the Gr/Al interface, resulting in the loss of load-carrying capacity. The Gr(0001) structure plays a dominant role in resisting plastic deformation through lattice distortion so as to promote the strength of Al matrix.

Atomic configuration diagrams of the AAA-Gr/Al interface structure: (a) side view, (b) top view, and stretching deformation evolution along (c) biaxial, (d) x-direction, and (e) y-direction.
Table 3 presents the bond length parameters of the AAA-Gr/Al interface structure under different strain conditions. For the in-plane spacing between Al atoms in the interfacial Al(111) layer, dAl16−Al17 and dAl17−Al18 exhibit the highest obvious bond length increment under biaxial stretching, followed by the x-direction, while the bond length along the y-direction remains relatively stable with minimal deformation. For the interlayer spacing dAl13−Al17 between Al atoms in the Al(111) matrix, no clear trend in bond length increment is observed in different tensile directions.
In the interfacial Gr(0001) layer, for the in-plane bond length between C atoms, the C1-C7 bond aligns with the x-axis. Consequently, under x-direction stretching, the dC1−C7 bond length continuously increases. Under biaxial stretching, the increase in dC1−C7 bond length is less pronounced than that in the x-direction, while under y-direction stretching, the dC1−C7 bond length remains nearly unchanged. Similarly, the C2-C7 and C7-C8 bonds are aligned with the C-C bonds in Gr(0001), and have an initial length of 1.42 Å. Under x-direction stretching, both dC2−C7 and dC7−C8 bond lengths increase; under biaxial stretching, the increase of bond length is less pronounced than that in the x-direction; while under y-direction stretching, the bond lengths remain nearly unchanged. According to44 that when the C-C distance exceeds 1.5 ~ 1.7 Å, the C-C bond can be considered breaking. In this paper, 1.7 Å is taken to assess if the C-C bond is broken. Therefore, when stretched along the x direction, the C-C bond of the Gr(0001) structure is broken during experiencing low strain. When stretched along y direction, the bond length of Gr(0001) structure is almost constant and the deformation resistance is strong, which makes the interface structure present relatively high toughness compared with the x-direction stretching. When stretched along the biaxial direction, the C-C bond fracture of Gr(0001) structure is slower than that in the x direction, and the strain range is larger. For the interlayer C atom spacing of Gr(0001) layers, when stretched along different directions, dC8−C10 and dC10−C12 both increase, with the amount of increase in the order of the biaxial, x- and y-directions from high to low. This indicates that the Gr layers tend to be stripped off during the stretching process and the van der Waals effect between layers is weak. According to these results, the mechanical properties the biaxial tensile direction is representative, further analysis of the interaction between atoms in the interface structure in this direction is performed to reveal the strengthening mechanism.
For the spacing of two adjacent Al atom and C atom at the interface of the Gr/Al structures, when stretched along different directions, dAl17−C1 and dAl17−C7 increase, with the amount of increase in the order of the x-, biaxial and y-directions from high to low. Although the bond length increases, the Gr structure at the interface still maintain the complete hexagonal honeycomb structure. This indicates that during the deformation process, the increasing of the C-C bond length and the layer spacing between each Gr layer can provide the stronger deformation resistance, so that the ideal strength of the interface structure is enhanced.
The electronic properties of the AAA-Gr/Al interface structure
Figure 5 presents the charge density difference of the AAA-Gr/Al interface structure under biaxial stretching at various strain levels. The three-dimensional charge density difference is visualized using isosurface set at 0.01 e/Bohr3, where yellow signifies electron accumulation and blue indicates electron depletion. In addition, two-dimensional charge density difference maps are plotted along the (001) planes containing the C1 and Al17 atoms to analyze charge redistribution at the atomic level.
In terms of Fig. 5, it is evident that as the Gr/Al interface structure undergoes biaxial stretching, the electronic properties of the Gr(0001) structure progressively transition from localization to delocalization with the increase of the strain. Moreover, the interaction between the Gr(0001) and Al(111) structures intensifies. Due to the physical adsorption between Gr(0001) and Al(111) structures and the van der Waals interaction between layers, no significant charge transfer was observed.
The observation of the cleavage surface of atom C1 reveals that as the strain increases, atom C1 experiences electron accumulation at the strain of 0.12, resulting in obvious electron localization. However, at the strain of 0.16, electron depletion occurs, leading to electron delocalization. Furthermore, at the strain of 0.18, the electron distribution among C atoms in the Gr(0001) structure is no longer uniform, thereby weakening the strong σ bonds. Similarly, analysis of the Al17 atomic cleavage surface indicates that with the increase of the strain, the Al17 atom undergoes electron depletion, leading to significant electron delocalization. Notably, lattice distortion in the Al(111) structure emerges since the elastic stage at the strain of 0.12, which can be further corroborated by the chaotic charge distribution around the Al17 atom.

Charge density difference diagrams of the AAA-Gr/Al interface structure under biaxial stretching at strain values of (a) 0, (b) 0.12, (c) 0.16, and (d) 0.18.
To provide insights into the electronic properties of the interface structure during the stretching process, Fig. 6 presents the total density of states (DOS) of the AAA-Gr/Al interface structure under different strains during biaxial stretching including the DOS of the AAA-Gr/Al interface (marked as Total), the 3-layer Gr(0001) structure (marked as C all), the 6-layer Al(111) structure (marked as Al all), Al17, C1, and C7 atoms at the interface. It is revealed that the DOS diagram presents a symmetric state near the Fermi level, indicating that the applied strain does not affect the magnetic properties of the interface structure.
Atomic orbitals describe the spatial distribution of electrons around the nucleus and are fundamental to understanding the electronic structure of atoms. Among them, the s orbital is spherically symmetric and has no directional preference, while p orbitals are dumbbell-shaped and oriented along specific Cartesian axes, denoted as pₓ, pγ, and p𝓏, corresponding to the x-, y-, and z-directions, respectively. These orbitals form the basis for constructing the electronic wavefunctions and play a crucial role in determining the bonding characteristics and charge distribution at the atomic scale. The DOS peaks of the Gr(0001) structure occur at −2 eV and 2 eV, primarily contributed by the pz orbitals. With the increase of the strain, the energy position of the highest peak gradually shifts towards 0 eV, that is, the pseudogap reduces and the bonding weakens. At the strain of 0.18, significant deformation in the Gr(0001) structure diminishes the influence of pz orbitals, making their strength comparable to that of the secondary peak associated with px orbitals. The DOS variations of C1 and C7 atoms at the interface follow a similar trend to that of the Gr(0001) structure.
For the Al(111) structure, the DOS peak appears at −5 eV, primarily contributed by the s orbitals. Additionally, a resonance peak with the px orbitals of C occurs at this position, indicating a weak hybridization by the low peak intensity. Different from that of the Al(111) structure, the DOS peak of the Al17 atom appears at −6 eV. However, with the increase of the strain, the DOS peak of the Al17 gradually shifts towards − 5 eV.
The DOS profile of the AAA-Gr/Al interface structure is predominantly influenced by the Gr(0001) structure, exhibiting two main peaks and four secondary peaks, in which the two main peaks and two of the secondary peaks primarily originate from the pz orbitals of graphene, while the remaining two secondary peaks are mainly influenced by the px orbitals. At the strain of 0.18, the deformation in both the Gr(0001) and Al(111) structures leads to a reduction in the total DOS of the AAA-Gr/Al interface structure.
In conclusion, during the stretching process, the pz orbitals of Gr play a dominant role in resisting deformation. Meanwhile, the weak hybridization between the px orbitals of Gr and the s orbitals of Al also contributes to deformation resistance. These two factors collectively enhancing the mechanical properties of the Gr/Al interface structure.

Density of states diagrams of the AAA-Gr/Al interface structure under biaxial stretching at strain values of (a) 0, (b) 0.12, (c) 0.16, and (d) 0.18.
Conclusion
This study employs first-principles calculations to investigate the ideal strength of four different interface structures (A-Gr/Al, AA-Gr/Al, AAA-Gr/Al, and V-Gr/Al) under biaxial and uniaxial deformation and the enhancement mechanism of AAA-Gr/Al interface structure is revealed by the analysis of the deformation evolution and electronic properties. The main conclusions are as follows.
(1) The calculated basic properties of the A-Gr/Al, AA-Gr/Al, and AAA-Gr/Al interface structures reveal that the number of graphene layers has nearly no influence on the lattice constants, the interlayer spacing and the interface binding energy of the materials. All the interface binding energy of the interface structures is below 0.1 eV, which suggests a weak physical adsorption process, characterized by van der Waals forces and easy desorption. Introducing vacancies into the graphene structure of the V-Gr/Al interface results in notable changes such as larger lattice constants, smaller interlayer spacing, and higher interface binding energy. These changes will further introduce the corresponding lattice distortion and then lead to an increasing work function and a decreasing Fermi level.
(2) With the increasing of graphene layers, the ideal strength of the four Gr/Al interface structures gradually increases, with the V-Gr/Al interface structure exhibiting the lowest ideal strength. The A-Gr/Al, AA-Gr/Al, and AAA-Gr/Al interface structures shows similar stress-strain relationships and the ideal strength ranking from high to low is σx > σy > σbiaxial when subjected to deformation along different directions.
(3) During stretching, the deformation evolution of the AAA-Gr/Al interface structure reveals that the elastic deformation stage is primarily resisted by the Al(111) structure, while the plastic deformation stage is mainly resisted by the Gr(0001) structure. Fracture initiates accompanying with the breaking of C-C bonds in the graphene layer, leading to severe distortion at the interface.
(4) Computational analyses of ideal strength are carried out for a series of multilayer Gr/Al structures. The strengthening mechanism of the AAA-Gr/Al interface is elucidated at the atomic orbital level. A qualitative correlation between interface adhesion and ideal strength is established. The study focused on in-plane tensile loading parallel to the interface, under which the structural integrity of graphene plays a decisive role in governing the interface mechanical performance. During biaxial stretching, the electronic properties of the AAA-Gr/Al interface structure indicate that the pz orbitals of graphene are pivotal in resisting deformation. In addition, the weak hybridization between the px orbitals of graphene and the s orbitals of aluminum also contributes to the enhancement of the mechanical properties of the Gr/Al interface structure. The present study focuses on the single-vacancy model, and for multiple-vacancy and other complex defects the strengthening mechanisms still need further detailed investigations.
Data availability
All data generated or analyzed during this study are included in this published article. Additional data are available from the corresponding author upon reasonable request.
References
Novoselov, K. S. & Geim, A. K. S.V. Morozov, ect, Electric Field Effect in Atomically Thin Carbon Films, science 10 (2004). https://doi.org/10.1126/science.1102896
Geim, A. & Novoselov, K. The rise of graphene. Nat. Mater. 6, 183–191. https://doi.org/10.1038/nmat1849 (2007).
Li, M. et al. Formation of multilayer interfaces and the load transfer in graphene nanoplatelets reinforced al matrix composites. Mater. Charact. 159 https://doi.org/10.1016/j.matchar.2019.110018 (2020).
Chak, V., Chattopadhyay, H. & Dora, T. L. Application of solid processing routes for the synthesis of graphene-aluminum composites- a review. Mater. Manuf. Processes. 36 (11), 1219–1235. https://doi.org/10.1080/10426914.2021.1914845 (2021).
Wang, J. et al. Reinforcement with graphene nanosheets in aluminum matrix composites. Scripta Mater. 66 (8), 594–597. https://doi.org/10.1016/j.scriptamat.2012.01.012 (2012).
Zhou, W. et al. Creation of individual few-layer graphene incorporated in an aluminum matrix. Compos. Part A: Appl. Sci. Manufac. 112, 168–177. https://doi.org/10.1016/j.compositesa.2018.06.008 (2018).
Zhang, H., Zhang, B., Gao, Q., Song, J. & Han, G. A review on microstructures and properties of graphene-reinforced aluminum matrix composites fabricated by friction stir processing. J. Manuf. Process. 68, 126–135. https://doi.org/10.1016/j.jmapro.2021.07.023 (2021).
Ju, B. et al. Effect of interfacial microstructure on the mechanical properties of GNPs/Al composites. Carbon 162, 346–355. https://doi.org/10.1016/j.carbon.2020.02.069 (2020).
Li, Z. et al. Enhanced mechanical properties of graphene (Reduced graphene Oxide)/Aluminum composites with a bioinspired nanolaminated structure. Nano Lett. 15 (12), 8077–8083. https://doi.org/10.1021/acs.nanolett.5b03492 (2015).
Shin, S. E., Choi, H. J., Shin, J. H. & Bae, D. H. Strengthening behavior of few-layered graphene/aluminum composites. Carbon 82, 143–151. https://doi.org/10.1016/j.carbon.2014.10.044 (2015).
Xie, Y., Meng, X., Huang, Y., Li, J. & Cao, J. Deformation-driven metallurgy of graphene nanoplatelets reinforced aluminum composite for the balance between strength and ductility. Compos. Part. B: Eng. 177 https://doi.org/10.1016/j.compositesb.2019.107413 (2019).
Gusmão, M. S., Ghosh, A. & Frota, H. O. Electronic transport properties of graphene/Al2O3 (0001) interface. Curr. Appl. Phys. 18 (1), 90–95. https://doi.org/10.1016/j.cap.2017.10.008 (2018).
Liu, P., Xie, J., Wang, A., Ma, D. & Mao, Z. First-principles prediction of enhancing graphene/Al interface bonding strength by graphene doping strategy. Appl. Surf. Sci. 517 https://doi.org/10.1016/j.apsusc.2020.146040 (2020).
Qi, Y., Hector, L. G., Ooi, N. & Adams, J. B. A first principles study of adhesion and adhesive transfer at Al(111)/graphite(0001). Surf. Sci. 581 (2–3), 155–168. https://doi.org/10.1016/j.susc.2005.02.048 (2005).
Wang, Y. et al. Preferred orientation at the Al/graphene interface: First-principles calculations and experimental observation. J. Alloys Compd. 900 https://doi.org/10.1016/j.jallcom.2021.163304 (2022).
Qiu, C. et al. Structural modelling and mechanical behaviors of graphene/carbon nanotubes reinforced metal matrix composites via atomic-scale simulations: A review. Compos. Part. C: Open. Access. 4 https://doi.org/10.1016/j.jcomc.2021.100120 (2021).
Zhang, X. & Wang, S. Interfacial strengthening of Graphene/Aluminum composites through point defects: A First-Principles study. Nanomaterials (Basel). 11 (3). https://doi.org/10.3390/nano11030738 (2021).
Restuccia, P. et al. Ideal adhesive and shear strengths of solid interfaces: A high throughput Ab initio approach. Comput. Mater. Sci. 154, 517–529. https://doi.org/10.1016/j.commatsci.2018.08.006 (2018).
Zhao, M. et al. Lateral size effect of graphene on mechanical properties of aluminum matrix nanolaminated composites. Scripta Mater. 139, 44–48. https://doi.org/10.1016/j.scriptamat.2017.06.018 (2017).
Cooper, R. C. et al. Nonlinear elastic behavior of two-dimensional molybdenum disulfide. Phys. Rev. B. 87 (3). https://doi.org/10.1103/PhysRevB.87.035423 (2013).
Pan, H. et al. Unusual mechanical and electronic behaviors of bulk layered hydrogen substituted Graphdiyne under biaxial strain. Appl. Surf. Sci. 513 https://doi.org/10.1016/j.apsusc.2020.145694 (2020).
Fu, Z. et al. Mechanistic quantification of thermodynamic stability and mechanical strength for Two-Dimensional Transition-Metal carbides. J. Phys. Chem. C. 122 (8), 4710–4722. https://doi.org/10.1021/acs.jpcc.8b00142 (2018).
Zhou, S., Han, J., Dai, S., Sun, J. & van der Srolovitz, D. J. Waals bilayer energetics: generalized stacking-fault energy of graphene, Boron nitride, and graphene/boron nitride bilayers. Phys. Rev. B. 92 (15). https://doi.org/10.1103/PhysRevB.92.155438 (2015).
Fu, Z. H. et al. Stabilization and strengthening effects of functional groups in two-dimensional titanium carbide. Phys. Rev. B. 94 (10). https://doi.org/10.1103/PhysRevB.94.104103 (2016).
Nazarov, R., Hickel, T. & Neugebauer, J. Vacancy formation energies in Fcc metals: influence of exchange-correlation functionals and correction schemes. Phys. Rev. B. 85 (14). https://doi.org/10.1103/PhysRevB.85.144118 (2012).
John, K. B., Perdew, P., Ernzerhof, M. & Simple, G. G. A. M. Phys. Rev. Lett. 78, 1396 https://doi.org/10.1103/PhysRevLett.77.3865 (1997).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate Ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132 (15), 154104. https://doi.org/10.1063/1.3382344 (2010).
Yang, H. G. et al. Anatase TiO2 single crystals with a large percentage of reactive facets. Nature 453 (7195), 638–641. https://doi.org/10.1038/nature06964 (2008).
Chunshan He. Work function of Fe2O3: a DFT calculation. Mater. Sci. https://doi.org/10.48550/arXiv.1709.04672 (2017).
Vitos a, L., Ruban a, A. V. & Skriver a, H. L. Kollár b. The surface energy of metals. Surf. Sci. 411, 1–2. https://doi.org/10.1016/S0039-6028(98)00363-X (1998).
Eastment, R. M. & Mee, C. H. B. Work function measurements on (100), (110) and (111) surfaces of aluminium. Journal of Physics F: Metal Physics. 3 1738. https://iopscience.iop.org/article/ (1973). https://doi.org/10.1088/0305-4608/3/9/016
Electron Work Function of The Elements. Washington State University. https://public.archive.wsu.edu/pchemlab/public_html/documents/Work-functionvalues.pdf
Huang, J. et al. A systematic study of interface properties and fracture behavior of graphene/aluminum: insights from a first-principles study. Vacuum 204, 111346. https://doi.org/10.1016/j.vacuum.2022.111346 (2022).
Polatoglou, H. M., Methfessel, M. & Scheffler, M. Vacancy-formation energies at the (111) surface and in bulk Al, Cu, Ag, and Rh. Phys. Rev. B Condens. Matter. 48 (3), 1877–1883. https://doi.org/10.1103/physrevb.48.1877 (1993).
Li, D., Gao, S., Li, L. & Yang, P. Improvement ofthe performance of graphene/Al(111) interface with defect mode and doped mode. Comput. Theor. Chem. 1217, 113931. https://doi.org/10.1016/j.comptc.2022.113931 (2022).
David, R. & Lide CRC Handbook of Chemistry and Physics. Former Director, Standard Reference Data National Institute of Standards and Technology. (2003).
Gföhler, M., Loicht, M. & Lugner, P. Exercise tricycle for paraplegics. Med. Biol. Eng. Comput. 36, 118–121. https://doi.org/10.1007/BF02522868 (1998).
Ooi, N., Rairkar, A. & Adams, J. B. Density functional study of graphite bulk and surface properties. Carbon 44 (2), 231–242. https://doi.org/10.1016/j.carbon.2005.07.036 (2006).
Gong, C. et al. First-principles study of metal–graphene interfaces. J. Appl. Phys. 108 (12). https://doi.org/10.1063/1.3524232 (2010).
Dou, Y. H., Bai, Q., Guo, W., Guo, Y. & Du, Y. Wettability of graphene coated on aluminum substrate with microstructure modification. Curr. Nanosci. 19 (2), 270–278. https://doi.org/10.2174/1573413718666220428114115 (2023).
Chen, Y. et al. The interface properties of defective graphene on aluminium: A first-principles calculation. Comput. Mater. Sci. 188, 110157. https://doi.org/10.1016/j.commatsci.2020.110157 (2021).
Liu, F., Ming, P. & Li, J. Ab initiocalculation of ideal strength and phonon instability of graphene under tension. Phys. Rev. B. 76 (6). https://doi.org/10.1103/PhysRevB.76.064120 (2007).
Wei, X., Fragneaud, B., Marianetti, C. A. & Kysar, J. W. Nonlinear elastic behavior of graphene:ab initiocalculations to continuum description. Phys. Rev. B. 80 (20). https://doi.org/10.1103/PhysRevB.80.205407 (2009).
Wang, S. et al. Mechanical properties and failure mechanisms of graphene under a central load. Chemphyschem 15 (13), 2749–2755. https://doi.org/10.1002/cphc.201402258 (2014).
Acknowledgements
The authors would like to thank the supports of the National Natural Science Foundation of China (Grant Nos. 52475169 and 52371014).
Funding
National Natural Science Foundation of China under Grant Nos. 52475169 and 52371014.
Author information
Authors and Affiliations
Contributions
Wei Wang and Lijie Chen contributed to the conceptual design, data curation, formal analysis, funding acquisition, investigation, methodology development, project administration, software application, supervision, validation, visualization, and manuscript preparation. Tieqiang Gang contributed equally to the study and was involved in all aspects of the research and manuscript preparation. Can Cui, Fangfang Xia, and Weiwei Xu provided administrative support and contributed to resource coordination during the research process. All authors discussed the results and commented on the manuscript at all stages. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, W., Cui, C., Xia, F. et al. A first principles investigation into the mechanical properties and the strengthening mechanism of the graphene/aluminum interface structure. Sci Rep 15, 38571 (2025). https://doi.org/10.1038/s41598-025-22443-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-22443-y
