
Abstract
The genus Bletilla Rchb.f., an endangered taxon of Orchidaceae Juss. with significant medicinal value, faces persistent controversies in taxonomic identification and phylogenetic relationships. In this study, we analyzed a dataset of 57 complete chloroplast genomes (CPGs) from five subfamilies of Orchidaceae, including five newly sequenced Bletilla CPGs and publicly available data. These CPGs were combined with corresponding nuclear ribosomal internal transcribed spacer (nrITS) sequences obtained from public databases to resolve taxonomic uncertainties and phylogenetic relationships within Bletilla. Our results revealed significant intraspecific variation in Bletilla CPGs with overlapping intraspecific and interspecific genetic distances (GDs), alongside non-monophyletic clades of B. striata, B. ochracea, and B. formosana in species-level phylogenies. Consequently, the CPG ‘super barcodes’—proposed as high-resolution markers for plant identification—demonstrated limited discriminatory power in Bletilla, challenging their universal reliability. Phylogenetic analysis revealed conflicting placements between Bletilla and B. sinensis in CPG and nrITS trees: while nrITS data supported a monophyletic Bletilla within the subtribe Coelogyninae Benth., CPG data placed B. sinensis outside Bletilla, forming a sister clade with Calanthe triplicata and Tainia dunnii. Collectively, this study provides new evidence for the taxonomic identification and phylogenetic relationships of Bletilla through chloroplast-nuclear genome integration, offering a preliminary framework for taxonomic re-evaluation of complex plant groups.
Similar content being viewed by others


Introduction
The genus Bletilla, classified within the subfamily Epidendroideae Kostel., tribe Arethuseae Lindl., and subtribe Coelogyninae, is a taxon of significant medicinal and economic value in the family Orchidaceae. This genus includes six recognized species, four of which are native to China: B. striata (Thunb.) H. G. Reichenbach, B. ochracea Schltr., B. formosana (Hayata) Schltr., and B. sinensis (Rolf) Schltr. These species are distributed across multiple provinces in the southwestern, central, and southern regions of China, particularly in Guizhou, Sichuan, Yunnan, and Tibet1. B. striata is a medicinal herb officially listed in the Chinese Pharmacopoeia, renowned for its diverse bioactive compounds. These include dihydrophenanthrene derivatives, polysaccharides, and 2-isobutyl malic acid, which contribute to its pharmacological properties2,3. It demonstrates many pharmacological properties, including antibacterial, anti-inflammatory, and tumor-inhibitory activities4,5,6,7. However, over-excavation of wild B. striata has resulted in its categorization as a critically endangered species. Therefore, resolving the phylogenetic relationships within Bletilla is crucial for its effective conservation, taxonomic clarity, and sustainable utilization.
The taxonomic classification and phylogenetic relationships of Bletilla have long been debated, primarily due to morphological similarities with Arethusa L. and Bletia Ruiz & Pav.8,9,10,11,12,13,14. This has resulted in inconsistent placements across different taxonomic systems. Recently, van den Berg et al.15 and subsequent studies16,17 assigned Bletilla to subtribe Coelogyninae (tribe Arethuseae), which is currently the most widely accepted classification. In addition to these taxonomic difficulties, several studies have found it challenging to determine the monophyly as well as the systematic position of B. sinensis. While some studies support the inclusion of B. sinensis within Bletilla using DNA barcode fragments and CPG sequences18,19, and confirm the monophyly of Bletilla within Arethuseae, others propose segregating B. sinensis from Bletilla into a new monotypic genus Mengzia S. C. Chen & J. W. Zhai based on CPG markers and morphological evidence20. Further, Han et al.21 found B. sinensis clustered separately at the base of the phylogenetic tree, distinct from other Bletilla species. These unresolved conflicts, attributable to limited taxon sampling and insufficient nuclear gene integration, impede coherent taxonomic treatment and conservation of this endangered medicinal complex. To clarify the phylogenetic and evolutionary relationships within Bletilla, it is essential to expand the sampling range and conduct more comprehensive analyses of genetic markers. To resolve these conflicts, this study aimed to: (1) sequence, assemble, and annotate the CPGs of three Bletilla species; (2) systematically investigate the phylogenetic relationships within Bletilla by integrating CPG and nrITS sequences; (3) evaluate the efficiency of CPG ‘super barcodes’ for species identification in Bletilla, providing a reliable basis for taxonomic research and conservation of Bletilla.
Due to extensive morphological variation in Bletilla, key diagnostic traits1,22,23—including petal coloration, labellum pattern, and leaf morphology—overlap across species, resulting in ambiguous delimitation and challenging species identification. Wu et al.24 accurately identified four Bletilla species using the nuclear ribosomal internal transcribed spacer (nrITS) and ycf1 sequences. However, as sampling expanded, Huang et al.20 found that short standard DNA barcodes were inadequate for effective discrimination, necessitating higher resolution methods to resolve interspecific phylogenetic relationships and species identification of Bletilla.
In recent years, rising market demand for B. striata has driven year-on-year price increases. Consequently, various adulterants of B. striata, which are difficult for ordinary consumers to recognize due to their similar appearance and traits, have appeared in the Chinese herbal medicine market25. Cremastra appendiculata, Pleione bulbocodioides, and Pleione formosana, are are commonly misused as substitutes26,27. The use of adulterated products has compromised the safety and efficacy of B. striata medication. The CPG sequences, also known as ‘super barcodes’, characterized by structural simplicity, maternal inheritance, and hypervariable loci, overcome single-gene barcode limitations28,29. CPG data provide sufficient variation with higher identification accuracy30,31 and have thus been successfully applied for phylogenetics and species discrimination in complex genera like Aconitum L.32, Seriphidium (Besser) Fourr.33, and Zingiber Mill.34. This motivates our use of CPG barcoding to resolve Bletilla taxonomic conflicts.
Results
CPG sequencing, assembly, and gene types
The CPGs of B. striata, B. ochracea, and B. formosana exhibited typical circular structures, ranging in size from 159,418 to 160,052 bp (Table 1). Each CPG had conservative quadrilateral structures, containing a pair of inverted repeats IRa and IRb (25,984–26,780 bp), a large single-copy region (LSC) (87,058–87,620 bp), and a small single-copy region (SSC) (18,768–20,464 bp). The overall GC content of the CPGs was approximately in the range of 37.16–37.23%.
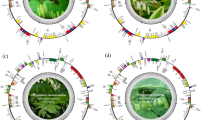
Genetic annotation of the assembled CPGs revealed a total of 127 annotated genes, including 83 protein-coding genes, 36 tRNA genes, and 8 rRNA genes (Fig. 1 and Supplementary Table S1). Similar to other angiosperms, such as Phalaenopsis stobartiana35, Polystachya concreta36, and Physalis angulata37, the Bletilla CPGs contained 17 intron-containing genes. These included five tRNA genes (trnV-UAC, trnK-UUU, trnL-UAA, trnE-UUC, trnA-UGC) and 12 protein-coding genes (rps16, atpF, rpoC1, petB, petD, rpl16, rps12, rpl2, ndhB, ndhA, ycf3, clpP). Among these, rps12, ycf3, and clpP each contained two introns. The rps12 gene exhibited a unique trans-splicing structure, with its 5′ and 3′ ends located in the LSC and IR regions, respectively, forming two independent transcription units. The CPGs of B. striata, B. ochracea, and B. formosana showed structural similarities.

The chloroplast genome map of Bletilla striata, B. ochracea, and B. formosana. The genes colored according to the functional group are represented in the outermost circle, the genes external to the circumference are transcribed in a counterclockwise direction, and the internal genes are transcribed in a clockwise direction.
Sequence repeats and Codon preference analysis
The simple sequence repeats (SSRs) were found in the different regions of the five samples. A total of 60, 56 and 65 of the SSRs were identified in the B. ochracea, B. formosana and B. striata CPGs, respectively. The most abundant mononucleotide repeat was A/T, which represents approximately 66.07% to 70.77% of the total number. The SSRs (T)13, (T)16, (ATAG)4, (AT)9, and (AGTATA)3 were unique to B. ochracea. (AT)7, (AAGA)3, and (CTATAT)3 were found only in B. striata. (A)13 were lost in B. formosana (Fig. 2a). These specific SSRs provide valuable information for Bletilla taxonomy.

The number of simple sequence repeats (SSRs) and relative synonymous codon usage (RSCU) of amino acids in B. striata, B. ochracea, and B. formosana. (a) Classified SSR repeat units, (b) Amino acid usage frequency calculated by RSCU.
Relative synonymous codon usage (RSCU) was used to evaluate the codon usage frequency. The RSCU value of each codon was nearly identical, suggesting that the codon preferences among the five samples were similar. Met and Trp were the only two codons exhibiting no bias (RSCU = 1.00) in the five CPGs. Arg, Leu, and Ser were the most frequent amino acids (Fig. 2b). There were 30 codons (one stop codon) with an RSCU > 1.00, indicating that the usage frequency of these codons was greater than that of other synonymous codons. Among these codons, 29 ended with the A/U base, suggesting that high-frequency codons tend to end in A/U bases.
CPG sequences divergence
To assess sequence divergence within the genus Bletilla, we investigated the global comparison of the 13 Bletilla CPGs using mVISTA software, with B. striata (accessions NC_028422) as the reference. The alignment revealed over 25 regions with less than 50% sequence identity, indicating substantial interspecific variation and genetic differentiation, particularly in non-coding regions (Fig. 3). Furthermore, significant intraspecific variations were observed among the CPGs of B. striata, B. ochracea, and B. formosana. The mVISTA plot revealed some regions indicating mutations or deletions, such as trnS-trnG, psbM-trnD, trnL-ndhJ, accD-psaI, and psbE-petG in B. striata. The regions of rps16-trnK, pbsI-trnG, psbM-trnD, psbB-psbT, and ndhF-rpl32 also showed low similarity in B. ochracea.

Global comparison of the Bletilla chloroplast genomes. Gray arrows, blue and red areas above the alignment indicate genes with their orientation, exon and noncoding region, respectively. The x-axis stands for sequence and the y-axis represents the percent identity ranging from 50 to 100%.
We used the sliding window in DnaSP to filter eight highly variable regions in the CPG, namely, rps16-trnQ, pbsI-trnG, psbM-trnD, trnE-trnT, accD-psaI, rps3-rpl16, ndhF-rpl32, and psaC-ndhE (Fig. 4). The average value of nucleotide diversity (π) among the Bletilla CPGs was 0.01193, with the psbM-trnD (π = 0.2450) and ndhF-rpl32 (π = 0.0899) variable regions exhibiting significantly higher π values compared to other regions (Supplementary Table S2). These highly variable regions may be potential molecular markers for the identification of Bletilla species.

Sliding window analysis of the Bletilla chloroplast genomes x-axis: position of the midpoint of a window; y-axis: nucleotide diversity of each window; Window length: 600 bp; step size: 200 bp.
Intraspecific and interspecific genetic distances overlap
Genetic distance analysis revealed substantial challenges for delineating species boundaries in Bletilla. While B. sinensis exhibited distant relationships to congeners (GDs > 0.022), we observed significant overlap between intraspecific and interspecific distances among B. striata, B. ochracea, and B. formosana (Table 2 and Supplementary Table S3). Critically, the maximum intraspecific GDs for these three species exceeded the minimum interspecific GDs. This was most notably observed in B. ochracea where the maximum intraspecific GDs (0.058476) surpassed the minimum interspecific GDs (0.000097). Such overlap indicates that genetic distance thresholds alone cannot reliably differentiate these taxa.
Cluster analysis and identification of Bletilla species based on CPG
Previous nrITS maximum likelihood (ML) phylogeny revealed non-monophyly of B. striata, B. ochracea, and B. formosana, indicating the nrITS data limited discriminatory power for Bletilla species identification (Supplementary Fig. S1). To assess the impact of expanded taxon sampling, we reconstructed a CPG-ML phylogeny using all 26 available Bletilla CPGs from GenBank alongside common adulterants (Fig. 5). All Bletilla (excluding B. sinensis) samples formed a strongly supported monophyletic clade (ML bootstrap support [MLBS] = 100); however, none of the species demonstrated monophyly at the species level. Notably, the nine B. striata samples were distributed across three distinct clades (MLBS = 100). This polyphyletic distribution pattern was similarly observed in both B. ochracea and B. formosana samples. The adulterant species, including P. chlorantha, C. appendiculata, P. bulbocodioides, P. formosana, and T. alba, were distinctly separated from the genus Bletilla clade (MLBS = 100), each forming independent branches on the phylogenetic tree.

Phylogenetic tree of the genus Bletilla and its adulterants based on chloroplast genomes using Maximum likelihood (ML) method. Numbers are ML bootstrap (MLBS) support.
Phylogenetic analyses of Bletilla based on CPG and nrITS
To gain a more comprehensive understanding of the evolutionary relationships within Bletilla, we utilized the CPG and the corresponding nrITS sequences from the Orchidaceae family for phylogenetic analyses. Our analysis incorporated newly sequenced Bletilla CPGs and 57 representative species from 11 subtribes of Orchidaceae, based on 58 protein-coding genes. Based on these two different types of sequences, the phylogenetic trees (the CPG-tree and the nrITS-tree) constructed using the ML and BI methods were largely congruent and exhibited similar topologies.
At the subfamily level, the clustering pattern of the five subfamilies of Orchidaceae exhibited a high degree of consistency. However, at the genus level, there was a discrepancy in the taxonomic position of Bletilla and B. sinensis. In the CPG-tree (Fig. 6a), Bletilla (excluding B. sinensis) was more closely related to Pleione, Thunia, and Pholidota (MLBS = 100, Bayesian posterior probability [PP] = 1.00), which were grouped together in the branch of the subtribe Coelogyninae and the tribe Arethuseae . B. sinensis formed a clade group with Calanthe triplicata and Tainia dunnii (MLBS = 100, PP = 1.00). This sister clade was positioned outside the tribe Arethuseae and was in close proximity to the tribe Collabieae. In contrast, in the nrITS-tree (Fig. 6b), Bletilla was monophyletic, with B. striata, B. ochracea, B. formosana, and B. sinensis clustered into one group and forming a sister clade with the branch of Thunia, Pleione, and Pholidota (MLBS = 100, PP = 0.99).

Phylogenetic tree based on the 57 Orchidaceae species using (a) 58 plastomic shared protein-coding genes, and (b) nuclear ribosomal internal transcribed spacer sequences. Numbers to the left of/are Maximum likelihood bootstrap (MLBS) support, and those to the right indicate the Bayesian posterior probabilities (PP).
Discussion
Genetic variation in plants, fundamental for adaptation and evolution, provides critical value for germplasm conservation and variety identification38,39. A substantial quantity of genetic variation exists within Bletilla CPGs. This phenomenon may arise from combined adaptive and neutral forces: the complexity of China’s ecological conditions and geographical patterns driving divergence, alongside high mutation rates, genetic drift, and hybridization. As a widely distributed wild plant, Bletilla species show distinct biogeographic patterns: B. striata, B. ochracea, and B. formosana inhabit diverse ecoregions across China (e.g., southwestern mountains, southeastern coasts, northwestern/southeastern plains), whereas B. sinensis shows a restricted endemic distribution40,41. Temperature gradients (ΔT = 14–26.2 °C), precipitation regimes (467–1,687 mm/yr), and soil types (e.g., Yellow vs. Cinnamon soil) have been identified as major drivers of B. striata ecotype differentiation42. These factors collectively could contribute to the formation of distinct ecotypes in Bletilla, which often harbor unique sequence information. For example, B. striata from Anhui and Zhejiang populations exhibit 3–5 nrITS single nucleotide polymorphisms (SNPs), while samples from Hunan and Jiangxi show unique mutation hotspots43. Future analyses of selection pressures are needed to evaluate the relative roles of adaptive versus neutral processes in shaping this diversity44,45.
The exceptionally high nucleotide diversity of Bletilla CPGs (π = 0.01193) exceeds that of related medicinal genera by 3- to tenfold (Epimedium L.: 0.0012146, Lycium L.: 0.0014747, Schisandra Michx.: 0.0036448), further indicating high genetic diversity within this genus. Chromosomal analyses further validated this result. Chromosome numbers of Bletilla from different provenances were different: B. striata 2n = 32, B. striata (safflower) mutant 2n = 34, and B. ochracea/ B. formosana 2n = 34–3649. These chromosomal characteristics correlate with labellum pigmentation and floral architecture, as exemplified by distinct traits in BJ1 (Yunnan) and BJ2 (Chongqing) accessions (Fig. 7). Crucially, in the B. striata species complex, the B. striata (safflower) mutant may have originated from natural hybridization between the B. ochracea and B. formosana. This mutant exhibits an intermediate chromosome number (2n = 34) and labellum patterning more similar to B. ochracea49. This hybrid genotype demonstrates how hybridization drives rapid trait innovation through gene exchange, expanding intraspecific variation beyond ecological adaptation alone.

Images of Bletilla striata, B. ochracea, and B. formosana. (a, d, g, j, m) The overall morphology of the plant. (b, e, h, k, n) The morphology of the flower. (c, f, i, l, o) The morphology of the fresh tuber.
Orchidaceae, the most species-rich family of angiosperms, comprises five subfamilies and over 28,000 species50. Extensive species diversity and complex morphological variation have resulted in ambiguous generic and specific boundaries, with persistent taxonomic uncertainties51,52. Although recent studies support separating B. sinensis from Bletilla20,21, its phylogenetic affinities remain unresolved. In our study, both CPG and nrITS phylogenetic trees classified Orchidaceae into five subfamilies, which was consistent with previous reports53,54. However, phylogeny trees showed differences between the monophyletic status of Bletilla and the taxonomic position of B. sinensis, indicating a significant nucleocytoplasmic conflict. Specifically, in nuclear gene (nrITS) phylogenies, B. sinensis clustered into a single branch with B. striata, B. ochracea, and B. formosana with high support, forming a monophyletic Bletilla group within the subtribe Coelogyninae. In contrast, CPG gphylogenies showed B. sinensis distantly related to Bletilla, instead forming a sister clade to the tribe Collabieae.
Despite differing phylogenetic placements of B. sinensis between our study and prior works20,21, both consistently support its separation from the genus Bletilla based specifically on CPG evidence. Methodologically, while previous analyses focused specifically on the Arethuseae tribe20,21, our expanded sampling across 11 subtribes of Orchidaceae (Fig. 6a) reveals novel affinities between B. sinensis and the tribe Collabieae. Thus, we speculate that compared to using selected the tribe Arethuseae samples, a broad taxonomic sampling range leads to changes the in phylogenetic topology and significant shifts in the position of B. sinensis. These findings collectively highlight the necessity for a taxonomic re-evaluation of B. sinensis. Based on the observations, we propose two non-exclusive hypotheses: B. sinensis either represents an independent lineage warranting taxonomic segregation or it should be retained in Bletilla but exhibits a hybrid genomic mosaic potentially derived from historical introgression between Bletilla and the tribe Collabieae ancestors. This speculation is consistent with the common phenomenon of nucleocytoplasmic conflicts caused by hybridization in Orchidaceae. To test these hypotheses, future studies should compare floral ontogeny, conduct D-statistic analyses of population admixture55, and perform reproductive isolation assays to clarify its taxonomic status. Moreover, irrespective of its final taxonomic classification, the endemic vulnerability and evolutionary distinctiveness of B. sinensis warrant its designation as a high conservation priority56. This approach aligns with conservation strategies emphasizing evolutionary distinctiveness.
Nucleocytoplasmic conflicts can arise through multiple mechanisms, with introgressive hybridization57 and chloroplast capture58,59 being common triggering factors. Such conflicts often manifest as gene tree discordance, particularly between maternally inherited CPG genes and biparentally inherited nuclear genes60,61. In angiosperms, limited interspecific nuclear gene exchange stands in sharp contrast to the maternal inheritance of CPG. This contrast not only facilitates chloroplast capture during hybridization but also generates hybrid genotypes. Notably, hybridization and gene introgression are pivotal for the formation of new genotypes. These mechanistic foundations lead us to hypothesize that hybridization and introgression may be the causes of the phylogenetic conflicts and difficulties in Bletilla species identification. First, hybridization-generated recombinant genotypes disrupt species monophyly, as evidenced by the non-monophyly of B. striata, B. ochracea, and B. formosana in phylogenetic trees. Second, chloroplast capture via introgression causes the discordant placement of B. sinensis. The CPG of B. sinensis was grouped with C. triplicata and T. dunnii (Fig. 6a), while the nrITS supported its phylogenetic affinity with Bletilla (Fig. 6b). Third, introgression widens intraspecific genetic variation, invalidating the ‘distance threshold’ method (Table 2). These processes collectively undermine conventional molecular delineation and prompt us to propose an integrative framework.
Furthermore, these pronounced genetic variations profoundly threaten species conservation and medicinal authentication for B. striata. CPG ‘super barcodes’ demonstrate efficacy in some taxa62,63, yet fail critically in Bletilla (Fig. 5, Supplementary Fig. S1). These failures mirror limitations in other taxa: CPG barcoding resolved only 60.00% of Lauraceae Juss. species64 and 27.27% of Schima Reinw. ex Blume species65. This limitation, compounded by overlapping intra-/inter-specific genetic distances and non-monophyly phylogeny relationships, prevents reliable species delineation. It also increases the adulteration risks from substitutes like C. appendiculata, thereby threatening medicinal safety and regional quality standards. To address this, we propose developing ecotype-specific SNP markers for geographic traceability66 and combining plastid hypervariable regions (psbM-trnD) with nrITS to overcome single-barcode limitations.
In conclusion, to achieve accurate phylogenetic inferences and species identification in Bletilla, it is essential to integrate multiple lines of evidence for a comprehensive re-evaluation. Consequently, an integrative framework is indispensable to reconcile discordant data types. It synergistically incorporates nuclear loci to resolve species boundaries obscured by chloroplast capture67, plastid hypervariable regions to detect hybridization signatures68, ecological niche data to identify hybrid zones and validate ecotype divergence69, and genome-wide markers to resolve recombinant genotypes while enabling the development of DNA signature and tissue-specific expression markers70,71. This comprehensive approach promises to enhance taxonomic resolution in Bletilla.
Conclusion
In this study, we assembled and annotated five novel CPGs of Bletilla (Orchidaceae), revealing conserved structural features alongside significant intraspecific variations. We identified eight hypervariable regions as potential molecular markers. The observed phylogenetic conflicts in the placement of Bletilla and B. sinensis, likely arising from introgressive hybridization, help explain the taxonomic controversies. The CPG ‘super barcodes’ show limited discriminatory power due to overlapping intraspecific distances and high genetic diversity, severely hindering reliable species identification. For practical applications, the evolutionary distinctiveness and unresolved taxonomy of B. sinensis necessitate its urgent designation as a high-priority conservation unit, while the failure of conventional barcodes severely threatens B. striata medicinal authentication, increasing adulteration risks. To address these, we propose an integrative framework combining nuclear loci, plastid hypervariable regions, ecological niche modeling, and whole-genome sequencing for developing ecotype-specific SNP markers for reliable herbal traceability and adulteration prevention. This framework enables the simultaneous protection of unique genetic diversity and safeguarding of medicinal product safety.
Materials and methods
Sample material and DNA extraction
We sequenced and analyzed five new CPGs from Bletilla species, comprising two B. striata, two B. ochracea, and one B. formosana. The plant materials of Bletilla species were collected from natural populations in Chongqing, Yunnan, Zhejiang, Shanxi, and Anhui provinces, China (Fig. 7), during the field investigation for ‘The Fourth National Survey of Chinese Material Medica Resources’ (Grant No. 201207002). All field collections were conducted under the supervision of and with prior permission from the local forestry departments, in strict compliance with relevant Chinese laws and ethical guidelines. The formal identification of the plant material was undertaken by Researcher Zhengyu Liu from the Chongqing Institute of Medicinal Plant Cultivation, based on examination of key morphological characteristics. Voucher specimens have been deposited in the Herbarium of the Chongqing Institute of Medicinal Plant Cultivation with the accession numbers CIMPC-RFM-20220401 to CIMPC-RFM-20220405 (see also Supplementary Table S4).
Before DNA extraction, healthy young leaves were surface-sterilized by wiping with 70% ethanol, followed by two to three rinses with sterile distilled water. Total genomic DNA was extracted using the plant genomic DNA kit (Tiangen Biotech, Beijing, China). DNA quality and concentration were evaluated using a NanoPhotometer® N60, with samples exhibiting an OD260/OD280 ratio between 1.8 and 2.0 considered high-quality. For each sample, the total amount of DNA was required to be no less than 0.5–1 µg. High-quality DNA samples were subsequently sent to Lingen Biotechnology Co., Ltd. (Shanghai, China) for sequencing and library preparation.
DNA sequencing and assembly annotation
The DNA library was constructed using the NEBNEXT® Ultra™ II DNA Library Prep Kit72, with an insertion fragment length of 350 bp. Sequencing was performed on the Illumina HiSeq X Ten platform (PE150) at Novogene (Beijing, China). Raw reads were processed using Trimmomatic73 to remove low-quality sequences and adapter contamination. The filtered reads (in FASTQ format) were used to assemble the CPG sequences by GetOrganelle74 (script: get_organelle_from_reads.py -1 R1.fq.gz -2 R2.fq.gz -o output -R 50 -k 21,45,65,85,105 -P 1000000 -F embplant_pt -t 50). The CPGAVAS275 was used for annotation, and a circular genome map was generated with OGDRAW76. The MISA77 was applied to predict simple sequence repeats (SSR) with parameter thresholds set at 1, 2, 3, 4, 5 and 6, and nucleotide parameters of 10, 5, 4, 3, 3 and 3. Relative synonymous codon usage (RSCU) was analyzed with codon W 1.4.2 (http://codonw.sourceforge.net/). The final annotated sequences were manually curated using Sequin software and deposited into the GenBank database (https://www.ncbi.nlm.nih.gov/).
Comparative genome analysis
The CPG of B. striata (accessions NC_028422) was used as a reference for comparative analysis. Full-length CPG sequences were compared using mVISTA78 with default parameters to identify structural variations and conserved regions. To assess nucleotide diversity and identify highly variable regions, the CPG sequences were first aligned using MAFFT v7.313. DNA polymorphism analysis was then performed with DnaSP v5.179 employing a sliding window approach (200 bp step size, 600 bp window length) based on the multiple sequence alignment (FASTA format). Genetic distances were calculated using the Kimura 2-parameter model in MEGA v11.0.
Phylogenetic analysis of Bletilla and B. sinensis
To resolve the phylogenetic controversies surrounding Bletilla, we retrieved the CPG sequences and corresponding nrITS sequences of 57 Orchidaceae species from the GenBank database, with Marchantia polymorpha selected as the outgroup. A total of 58 shared protein-coding genes were extracted from these CPGs. These homologous regions were aligned using MAFFT v7.313 (script: mafft --auto ingroup_outgroup.fas > ingroup_outgroup_mafft.fasta)80. Two different phylogenetic algorithms were employed in this analysis: Maximum likelihood (ML) method and Bayesian inference (BI) method. ML analysis containing 1000 bootstrap repeats was constructed by IQTree v1.6.681 (script: iqtree -s ingroup_outgroup_mafft.fasta -m MFP -bb 1000 -bnni) under the GTR + F + I + R4 (CPG) and TNe + I + G4 (nrITS) model selected by ModelFinder82. BI analysis was performed using MrBayes v3.2.683 under GTR + G + I models selected by MrModeltest 2.384, with four Markov chains and two parallel runs. Each run consisted of 3,000,000 generations, sampling every 1000 generations. Convergence was assessed to ensure reliable results. The phylogenetic tree was visualized using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/). All sequence data used in this phylogenetic analysis were sourced from the NCBI database and publicly available literature, with detailed information provided in Supplementary Table S5.
Identification of Bletilla and its adulterants
To evaluate the identification efficiency of CPG for the genus Bletilla and its common adulterants, we conducted a cluster analysis on six adulterant species (Platanthera chlorantha, Cremastra appendiculata, Pleione bulbocodioides, Pleione formosana, Pleione yunnanensis, Thunia alba) and the genus Bletilla, with Nymphaea nouchali and Barclaya longifolia85 selected as the outgroups. CPG sequences were aligned using the MAFFT v7.31380, and a phylogenetic tree was constructed using the IQTree v1.6.681 with the same parameters as described in “Phylogenetic analysis of Bletilla and B. sinensis” section, under the TVM + F + R3 model selected by ModelFinder82. Successful species identification was defined by the clustering of samples from the same species into a monophyletic group in the ML tree. All sequence data used in this identification analysis were sourced from the NCBI database and publicly available literature, with detailed information provided in Supplementary Table S6.
Data availability
The datasets generated in this study are available in GenBank (https://www.ncbi.nlm.nih.gov/) under NCBI BioProject PRJNA1146888, with chloroplast genome sequences deposited as follows: HHBJ1 (accession OQ679855; BioSample SAMN43276416; SRA SRR30355760), HHBJ2 (OQ679856; SAMN43276416; SRR30355760), XBJ1 (OQ679857; SAMN43268163; SRR30321185), BJ1 (OQ679858; SAMN46362170; SRR32108698), and BJ2 (OQ679859; SAMN46362170; SRR32108698).
References
Flora of China Editorial Committee. Flora of China, Volume 25: Orchidaceae. (Science Press, Beijing; Missouri Botanical Garden Press, St. Louis, 2009)
Jiang, S. et al. The medicinal uses of the genus Bletilla in traditional Chinese medicine: A phytochemical and pharmacological review. J. Ethnopharmacol. 280, 114263 (2021).
Xu, D., Pan, Y. & Chen, J. Chemical constituents, pharmacologic properties, and clinical applications of Bletilla striata. Front. Pharmacol. 10, 1168 (2019).
Yue, Z. et al. Bletilla striata polysaccharide-coated andrographolide nanomicelles for targeted drug delivery to enhance anti-colon cancer efficacy. Front. Immunol. 15, 1380229 (2024).
He, X. et al. Exploration of the anti-inflammatory, analgesic, and wound healing activities of Bletilla Striata polysaccharide. Int. J. Biol. Macromol. 261, 129874 (2024).
Bai, L. et al. Dual properties of pharmacological activities and preparation excipient: Bletilla striata polysaccharides. Int. J. Biol. Macromol. 254, 127643 (2024).
He, X. et al. Bletilla striata: Medicinal uses, phytochemistry and pharmacological activities. J. Ethnopharmacol. 195, 20–38 (2017).
Bentham, & George,. Genera plantarum ad exemplaria imprimis in herbariis Kewensibus seivata definita. Nature 28, 485–487 (1883).
Schlechter, R. Das system der Orchidaceen. Notizblatt des Königl. Botanischen Gartens und Museums zu Berlin 9, 563 (1926).
Mansfeld, R. Üeber das system der Orchidaceae. Blumea Supplement 1, 25–37 (1937).
Dressler, R. L. Phylogeny and Classification of the Orchid Family (Dioscorides Press, Portland, 1993).
Mansfeld, R. Über die verteilung der merkmaleinnerhalb der Orchidaceae-monandrae. Flora oder Allgemeine Botanische Zeitung 142, 65–80 (1954).
Szlachetko, D. L. On the Lectotypification of Deiregyne [Orchidaceae, Spiranthinae]. Fragm. Flor. Geobot 40, 785–795 (1995).
Dressler, R. L. The Orchids: Natural History and Classification (Harvard University Press, Cambridge, 1981).
van den Berg, C. et al. An overview of the phylogenetic relationships within Epidendroideae inferred from multiple DNA regions and recircumscription of Epidendreae and Arethuseae (Orchidaceae). Am J Bot 92, 613–624 (2005).
Chase, M. W. et al. An updated classification of Orchidaceae. Bot. J. Linn. Soc. 177, 151–174 (2015).
Freudenstein, J. V. & Chase, M. W. Phylogenetic relationships in Epidendroideae (Orchidaceae), one of the great flowering plant radiations: Progressive specialization and diversification. Ann. Bot. 115, 665–681 (2015).
Feng, H. et al. Characterization of the complete chloroplast genome of Bletilla striata (Orchidaceae: Bletilla), the herb in China. Mitochondrial DNA Part B 4, 3542–3543 (2019).
Li, L. et al. Thuniopsis: A new Orchid genus and phylogeny of the Tribe Arethuseae (Orchidaceae). PLoS ONE 10, e0132777 (2015).
Huang, W. et al. Phylogenetic analysis and character evolution of tribe Arethuseae (Orchidaceae) reveal a new genus Mengzia. Mol. Phylogenet. Evol. 167, 107362 (2022).
Han, S. et al. Plastomes of Bletilla (Orchidaceae) and phylogenetic implications. IJMS 23, 10151 (2022).
Su, Z. et al. Comprehensive evaluation of different germplasm resources of Bletilla. J. Shanxi Agric. Sci. 47, 1823–1827+1852 (2019).
Zhang, Y. et al. Comparative observation on floral characteristics and pollen morphology of three species in Bletilla by scanning electron microscope. Southwest China J. Agric. Sci. 32, 2942–2948+2979 (2019).
Wu, J. S. et al. DNA barcoding research and its application on medicinal plants of Bletilla H. G. Reichenbach. Acta Pharmaceutica Sinica 49, 1466–1474 (2014).
Jin, L. et al. Research progress on molecular identification of chinese medicinal material Bletilla. Mol. Plant Breed. 22, 446–452 (2024).
Luo, Y. et al. Identification of Bletilla striata and its adulterant Bletilla microphylla based on ITS2 sequence. Mod. Tradit. Chin. Med. Materia Medica World Sci. Technol. 19, 841–845 (2017).
Chen, M. et al. Comparison and identification of secondary structures of ITS2 sequences between Bletilla striata and its adulterants. Chin. J. Exp. Tradit. Med. Formulae 23, 46–52 (2017).
Letsiou, S. et al. DNA barcoding as a plant identification method. Appl. Sci. Basel 14, 12 (2024).
Coissac, E. et al. From barcodes to genomes: extending the concept of DNA barcoding. Mol. Ecol. 25, 1423–1428 (2016).
Cai, J. et al. Current advances in DNA barcoding of medicinal plants. Plant Sci. J. 35, 452–464 (2017).
Hollingsworth, P. M. et al. Telling plant species apart with DNA: From barcodes to genomes. Phil. Trans. R. Soc. B 371, 20150338 (2016).
Wang, G. et al. Molecular marker development and phylogenetic analysis of Aconitum species based on chloroplast genomes. Ind. Crops Prod. 221, 119386 (2024).
Jin, G. et al. Comparative analysis of complete Artemisia subgenus Seriphidium (Asteraceae: Anthemideae) chloroplast genomes: Insights into structural divergence and phylogenetic relationships. BMC Plant Biol. 23, 136 (2023).
Jiang, D. et al. Correction: Complete chloroplast genomes provide insights into evolution and phylogeny of Zingiber (Zingiberaceae). BMC Genomics 24, 397 (2023).
Tao, L. et al. Complete chloroplast genome structural characterization of two Phalaenopsis (Orchidaceae) species and comparative analysis with their alliance. BMC Genomics 24, 359 (2023).
Jiang, H. et al. Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 22, 177 (2022).
Feng, S. et al. Complete chloroplast genomes of four Physalis species (Solanaceae): Lights into genome structure, comparative analysis, and phylogenetic relationships. BMC Plant Biol. 20, 242 (2020).
Hasnain, H. & Mehvish, N. Assessment of plant genetic variations using molecular markers: A review. J. Appl. Biol. Biotech 8, 99–109 (2020).
Duran, C., Appleby, N., Edwards, D. & Batley, J. Molecular genetic markers: Discovery, applications, data storage and visualisation. CBIO 4, 16–27 (2009).
Gong, Y. et al. Potential distribution of Bletilla striata (Orchidaceae) in China and its climate characteristics. Plant Divers. Resour. 36, 237–244 (2014).
Su, T., Qiu, B. & Li, Y. Resource Investigation and market evaluation of Bletilla produced in Yunnan. Chin. Wild Plant Resour. 33, 49–52 (2014).
Chen, L. Study on Suitability and Quality Evaluation of Bletilla Striata in Northwest Hubei (Hubei University of Chinese Medicine, 2014).
Ren, F. et al. Study on genetic diversity of Bletilla striata from different localities based on nrDNA ITS. Mod. Tradit. Chin. Med. Materia Medica-World Sci. Technol. 21, 1189–1195 (2019).
Luikart, G. et al. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 4, 981–994 (2003).
Beaumont, M. A. & Balding, D. J. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13, 969–980 (2004).
Guo, M. et al. Development of plastid genomic resources for discrimination and classification of Epimedium wushanense (Berberidaceae). IJMS 20, 4003 (2019).
Cui, Y. et al. Complete chloroplast genome and comparative analysis of three Lycium (Solanaceae) species with medicinal and edible properties. Gene Rep. 17, 100464 (2019).
Wei, X. et al. Comparing chloroplast genomes of traditional Chinese herbs Schisandra sphenanthera and S. chinensis. Chin. Herbal Med. 12, 247–256 (2020).
Huan, J. et al. The genetic diversity of Bletilla spp. based on SLAF-seq and oligo-FISH. Genes 13, 1118 (2022).
Christenhusz, M. J. M. & Byng, J. W. The number of known plants species in the world and its annual increase. Phytotaxa 261, 201 (2016).
Wettewa, E., Bailey, N. & Wallace, L. E. Comparative analysis of genetic and morphological variation within the Platanthera hyperborea complex (Orchidaceae). Syst. Bot. 45, 767–778 (2020).
Freudenstein, J. V. Orchid phylogenetics and evolution: history, current status and prospects. Ann. Bot. 135, 805–822 (2025).
Wang, C. W. Phylogenomics of Orchidaceae Based on Plastid Genomes (University of Nanchang, 2019).
Kim, Y. et al. Plastome evolution and phylogeny of Orchidaceae, with 24 new sequences. Front. Plant Sci. 11, 22 (2020).
Green, R. E. et al. A draft sequence of the neandertal genome. Science 328, 710–722 (2010).
Wilson, K. A. et al. Conserving biodiversity efficiently: What to do, where, and when. PLoS Biol. 5, e223 (2007).
McLay, T. G. B. et al. Phylogenomics reveals extreme gene tree discordance in a lineage of dominant trees: Hybridization, introgression, and incomplete lineage sorting blur deep evolutionary relationships despite clear species groupings in Eucalyptus subgenus Eudesmia. Mol. Phylogenet. Evol. 187, 107869 (2023).
Yi, T., Jin, G. & Wen, J. Chloroplast capture and intra- and inter-continental biogeographic diversification in the Asian—New world disjunct plant genus Osmorhiza (Apiaceae). Mol. Phylogenet. Evol. 85, 10–21 (2015).
Yang, Y. et al. Plastid phylogenomic analyses of fagales reveal signatures of conflict and ancient chloroplast capture. Mol. Phylogenet. Evol. 163, 107232 (2021).
Morales-Briones, D. F. et al. Disentangling sources of gene tree discordance in phylogenomic data sets: Testing ancient hybridizations in Amaranthaceae s.l. Syst. Biol. 70, 219–235 (2021).
Li, X. et al. Hybridization and introgression in sympatric and allopatric populations of four oak species. BMC Plant Biol. 21, 266 (2021).
Wu, L. et al. Plant super-barcode: A case study on genome-based identification for closely related species of Fritillaria. Chin Med 16, 52 (2021).
Fu, C. et al. Prevalence of isomeric plastomes and effectiveness of plastome super-barcodes in yews (Taxus) worldwide. Sci. Rep. 9, 2773 (2019).
Liu, Z. et al. Can plastid genome sequencing be used for species identification in Lauraceae?. Bot. J. Linn. Soc. 197, 1–14 (2021).
Yu, X. et al. Species discrimination in Schima (Theaceae): Next-generation super-barcodes meet evolutionary complexity. Mol. Ecol. Resour. 22, 3161–3175 (2022).
Sun, J. et al. Tracing the geographic origin of endangered plant species using transcriptome-derived SNPs: An example of Cathaya argyrophylla. Mol. Ecol. Resour. 23, 844–854 (2023).
Liu, L.-X. et al. Plastome evolution in Saxifragaceae and multiple plastid aapture events involving Heuchera and Tiarella. Front. Plant Sci. 11, 361 (2020).
Soorni, A. & Golchini, M. M. Complete chloroplast genome of Mentha aquatica reveals hypervariable regions and resolves phylogenetic position within the genus Mentha. Mol. Biol. Rep. 52, 677 (2025).
Tysklind, N. et al. Microgeographic local adaptation and ecotype distributions: The role of selective processes on early life-history traits in sympatric, ecologically divergent Symphonia populations. Ecol. Evol. 10, 10735–10753 (2020).
Hua, Z. et al. Accurate identification of taxon-specific molecular markers in plants based on DNA signature sequence. Mol. Ecol. Resour. 23, 106–117 (2023).
Istanbuli, T. et al. Genome-wide association study reveals SNP markers controlling drought tolerance and related agronomic traits in chickpea across multiple environments. Front. Plant Sci. 15, 1260690 (2024).
Emerman, A. B. et al. NEBNext Direct: A novel, rapid, hybridization-based approach for the capture and library conversion of genomic regions of interest. Curr. Protoc. Mol. Biol. 119, 7–30 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Jin, J.-J. et al. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241 (2020).
Shi, L. et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73 (2019).
Lohse, M., Drechsel, O. & Bock, R. Organellar Genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274 (2007).
Beier, S. et al. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Frazer, K. A. et al. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Katoh, K. & Standley, D. M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Trifinopoulos, J. et al. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Kalyaanamoorthy, S. et al. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Nylander, J. A. A. Mrmodeltest v2. Evolutionary Biology Centre, Uppsala University (2004).
Givnish, T. J. et al. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. R. Soc. B. 282, 20151553 (2015).
Acknowledgements
This research was supported by Basic Research Expenses of Chongqing Science and Technology Bureau (2024jbky-032), Chongqing Talent Program (cstc2022ycjh-bgzxm0029, CQYC20210309793), Chongqing Traditional Chinese Medicine Technology Projec (ZY201402128).
Author information
Authors and Affiliations
Contributions
Conceptualization, W.Z. and F.R.; methodology, J.Z.; software, L.W.; validation, L.W.; formal analysis, S.L. and G.Z.; investigation, B.X.; resources, F.R.; data curation, Y.L.; writing—original draft preparation, W.Z. and Y.L.; writing—review and editing, W.Z. and J.Q.; visualization, N.Z. and C.X.; supervision, M.Y.; project administration, W.Z.; funding acquisition, F.R. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhuo, W., Liu, Y., Wang, H. et al. Chloroplast genome analysis reveals intraspecific variation and phylogenetic conflicts in Bletilla. Sci Rep 15, 40297 (2025). https://doi.org/10.1038/s41598-025-24120-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-24120-6
