
Abstract
Sporotrichosis is a chronic subcutaneous infection caused by the Sporothrix complex, a dimorphic fungus that exhibits significant clinical heterogeneity. However, the molecular targets and underlying mechanisms of Sporothrix globosa infection remain unclear. Therefore, in this study, we aimed to integrate whole-transcriptome sequencing analysis of skin lesion tissues from patients with sporotrichosis and healthy controls. In addition, differential gene expression screening, protein-protein interaction network construction, and functional enrichment analysis. We systematically identified the key immune-related genes, Cluster of Differentiation 247 (CD247), Signal Transducer and Activator of Transcription 1 (STAT1), and Lymphocyte-Specific Tyrosine Kinase (LCK), which are majorly upregulated during infections. The high expression of these genes in the disease group was consistently validated by western blot, quantitative polymerase chain reaction, and immunohistochemistry. Mechanistic analysis revealed the contribution of these genes to sporotrichosis-dysregulated immune response. This occurs by causing excessive activation of the interferon signaling pathway, lymphocyte overactivation, and the JAK-STAT signaling pathway, which leads to the host immune imbalance. This suggests their central role in immune evasion and overactivation during sporotrichosis. In this novel study, we revealed the pathological regulatory role of the CD247-STAT1-LCK molecular network in sporotrichosis, which provided novel insights into fungal-host interactions and identified potential therapeutic targets for immune-modulating antifungal treatments.
Similar content being viewed by others


Introduction
Sporotrichosis is a chronic infectious disease caused by the Sporothrix complex, and has a global distribution. The disease primarily affects the skin, subcutaneous tissue, mucous membranes, and local lymphatic system and disseminates systemically, causing systemic damage1. Epidemiological surveys show the high incidence of this disease in China, particularly in the northeastern region. Sporotrichosis exhibits distinct occupational clustering in agricultural workers, a phenomenon that may be closely related to pathogenic fungi application in humus-rich soils and occupational exposure to skin trauma, which is a significant public health problem2,3,4.
Currently, clinical treatment relies primarily on traditional antifungal medications, including saturated potassium iodide solution and amphotericin B. However, these conventional treatments have significant limitations: Potassium iodide therapy usually requires a 3–6 month course, and approximately 15–20% of patients experience adverse drug reactions, such as thyroid dysfunction, whereas amphotericin B is effective for deep infections, and is unsuitable for long-term use due to its significant nephrotoxicity5. Recently, treatment failures in sporotrichosis have increased, and strains with low itraconazole sensitivity have been reported6,7. This highlights the urgent need to elucidate the pathogenic mechanisms and develop novel therapeutic strategies.
Multiomic technologies based on transcriptomics, proteomics, and metabolomics provide powerful tools for systematically analyzing dynamic changes in biological molecular networks. This approach enables the precise identification of the key regulatory molecules or pathways involved in disease progression. Gene expression profiling using omics data has multiple applications in the biomedical field, which enables the identification of disease-specific biomarkers for diagnosis and facilitates the construction of molecular subtyping models for disease progression prediction and prognostic evaluation. Furthermore, during treatment, it allows dynamic monitoring of temporal changes in gene expression, providing molecular evidence for evaluating efficacy and individualized therapeutic strategies3,8. Recent studies have revealed that Sporothrix can modify the host immune microenvironment through Toll-like receptor signaling and cytokine-mediated immune responses9,10,11,12. The regulatory network of T cell-related genes and their functional impact on sporotrichosis remain incompletely understood despite existing research elucidating the aberrant activation of innate immunity13. This suggests that a systematic analysis of host-pathogen interactions using transcriptomics, particularly identifying differentially expressed genes to identify key immune regulatory targets, has significant scientific value. Transcriptomics-based systematic analysis offers a new perspective for exploring host-pathogen interactions. Key targets for immune regulation can be recognized by identifying the differentially expressed genes. In this study, we focused on CD247, STAT1, and LCK in the T-cell receptor signaling pathway, which is significantly associated with immune activation in preliminary research. We aimed to elucidate the regulatory mechanisms of these genes in sporotrichosis by integrating transcriptomic sequencing and experimental validation, thereby providing a theoretical basis for exploring new immune intervention strategies.
Materials and methods
Identification of disease-specific targets
This study enrolled 10 patients with sporotrichosis diagnosed between December 2023 and December 2024 (confirmed by histopathological and mycological cultures, identified as Sporothrix globosa via phase-conversion culture and molecular methods). The intervention group included patients aged ≥ 18 years with fixed or lymphocutaneous sporotrichosis, excluding those with disseminated/extracutaneous forms, comorbidities, major organ diseases, or recent use of steroids/antifungals/immunosuppressants. Control samples were normal skin tissues from 10 blepharoplasty patients, excluding individuals with chronic skin/systemic diseases. For comparative analysis, formalin-fixed paraffin-embedded (FFPE) tissue blocks from 10 histopathologically confirmed non-tuberculous mycobacterial (NTM) infection cases (2023–2024) were included as disease controls. All NTM cases were culture-positive for Mycobacterium avium complex and exhibited similar granulomatous histopathological features to sporotrichosis. We identified disease modules and differentially expressed genes in sporotrichosis by sequencing patient samples and applying Weighted Gene Co-expression Network Analysis (WGCNA) and Limma. Intersecting genes considered disease-associated genes were extracted using the Venn package(versions 1.12 ,https://www.rdocumentation.org/packages/venn/versions/1.12).
Identification of disease molecular network
We imported the intersecting genes obtained from the Venn package into the STRING database (https://STRING-db.org/) and selected “Homo sapiens” as the species, with a score threshold of > 0.4. Furthermore, the unconnected proteins were removed. We visualized the resulting data using the Cytoscape software (version 3.82). The Cytoscape network centrality analysis (CytoNCA) plugin was used to calculate six parameters: betweenness centrality (BC), closeness centrality (CC), degree centrality (DC), eigenvector centrality (EC), local average connectivity-based method (LAC), and network centrality. Gene values above average were extracted and used as core genes.
Functional enrichment analysis of key gene clusters
Functional enrichment analyses were performed for drug-related, disease-associated, and hub genes to observe better the potentially enriched pathways of the core gene clusters. The analyses included Gene Ontology (GO), which comprised biological process (BP), cellular component (CC), and molecular function (MF) categories, as well as KEGG pathway and Disease Ontology (DO) analyses. The analysis was conducted using the clusterProfiler, org.Hs.eg.db, enrichplot, circlize (Version 0.4.16,https://cran.r-project.org/web/packages/circlize/index.html), RColorBrewer, and ComplexHeatmap R packages, with gene significance thresholds set at p-value < 0.05 and q-value < 0.05.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
We extracted total RNA from the collected tissue samples using TRIzol reagent (Thermo Fisher, Waltham, MA, USA) following the manufacturer’s instructions. Complementary DNA synthesis was performed using the PrimeScript™ RT reagent kit (TAKARA, Tokyo, Japan). Quantitative PCR was performed using a QuantStudio 5 Real-Time PCR System (ABI, USA) with HieffTM qPCR SYBR Green Master Mix (No Rox) (Yeasen, Shanghai, China). The reaction program was as follows: initial denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The primer sequences used are listed in supplementary Table 1. Relative mRNA expression was calculated using the 2−ΔΔCt method, with Glyceraldehyde 3 phosphate dehydrogenase (GAPDH) as the internal control. The amplification efficiency of all primer pairs was validated using the standard curve method. All primer pairs demonstrated amplification efficiencies between 90% and 110% (specific values are provided in supplementary Table 2).
Western blot (WB) analysis
Tissue samples were lysed using RIPA lysis buffer (Servicebio, Wuhan, China), which contained a protease inhibitor cocktail. Proteins were separated using 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Subsequently, they were transferred to a nitrocellulose membrane (ServiceBio). After the membranes were blocked using a blocking solution, the membrane was incubated overnight at 4 °C with primary antibodies against CD247, LCK, STAT1 (dilution 1:1000), and GAPDH (dilution 1:2000). Thereafter, the membrane was incubated with horseradish peroxidase-conjugated secondary antibody (Servicebio; dilution 1:5000) at room temperature for 1 h. Protein bands were detected using an enhanced chemiluminescence kit (Servicebio) and imaged using the CLINX 6100 (CLINX, Shanghai, China). Finally, quantitative analysis of protein band intensity was performed using AIWBwell™ software (version 1.0.0, http://pan.service-bio.com:10443/l/YJ21Jq), which automatically identified band regions and calculated grayscale values with background subtraction algorithms to ensure data accuracy, following the standardized analytical procedures predefined by the software. The protein expression levels were normalized against GAPDH as an internal control. Each experiment was independently repeated three times, with data presented as relative expression values (calculated by the ratio of target protein to GAPDH grayscale values).
Immunohistochemical staining
Skin tissue samples were fixed in 10% formalin for 24 h, dehydrated, and embedded in paraffin. Tissue sections were deparaffinized and rehydrated, followed by antigen retrieval with EDTA buffer (pH 8.0) using a high-pressure treatment. The sections were incubated overnight at 4 °C after blocking the endogenous peroxidase activity with primary antibodies against CD247 (1:100), LCK (1:100), and STAT1 (1:400), followed by DAB staining. Hematoxylin counterstaining was performed, and the positively stained areas were quantified using ImageJ software(Version13.0.6,https://imagej.net/ij/download.html).
Statistical analysis
Data are expressed as mean ± standard deviation (mean ± SD). Intergroup comparisons were performed using the t-test for two-group comparisons, while one-way ANOVA with Tukey’s post-hoc test was applied for multi-group comparisons. Correlation analysis between gene expression and clinical phenotypes was performed using Pearson’s correlation coefficient. All statistical analyses were performed using GraphPad Prism 10.0 software, with statistical significance set at p < 0.05.
Informed consent
This study was a retrospective analysis, and all patient identities were anonymized.
Institutional review board statement
This study was approved by the Ethics Committee of the Second Hospital of Jilin University (Approval No: 2025[135]), which waived the need for written informed consent. All the experiments were performed in accordance with the Declaration of Helsinki.
Results
Identification of disease genes
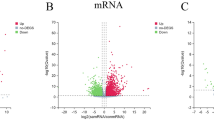
First, we standardized the unprocessed sporotrichosis patient samples (Fig. 1A) and subsequently normalized them (Fig. 1B). Disease-associated genes were obtained by applying the criteria p < 0.05 and logFC > 1, yielding 1120 differentially expressed genes (Fig. 1C, supplementary Table 3). WGCNA further partitioned the disease into modules, with the soft thresholding values detailed (Fig. 1D–E). The cut-off was β = 20 (scale-free R2 = 0.7), merging modules with 75% similarity (Fig. 1F–G). The combined modules were associated with the clinical features, resulting in the identification of seven modules (Fig. 1H–I). We selected the light cyan and blue modules with p < 0.05 (Fig. 1J–K). The intersection of disease module genes and differentially expressed genes resulted in 1025 disease-associated genes (Fig. 1L). These genes were imported into the STRING database to generate an initial molecular network (supplementary figure). Furthermore, we scored the genes based on several parameters using the CytoNCA plugin. The first analysis yielded 348 genes from module one (supplementary Table 4). The second part of the genes was selected as the core gene, and a network diagram was created (Fig. 1M).

Integrative analysis of transcriptomic data and modular networks in sporotrichosis. (A) Raw expression profiles of sporotrichosis samples prior to normalization. (B) Normalized expression profiles of sporotrichosis samples. (C) Volcano plot illustrating differentially expressed genes (DEGs) in sporotrichosis. (D) Scale-free topology fit index analysis across candidate soft-thresholding powers. (E) Optimization of soft-thresholding power: X-axis indicates soft-thresholding power; left Y-axis represents the scale-free fit index (R²), and right Y-axis denotes mean connectivity (degree). (F) Hierarchical clustering dendrogram of co-expression modules, with color bars indicating module assignments. (G) Module merging dendrogram demonstrating consolidation of highly correlated gene clusters. (H) Heatmap displaying correlations between module eigengenes (rows) and clinical traits of sporotrichosis (columns). (I) Module-trait relationship heatmap, highlighting inter-module correlations. (J) Lightcyan module: Gene significance vs. module membership correlation for disease-associated traits. (K) Blue module: Gene significance vs. module membership correlation for disease-associated traits. (L) Venn diagram intersecting co-expressed module genes and DEGs. (M) Molecular interaction network of hub genes, with node sizes proportional to connectivity degrees.
GO analysis showed that the pathogenic mechanisms of these genes were related to the following pathways: type II interferon production (GO:0032609), regulation of T cell activation (GO:0050863), and immune response activation (GO:0002253) (Fig. 2A). KEGG pathway analysis suggested that sporotrichosis development was associated with Th1, Th2, and Th17 cell differentiation (hsa04658) and the JAK-STAT signaling pathway (hsa04630) (Fig. 2B)14.

Functional enrichment and STAT1-associated gene network analysis in sporotrichosis. (A) Gene Ontology (GO) enrichment analysis of disease-associated targets, categorizing significant terms into biological processes (BP), molecular functions (MF), and cellular components (CC). (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, highlighting immune-related and metabolic pathways enriched in sporotrichosis. (C) Correlation network of STAT1 with co-expressed genes.
We performed multi-platform validation in an expanded sample cohort that included skin lesion tissues from three patients with sporotrichosis and healthy controls, respectively, to validate the differential expression patterns of the candidate genes obtained from the initial screening. The validation methods included quantitative reverse transcription polymerase chain reaction (qRT-PCR), western blotting, and immunohistochemistry to ensure comprehensive detection at both the transcriptional and protein levels.
Quantitative analysis
All three target genes demonstrated statistically significant upregulation in sporotrichosis lesions compared to normal controls (CD247: 4.1-fold, p = 0.0003; LCK: 2.3-fold, p = 0.0009; STAT1: 3.4-fold, p < 0.0001), with all genes also exhibiting significant differential expression when compared to nontuberculous mycobacterial (NTM) infection tissues (p < 0.05 for all comparisons) (Fig. 3A). All comparisons were statistically significant at α = 0.01 (two-tailed Student’s t-test). Western blot quantification confirmed these results, with protein expression levels normalized to GAPDH showing increases of 2.3-, 1.7-, and 1.6-fold for CD247, LCK, and STAT1, respectively (all comparisons, p < 0.05) (Fig. 3B–C).

(A) mRNA expression levels of CD247, LCK, and STAT1 in skin tissues from patients with sporotrichosis, nontuberculous mycobacteria (NTM) infection, and normal controls, as determined by RT-qPCR (normalized to GAPDH). (B) Western blot (WB) bands of CD247, LCK, and STAT1 proteins. Lanes A1-3: normal tissues; Lanes B1-3: sporotrichosis-affected tissues. (C) Protein expression levels of the three genes normalized to GAPDH. (D) Quantitative analysis of IHC-positive areas for the three genes compared to controls. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (E) Representative immunohistochemical (IHC) staining of CD247, LCK, and STAT1 in normal and diseased tissues. Scale bar: 150 μm (lower left corner).
Immunohistochemical staining
Immunohistochemical staining provided spatial validation of the results. The lesion sections showed strong membrane/cytoplasmic staining for CD247, LCK, and STAT1 in dermal lymphocytes compared to the weak expression in control tissues. The area percentage of staining in the lesions was more than five times higher than that in the control tissues (all comparisons, p < 0.05) (Fig. 3D–E).
Multimodal validation confirmed the persistent overexpression of T-cell receptor signaling components in sporotrichosis lesions, indicating their potential involvement in host-pathogen interactions and skin immune responses during chronic fungal infection. The consistency between the transcriptional and translational data across different detection platforms further validated these findings.
Discussion
In this novel study, we revealed the synergistic overexpression of CD247, STAT1, and LCK in fixed-type sporotrichosis skin lesion tissues by integrating transcriptomic data with experimental validation. Furthermore, we preliminarily elucidated their potential mechanisms in regulating T cell activation, interferon responses, and the Janus kinase (JAK)/Signal transducer and activator of transcription (STAT) signaling pathway in host antifungal immunity. The study was limited by the sample size and the lack of clinical subtype analysis; however, the systematic comparison between patients and healthy controls, along with multidimensional validation, led to the following core conclusions:
Our findings point to the continuous activation of T-cell pathways as a core mechanism driving immune pathological damage in sporotrichosis. The coordinated upregulation of CD247 (CD3ζ chain), LCK, and STAT1 indicated that T cell-related signaling is abnormally active during the disease progression. The upregulation of CD247, as a core signaling component of the T cell receptor (TCR) complex15,16,17, may enhance the phosphorylation of Zeta-chain-associated protein kinase 70 and phospholipase C gamma 1, promoting the release of pro-inflammatory cytokines such as interleukin-2 and interferon-gamma (IFN-γ), and creating a local inflammatory microenvironment18,19. Significant infiltration of CD3+ T cells was observed in the skin lesions of patients, which is consistent with the observations of MORGADO F N20. In line with this, CD3+ T cell infiltration in the lesions of sporotrichosis and leishmaniasis correlated positively with disease severity, and patients with lymphangitis exhibited more severe inflammatory responses. Therefore, excessive T-cell activation may increase tissue destruction and promote pathogen dissemination. Similar mechanisms have been observed in fungal infections with Th1/Th17 imbalance21,22. As shown in Table 1, significant differences exist in T cell pathway activation across various fungal infections. For example, RAI G et al. 23noted that Aspergillus flavus induces Th17 differentiation and suppresses Treg function by activating CD3+ and CD4+ T cells. Notably, LCK, a Src family tyrosine kinase, is major in initiating TCR signaling cascades and can activate downstream pathways by phosphorylating the immunoreceptor tyrosine-based activation motif domain of CD3 chains24,25. This study did not directly investigate Sporothrix schenckii regulation of T cells; however, previous studies on other pathogens have provided clues. DAUD M et al. 26showed that Candida albicans activates LCK in CD4+ T cells by binding to enolase CaEno1, inducing Th2 polarization, and inhibiting memory T cell responses. KONING F 27 suggested that Mycobacterium tuberculosis inhibits LCK synaptic translocation through ManLAM to suppress TCR signaling. This contradiction may be due to differences in the pathogen species and infection site; nevertheless, it underscores the importance of LCK in host–pathogen interactions. In our study, LCK upregulation may represent the host’s attempt to resist fungal invasion by enhancing TCR signaling. However, excessive activation of this compensatory response can induce T-cell exhaustion or autoimmune damage, creating a vicious cycle of “inflammation-tissue destruction-fungal dissemination.” This hypothesis is supported by clinical cases28 in which monoclonal anti-CD3 therapy led to T-cell exhaustion and a significantly higher incidence of invasive aspergillosis. In our study, qPCR analysis further demonstrated that while CD247, STAT1, and LCK were significantly upregulated in sporotrichosis lesions compared to normal skin (all p < 0.001), their expression patterns distinctly differed from those in nontuberculous mycobacterial (NTM) infections. Notably, LCK expression was significantly suppressed in NTM lesions (p < 0.05), whereas CD247 and STAT1 showed no statistically significant changes compared to normal controls. This divergent transcriptional profile suggests that the coordinated overexpression of CD247-STAT1-LCK may represent a sporotrichosis-specific immune signature, potentially reflecting unique host-pathogen interactions in Sporothrix globosa infection that are not observed in other granulomatous diseases such as NTM infections.
Sporothrix schenckii mediates its infection through interactions between fungal ligands (such as β-glucans and melanin) and host pattern recognition receptors (PRRs) including TLRs and CLRs. The fungal cell wall β-glucans may activate immune responses via host CLRs (e.g., Dectin-1). Upon recognizing β-glucans, Dectin-1 triggers the Syk-CARD9-NF-κB signaling pathway, promoting the release of pro-inflammatory cytokines (e.g., IL-6, TNF-α) while synergistically enhancing inflammatory responses through TLR2 cooperation29,30. Similarly, Candida albicans induces Th17 responses through Dectin-1-mediated β-glucan recognition, with TLR2/4 acting synergistically to amplify inflammation31,32. Aspergillus fumigatus can rapidly activate iNKT cells in vivo and in vitro. Although CD1d recognition is required, the antifungal iNKT cell response is independent of fungal lipids but rather depends on Dectin-1 and MyD88-mediated β-1,3-glucan recognition, which induces APCs to produce IL-12, thereby stimulating iNKT cells to secrete IFN-γ33. We hypothesize that the CD247, STAT1, and LCK network may also be activated through fungal ligand-host PRR interactions and function cooperatively. Investigating this potential mechanism represents a key direction for our future research.
Notably, the interaction between LCK and CD146 enhances TCR signaling sensitivity; CD146 dimerization recruits LCK, promoting its autophosphorylation and thereby amplifying inflammatory signals in CD4+ T cells in a positive feedback loop34. The amplification may be involved in the pathological processes of various inflammatory diseases, including cancer, asthma, diabetes, rheumatoid arthritis, psoriasis, Crohn’s disease, ulcerative colitis, and atherosclerosis35,36. Targeting the LCK-CD146 signaling axis could be a new strategy for regulating T cell-mediated immune pathology. Furthermore, the association between CD247 polymorphism and susceptibility to autoimmune diseases supports its dual role in immune regulation15,17,37. This highlights the major role of TCR signaling overactivation in immune homeostasis imbalances. In summary, the abnormal stimulation of T-cell activation pathways in sporotrichosis may be a double-edged sword in immune defense and pathological damage, suggesting that precise modulation of this pathway could be the key to therapeutic breakthroughs.
Beyond T-cell activation, our study also reveals a critical dysregulation of interferon responses and JAK/STAT pathway signaling that may disrupt the balance between immune defense and pathological damage. STAT1, as the core transcription factor of the interferon (IFN) signaling pathway, is abnormally overexpressed, highlighting the dual role of the IFN-γ signaling pathway in sporotrichosis. STAT1 enhances antifungal effects by activating macrophage-inducible nitric oxide synthase (iNOS)38,39, whereas persistent activation induces suppressor of cytokine signaling 1 expression, inhibiting the JAK-STAT pathway and promoting immune tolerance40. The JAK-STAT pathway is a double-edged sword, and its appropriate activation helps resist invading pathogens, while its excessive activation has the opposite effect41,42. This paradoxical effect is particularly evident in patients with STAT1 gain-of-function (GOF) mutations, who are highly susceptible to invasive fungal infections, suggesting that the dynamic balance of STAT1 activity is crucial for immune homeostasis43,44. Our protein interaction network analysis revealed that STAT1 forms a tight regulatory module with interferon-inducible genes such as interferon regulatory factor 1 and CXCL10 (Fig. 2C). Notably, STAT1 expression showed a significant correlation with CD247 and LCK (Pearson r > 0.65), potentially forming a positive feedback cascade network of “TCR activation-LCK phosphorylation-STAT1 nuclear translocation-IFN-γ secretion.” Our previous study revealed a significant increase in IL-6 levels in the skin lesions of patients with sporotrichosis, accompanied by sustained activation of the JAK3/STAT3 signaling axis and upregulation of SOCS3, with enhanced phosphorylation of JAK3 and STAT3 protein expression3. These observations suggest that the coordinated activation of different nodes in the JAK-STAT pathway could be an important molecular marker of the disease combined with the findings of STAT1 aberrant activation in this study. This cross-pathway interaction may explain the coexistence of inflammation and immune suppression in the disease phenotype: STAT1-driven IFN-γ responses enhanced early antifungal defense; however, negative feedback inhibition following chronic activation may create opportunities for immune evasion by fungi. This mechanism notably contrasts with the immune exhaustion model observed in invasive candidiasis, which is predominantly characterized by IL-10/TGF-β-mediated immune suppression45.
From a translational perspective, targeting immune homeostasis emerges as a potential strategy for the precise treatment of sporotrichosis. The discovery of the CD247-LCK-STAT1 network provides a theoretical basis for intervention strategy design. For example, LCK inhibitors such as nilotinib can balance immune defense and tissue damage by modulating the TCR signaling intensity. Clinical applications of nilotinib in chronic graft-versus-host disease have selectively inhibited overactivated T-cells without affecting initial T-cell function46. This finding suggests that similar strategies may be applied in immune modulation for sporotrichosis. JAK inhibitors uniquely regulate immune imbalance. Furthermore, by targeting the JAK-STAT pathway, the drugs have been successfully used in the clinical treatment of rheumatoid arthritis, systemic lupus erythematosus, and psoriasis47. Research on JAK inhibitors in patients with STAT1-GOF mutations has demonstrated that these drugs alleviate autoimmune symptoms and effectively control severe infection progression, offering important insights into the treatment of sporotrichosis43. However, caution is needed regarding the potential risk of weakening early antifungal responses.
Our study demonstrates the coordinated upregulation of CD247, STAT1, and LCK in sporotrichosis lesions, suggesting their potential utility as diagnostic or prognostic biomarkers. As core components of T-cell activation and interferon signaling pathways, their expression levels (Pearson r > 0.65) correlate with disease activity. The STAT1-mediated interferon signature may be particularly valuable for differentiating sporotrichosis from other granulomatous dermatoses. Furthermore, the association between CD3+ T cell infiltration (linked to CD247/LCK activity) and lymphangitic spread indicates this gene signature could identify high-risk patients requiring aggressive therapy20. For clinical translation, key challenges include establishing standardized assays, validating dynamic monitoring approaches, and conducting multicenter studies. Future research should explore their applications in disease stratification, treatment response assessment, and non-invasive detection methods.
To our knowledge, this is the first study using transcriptomics to reveal the essential roles of CD247, STAT1, and LCK in sporotrichosis. However, these are the following limitations: first, the sample size was small and lacked comparative analysis of different clinical subtypes; second, we focused on mRNA expression and did not deeply explore protein modifications and epigenetic regulatory mechanisms; third, the three gene interaction network and their specific impact on fungal immune evasion require functional experimental validation. Fourth, while our comparative analysis with non-tuberculous mycobacterial (NTM) infections has revealed a distinct immunological signature in sporotrichosis, the range of disease controls remains limited. Future studies should incorporate broader comparators, such as leishmaniasis and other deep fungal infections, to further validate the specificity of the identified molecular markers (the CD247/STAT1/LCK activation pattern) and to determine whether this pattern is unique to Sporothrix globosa infection or may also be observed in infections caused by other fungal pathogens (e.g., Candida, Aspergillus). This will help clarify the broader applicability of our findings across the spectrum of fungal diseases. Future studies could expand the sample cohort, integrate single-cell sequencing and spatial transcriptomics to analyze immune microenvironment heterogeneity, and use gene editing to construct a Sporothrix globosa–host interaction model to elucidate the regulatory mechanisms of key genes. In addition, preclinical evaluation of targeted drugs, such as JAK inhibitors, can be conducted to advance the translation of basic research into clinical applications.
In this study, we systematically investigated the expression profiles and regulatory networks of CD247, STAT1, and LCK in sporotrichosis, providing novel insights into host-pathogen interaction mechanisms. The “immune regulation triangle” formed by these three genes may collectively determine infectious outcomes by influencing processes including T cell activation, interferon responses, and signal transduction. Further research is needed to unravel the molecular interaction mechanisms that will lay the foundation for developing immunomodulatory therapies.
Data availability
The raw sequence data have been deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA003986) and are publicly accessible at [https://ngdc.cncb.ac.cn/gsa-human](mailto: https:/ngdc.cncb.ac.cn/gsa-human) .The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
References
Lopes-Bezerra, L. M. et al. Sporotrichosis between 1898 and 2017: the evolution of knowledge on a changeable disease and on emerging etiological agents. Med. Mycol. 56, 126–143. https://doi.org/10.1093/mmy/myx103 (2018).
Queiroz-Telles, F., Buccheri, R. & Benard, G. Sporotrichosis in immunocompromised hosts. J. Fungi (Basel). 5 https://doi.org/10.3390/jof5010008 (2019).
Liu, Z. et al. Whole transcriptome sequencing for revealing the pathogenesis of sporotrichosis caused by sporothrix globosa. Sci. Rep. 14, 359. https://doi.org/10.1038/s41598-023-50728-7 (2024).
Lv, S. et al. Clinical epidemiology of sporotrichosis in Jilin Province, China (1990–2019): A series of 4969 cases. Infect. Drug Resist. 15, 1753–1765. https://doi.org/10.2147/idr.S354380 (2022).
Rodrigues, A. M. et al. Current progress on Epidemiology, Diagnosis, and treatment of sporotrichosis and their future trends. J. Fungi (Basel). 8 https://doi.org/10.3390/jof8080776 (2022).
Fischman Gompertz, O. et al. Atypical clinical presentation of sporotrichosis caused by sporothrix globosa resistant to Itraconazole. Am. J. Trop. Med. Hyg. 94, 1218–1222. https://doi.org/10.4269/ajtmh.15-0267 (2016).
Poester, V. R. et al. Treatment of human sporotrichosis caused by sporothrix Brasiliensis. J. Fungi (Basel). 8 https://doi.org/10.3390/jof8010070 (2022).
Kaforou, M. et al. Transcriptomics for child and adolescent tuberculosis. Immunol. Rev. 309, 97–122. https://doi.org/10.1111/imr.13116 (2022).
Gonçalves, A. C. et al. The NLRP3 inflammasome contributes to host protection during sporothrix schenckii infection. Immunology 151, 154–166. https://doi.org/10.1111/imm.12719 (2017).
Yan, T., Li, F., Li, J. & Chen, F. Antifungal activity of a Neodymium-Doped yttrium aluminum Garnet 1,064-Nanometer laser against sporothrix globosa by inducing apoptosis and pyroptosis via the NLRP3/Caspase-1 signaling pathway: in vitro and in vivo study. Microbiol. Spectr. 9, e0136421. https://doi.org/10.1128/Spectrum.01364-21 (2021).
Li, M., Chen, Q., Sun, J., Shen, Y. & Liu, W. Inflammatory response of human keratinocytes triggered by sporothrix schenckii via Toll-like receptor 2 and 4. J. Dermatol. Sci. 66, 80–82. https://doi.org/10.1016/j.jdermsci.2012.01.003 (2012).
Gómez-Gaviria, M., Martínez-Duncker, I., García-Carnero, L. C. & Mora-Montes, H. M. Differential recognition of sporothrix schenckii, sporothrix brasiliensis, and sporothrix globosa by human Monocyte-Derived macrophages and dendritic cells. Infect. Drug Resist. 16, 4817–4834. https://doi.org/10.2147/idr.S419629 (2023).
Lin, P. et al. Innate immune responses to sporothrix schenckii: recognition and elimination. Mycopathologia 188, 71–86. https://doi.org/10.1007/s11046-022-00683-0 (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Weissman, A. M. et al. Molecular cloning and chromosomal localization of the human T-cell receptor zeta chain: distinction from the molecular CD3 complex. Proc. Natl. Acad. Sci. U S A. 85, 9709–9713. https://doi.org/10.1073/pnas.85.24.9709 (1988).
Koning, F., Maloy, W. L. & Coligan, J. E. The implications of subunit interactions for the structure of the T cell receptor-CD3 complex. Eur. J. Immunol. 20, 299–305. https://doi.org/10.1002/eji.1830200211 (1990).
Dexiu, C., Xianying, L. & Yingchun, H. Jiafu, L. Advances in CD247. Scand. J. Immunol. 96, e13170. https://doi.org/10.1111/sji.13170 (2022).
Castillo, P. et al. A new method for reactivating and expanding T cells specific for rhizopus oryzae. Mol. Ther. Methods Clin. Dev. 9, 305–312. https://doi.org/10.1016/j.omtm.2018.03.003 (2018).
Nambiar, M. P. et al. Reconstitution of deficient T cell receptor zeta chain restores T cell signaling and augments T cell receptor/CD3-induced interleukin-2 production in patients with systemic lupus erythematosus. Arthritis Rheum. 48, 1948–1955. https://doi.org/10.1002/art.11072 (2003).
Morgado, F. N. et al. Unbalanced inflammatory reaction could increase tissue destruction and worsen skin infectious diseases - a comparative study of leishmaniasis and sporotrichosis. Sci. Rep. 8, 2898. https://doi.org/10.1038/s41598-018-21277-1 (2018).
de Bernardis, F. et al. Local anticandidal immune responses in a rat model of vaginal infection by and protection against Candida albicans. Infect. Immun. 68, 3297–3304. https://doi.org/10.1128/iai.68.6.3297-3304.2000 (2000).
Richardson, J. P. & Moyes, D. L. Adaptive immune responses to Candida albicans infection. Virulence 6, 327–337. https://doi.org/10.1080/21505594.2015.1004977 (2015).
Rai, G. et al. Phenotypic and functional profile of Th17 and Treg cells in allergic fungal sinusitis. Int. Immunopharmacol. 57, 55–61. https://doi.org/10.1016/j.intimp.2018.02.009 (2018).
Bommhardt, U., Schraven, B., Simeoni, L., Beyond, T. C. R. & Signaling Emerging functions of Lck in cancer and immunotherapy. Int. J. Mol. Sci. 20 https://doi.org/10.3390/ijms20143500 (2019).
Lanz, A. L. et al. A novel biallelic LCK variant resulting in profound T-Cell immune deficiency and review of the literature. J. Clin. Immunol. 44, 1. https://doi.org/10.1007/s10875-023-01602-8 (2023).
Daud, M. et al. Enolase 1 of Candida albicans binds human CD4(+) T cells and modulates naïve and memory responses. Eur. J. Immunol. 53, e2250284. https://doi.org/10.1002/eji.202250284 (2023).
Mwebaza, I. et al. Impact of Mycobacterium tuberculosis glycolipids on the CD4 + T Cell-Macrophage immunological synapse. J. Immunol. 211, 1385–1396. https://doi.org/10.4049/jimmunol.2300107 (2023).
Kyriakidis, I. et al. Invasive fungal diseases in children with hematological malignancies treated with therapies that target cell surface antigens: monoclonal Antibodies, immune checkpoint inhibitors and CAR T-Cell therapies. J. Fungi (Basel). 7 https://doi.org/10.3390/jof7030186 (2021).
García-Carnero, L. C., Martínez-Duncker, I., Gómez-Gaviria, M. & Mora-Montes, H. M. Differential recognition of clinically relevant sporothrix species by human mononuclear cells. J. Fungi (Basel). 9 https://doi.org/10.3390/jof9040448 (2023).
López-Ramírez, L. A., Martínez-Álvarez, J. A., Martínez-Duncker, I., Lozoya-Pérez, N. E. & Mora-Montes, H. M. Silencing of sporothrix schenckii GP70 reveals its contribution to fungal Adhesion, Virulence, and the Host-Fungus interaction. J. Fungi (Basel). 10 https://doi.org/10.3390/jof10050302 (2024).
Kashem, S. W. et al. Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity 42, 356–366. https://doi.org/10.1016/j.immuni.2015.01.008 (2015).
Pinke, K. H., Lima, H. G., Cunha, F. Q. & Lara, V. S. Mast cells phagocyte Candida albicans and produce nitric oxide by mechanisms involving TLR2 and Dectin-1. Immunobiology 221, 220–227. https://doi.org/10.1016/j.imbio.2015.09.004 (2016).
Cohen, N. R. et al. Innate recognition of cell wall β-glucans drives invariant natural killer T cell responses against fungi. Cell. Host Microbe. 10, 437–450. https://doi.org/10.1016/j.chom.2011.09.011 (2011).
Duan, H. et al. CD146 bound to LCK promotes T cell receptor signaling and antitumor immune responses in mice. J. Clin. Invest. 131 https://doi.org/10.1172/jci148568 (2021).
Kumar Singh, P., Kashyap, A. & Silakari, O. Exploration of the therapeutic aspects of lck: A kinase target in inflammatory mediated pathological conditions. Biomed. Pharmacother. 108, 1565–1571. https://doi.org/10.1016/j.biopha.2018.10.002 (2018).
Elkamhawy, A., Ali, E. M. H. & Lee, K. New horizons in drug discovery of lymphocyte-specific protein tyrosine kinase (Lck) inhibitors: a decade review (2011–2021) focussing on structure-activity relationship (SAR) and Docking insights. J. Enzyme Inhib. Med. Chem. 36, 1574–1602. https://doi.org/10.1080/14756366.2021.1937143 (2021).
Fernandez, D. & Perl, A. Metabolic control of T cell activation and death in SLE. Autoimmun. Rev. 8, 184–189. https://doi.org/10.1016/j.autrev.2008.07.041 (2009).
Clark, D. N., Begg, L. R. & Filiano, A. J. Unique aspects of IFN-γ/STAT1 signaling in neurons. Immunol. Rev. 311, 187–204. https://doi.org/10.1111/imr.13092 (2022).
Zhang, B. et al. Macrolide derivatives reduce Proinflammatory macrophage activation and macrophage-mediated neurotoxicity. CNS Neurosci. Ther. 25, 591–600. https://doi.org/10.1111/cns.13092 (2019).
Inagaki-Ohara, K., Kondo, T., Ito, M. & Yoshimura, A. SOCS, inflammation, and cancer. Jakstat 2, e24053. https://doi.org/10.4161/jkst.24053 (2013).
Xu, J., Zhang, J., Mao, Q. F., Wu, J. & Wang, Y. The interaction between autophagy and JAK/STAT3 signaling pathway in tumors. Front. Genet. 13, 880359. https://doi.org/10.3389/fgene.2022.880359 (2022).
Asano, T., Noma, K., Mizoguchi, Y., Karakawa, S. & Okada, S. Human STAT1 gain of function with chronic mucocutaneous candidiasis: A comprehensive review for strengthening the connection between bedside observations and laboratory research. Immunol. Rev. 322, 81–97. https://doi.org/10.1111/imr.13300 (2024).
Okada, S. et al. Human STAT1 Gain-of-Function heterozygous mutations: chronic mucocutaneous candidiasis and type I interferonopathy. J. Clin. Immunol. 40, 1065–1081. https://doi.org/10.1007/s10875-020-00847-x (2020).
Break, T. J. et al. Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science 371 https://doi.org/10.1126/science.aay5731 (2021).
Ahmadi, N. et al. Systemic infection with Candida albicans in breast tumor bearing mice: cytokines dysregulation and induction of regulatory T cells. J. Mycol. Med. 29, 49–55. https://doi.org/10.1016/j.mycmed.2018.10.006 (2019).
Marinelli Busilacchi, E. et al. Immunomodulatory effects of tyrosine kinase inhibitor in vitro and in vivo study. Biol. Blood Marrow Transpl. 24, 267–275. https://doi.org/10.1016/j.bbmt.2017.10.039 (2018).
Jamilloux, Y. et al. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 18, 102390. https://doi.org/10.1016/j.autrev.2019.102390 (2019).
Nasiri-Jahrodi, A., Barati, M., Ahmadabad, N., Badali, H., Morovati, H. & H. & A comprehensive review on the role of T cell subsets and CAR-T cell therapy in Aspergillus fumigatus infection. Hum. Immunol. 85, 110763. https://doi.org/10.1016/j.humimm.2024.110763 (2024).
Kumaresan, P. R. et al. A novel lentiviral vector-based approach to generate chimeric antigen receptor T cells targeting Aspergillus fumigatus. mBio 15, e0341323. https://doi.org/10.1128/mbio.03413-23 (2024).
Puerta-Arias, J. D., Mejía, S. P. & González, Á. The role of the Interleukin-17 axis and neutrophils in the pathogenesis of endemic and systemic mycoses. Front. Cell. Infect. Microbiol. 10, 595301. https://doi.org/10.3389/fcimb.2020.595301 (2020).
Wang, H. et al. C-type lectin receptors differentially induce th17 cells and vaccine immunity to the endemic mycosis of North America. J. Immunol. 192, 1107–1119. https://doi.org/10.4049/jimmunol.1302314 (2014).
Borghi, M. et al. Targeting the Aryl hydrocarbon receptor with Indole-3-Aldehyde protects from vulvovaginal candidiasis via the IL-22-IL-18 Cross-Talk. Front. Immunol. 10, 2364. https://doi.org/10.3389/fimmu.2019.02364 (2019).
Hernández-Santos, N. et al. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. 6, 900–910. https://doi.org/10.1038/mi.2012.128 (2013).
Angkasekwinai, P. et al. Cryptococcus Gattii infection dampens Th1 and Th17 responses by attenuating dendritic cell function and pulmonary chemokine expression in the immunocompetent hosts. Infect. Immun. 82, 3880–3890. https://doi.org/10.1128/iai.01773-14 (2014).
Beenhouwer, D. O., Shapiro, S., Feldmesser, M., Casadevall, A. & Scharff, M. D. Both Th1 and Th2 cytokines affect the ability of monoclonal antibodies to protect mice against Cryptococcus neoformans. Infect. Immun. 69, 6445–6455. https://doi.org/10.1128/iai.69.10.6445-6455.2001 (2001).
Viriyakosol, S., Jimenez Mdel, P., Gurney, M. A., Ashbaugh, M. E. & Fierer, J. Dectin-1 is required for resistance to coccidioidomycosis in mice. mBio 4, e00597–e00512. https://doi.org/10.1128/mBio.00597-12 (2013).
Deepe, G. S. Jr. et al. Vaccination with an alkaline extract of Histoplasma capsulatum packaged in glucan particles confers protective immunity in mice. Vaccine 36, 3359–3367. https://doi.org/10.1016/j.vaccine.2018.04.047 (2018).
Kroetz, D. N. & Deepe, G. S. Jr. CCR5 deficiency mitigates the deleterious effects of tumor necrosis factor α antagonism in murine histoplasmosis. J. Infect. Dis. 205, 955–963. https://doi.org/10.1093/infdis/jir869 (2012).
Silva, L. B. R. et al. Experimental therapy of paracoccidioidomycosis using P10-Primed Monocyte-Derived dendritic cells isolated from infected mice. Front. Microbiol. 10, 1727. https://doi.org/10.3389/fmicb.2019.01727 (2019).
Linke, M. J., Ashbaugh, A., Collins, M. S., Lynch, K. & Cushion, M. T. Characterization of a distinct host response profile to Pneumocystis Murina Asci during clearance of Pneumocystis pneumonia. Infect. Immun. 81, 984–995. https://doi.org/10.1128/iai.01181-12 (2013).
de la Rua, N. M., Samuelson, D. R., Charles, T. P., Welsh, D. A. & Shellito, J. E. CD4(+) T-Cell-Independent secondary immune responses to Pneumocystis pneumonia. Front. Immunol. 7, 178. https://doi.org/10.3389/fimmu.2016.00178 (2016).
Galdino, N. A. L. et al. Depletion of regulatory T cells in ongoing paracoccidioidomycosis rescues protective Th1/Th17 immunity and prevents fatal disease outcome. Sci. Rep. 8, 16544. https://doi.org/10.1038/s41598-018-35037-8 (2018).
Tang, Y. et al. Dendritic cells promote Treg expansion but not Th17 generation in response to talaromyces Marneffei yeast cells. Infect. Drug Resist. 13, 805–813. https://doi.org/10.2147/idr.S239906 (2020).
Funding
This work was supported by National Key Research and Development Program of China (2022YFC2504800).
Author information
Authors and Affiliations
Contributions
LZ and WS contributed to the conception and design of the study and data analysis. TZH contributed to design of the study and wrote the first draft of the manuscript. LS and LSS contributed to data collection. LFQ contributed to the revision of the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
This study was approved by the Ethics Committee of the Second Hospital of Jilin University (Approval No: 2025[135]).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, Z., Lv, S., Li, S. et al. Immune genes CD247 STAT1 and LCK mediate host–pathogen interactions in sporotrichosis. Sci Rep 15, 41358 (2025). https://doi.org/10.1038/s41598-025-25337-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-25337-1
