
Abstract
Viral infections have been linked to myocarditis and may contribute to the development of cardiomyopathy. However, large-scale population-based evidence remains limited. This study aimed to investigate the association between viral infections and the subsequent risk of cardiomyopathy using Taiwan’s Longitudinal Generation Tracking Database (LGTD). We conducted a nationwide, retrospective cohort study using Taiwan’s National Health Insurance Research Database/LGTD. Individuals with a first recorded viral infection were matched to unexposed controls using 1:4 propensity scores incorporating demographics, comorbidities, medications, healthcare utilization, and index year. Incident cardiomyopathy was ascertained from ICD-9-CM diagnosis codes in claims data, without clinical adjudication or imaging confirmation. Risks were estimated using Cox proportional hazards models to obtain adjusted hazard ratios (aHRs) and 95% confidence intervals (CIs). Robustness was assessed via Fine–Gray competing-risk models for death, proportional-hazards diagnostics, alternative exposure/lag definitions, and prespecified subgroup analyses by age, sex, comorbidity burden, and virus categories. Over a mean follow-up of approximately 10 years, the incidence rate of cardiomyopathy was higher in the viral infection group (98.42 per 100,000 person-years) than in the control group (45.69 per 100,000 person-years). Viral infection was significantly associated with increased risk of cardiomyopathy (aHR = 2.915, 95% CI 1.177–4.828, p < 0.001). Subgroup analyses showed consistent risk elevation across sex (male aHR = 2.775; female aHR = 3.097), age groups, income levels, seasons, and urbanization levels (all p < 0.05). Among viral subtypes, viral hepatitis (aHR = 3.435), influenza (aHR = 3.002), and viral pneumonia (aHR = 3.091) were most strongly associated with cardiomyopathy. In this population-based cohort, viral infection was associated with increased long-term risk of cardiomyopathy. Given that outcomes were identified from administrative codes without clinical or imaging validation, causal inference is not warranted; however, the findings support post-infection cardiovascular surveillance and prevention strategies. Further studies integrating adjudicated clinical data, imaging, and biomarkers are needed to clarify mechanisms and refine risk stratification.
Similar content being viewed by others
Introduction
Cardiomyopathy comprises heterogeneous myocardial disorders that culminate in adverse remodeling, heart failure, and excess mortality1. Viral infections are established precipitants of myocarditis and may trigger or accelerate fibrotic remodeling; however, the long-term association between clinically recorded viral infection and incident cardiomyopathy at the population level remains uncertain2,3,4. Prior investigations have often been limited by small or selected samples, short follow-up, or incomplete adjustment for comorbidity and healthcare utilization5.
Using Taiwan’s National Health Insurance Research Database (NHIRD) -specifically the Longitudinal Generation Tracking Database (LGTD)—we assembled a nationwide, retrospective cohort6,7. Individuals with a first qualifying viral infection were matched to unexposed controls using 1:4 propensity scores constructed from demographics, baseline comorbidities, medications, healthcare utilization, and index year8. The primary outcome was incident cardiomyopathy defined by validated claims-based algorithms9. We estimated adjusted hazard ratios (aHRs) with Cox proportional hazards models, evaluated competing risk of death using Fine–Gray sub distribution models, and performed prespecified subgroup and sensitivity analyses, including lag/washout definitions, virus categories, and proportional-hazards diagnostics10,11.
Therefore, we hypothesized that virus and cardiomyopathy have related risk effects, and we used Taiwan’s LGTD from 2000 to 2015 to analyze the impact of virus and cardiomyopathy through long-term follow-up. This design enables an assessment of long-term risk and generalizability in routine care. By quantifying the association between viral infection and subsequent cardiomyopathy across clinical subgroups and virus categories, our study aims to inform post-infection cardiovascular surveillance and prevention strategies while highlighting priorities for mechanistic, imaging-based, and genomic research12.
Materials and methods
Data source
This retrospective cohort study utilized data from the LGTD 2000, a subset of Taiwan’s NHIRD, which includes data of 2 million beneficiaries randomly sampled from the 2000 registry and followed until 2015. The NHIRD contains de-identified information on outpatient and inpatient services, diagnostic codes, procedures, and prescriptions. Diseases were classified using the International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM).
Study design and population
A total of 1,936,512 individuals from the NHIRD were assessed for eligibility. Patients were included in the virus-infected cohort if they had a newly diagnosed viral infection (ICD-9-CM codes 001–139, excluding HIV: 042, V08) between January 1, 2001, and December 31, 2015. The index date was defined as the date of the first documented viral infection during the study period.
Exclusion criteria were
-
1.
Viral infection diagnosis prior to the year 2000.
-
2.
Pre-existing diagnosis of cardiomyopathy (ICD-9-CM: 425) before the index date.
-
3.
Incomplete or missing follow-up data.
-
4.
Age < 20 years at the index date.
-
5.
Unknown sex.
-
6.
documented HIV infection (ICD-9-CM 042 or V08) at or before the index date.
The exposure of interest was acute/episodic non-HIV viral infections with a definable index encounter. Chronic HIV infection is etiologically distinct and cannot be aligned to an acute index date; HIV disease severity (CD4 count/viral load), antiretroviral regimens, and opportunistic coinfections are unavailable in claims and would confound cardiomyopathy risk estimation. Therefore, PLWH were excluded a priori to preserve construct validity of the exposure and internal validity of effect estimates.
After applying exclusion criteria (n = 2137), a total of 6456 eligible individuals were included in the virus infection cohort.
A comparison cohort without any recorded viral infections throughout the entire observation period was generated using fourfold propensity score matching (1:4) based on age, sex, index year, and comorbidities. The same exclusion criteria were applied to this group. After matching, 25,824 individuals were included in the comparison cohort. A flowchart of the study design is depicted in Fig. 1.

The flowchart of study sample selection.
Outcome definition
The primary outcome was incident cardiomyopathy, defined as having at least one inpatient or two outpatient claims for cardiomyopathy (ICD-9-CM: 425) during follow-up. Subtypes such as dilated (425.4) and hypertrophic (425.1) cardiomyopathy were also considered in subgroup analysis. The ICD codes for cardiomyopathy are listed in Table S1. Cardiomyopathy ascertainment was based on ICD-9-CM diagnosis codes (425.xx) in claims data; therefore, phenotypic differentiation (e.g., dilated vs hypertrophic vs restrictive cardiomyopathy) was not possible in this dataset. Subtypes occasionally listed in code descriptions were not adjudicated against clinical records or imaging and were analyzed under the composite cardiomyopathy outcome.
Statistical analysis
Participants were followed from the index date until the first occurrence of cardiomyopathy, death, withdrawal from the insurance program, or December 31, 2015, whichever came first. Kaplan–Meier survival analysis and log-rank tests were used to compare the cumulative incidence of cardiomyopathy. Cox proportional hazards regression models were used to estimate adjusted hazard ratios (aHRs) and 95% confidence intervals (CIs) for the association between viral infection and cardiomyopathy. Subgroup analyses were performed based on sex, age group, and virus type. In addition to sex, age group, and virus category, we evaluated heterogeneity across the following prespecified factors derived from NHIRD/LGTD metadata:
Season of index encounter (winter, spring, summer, autumn, defined by calendar quarters; used to capture seasonal circulation of respiratory viruses).
Monthly insured premium (NHIRD income-based insurance brackets as a proxy for socioeconomic status; categories per NHIRD standard groupings; exact codes in Table S2).
Urbanization level (NHIRD 4-level classification, from most to least urbanized; code mapping in Table S2).
Level of care at index (medical center, regional hospital, district hospital/clinic; reflects care intensity and infection severity proxy).
Within each stratum, we estimated adjusted hazard ratios (aHRs) from Cox models using the same covariate set as the primary analysis. We assessed effect heterogeneity with exposure-by-subgroup interaction terms and two-sided Wald tests (reported as p for interaction).
Subgroup analyses were prespecified and exploratory; we present stratum-specific aHRs with 95% CIs in forest plots. For multiple comparisons, we emphasize effect sizes and consistency across strata; exact p values are shown without formal multiplicity adjustment for interactions, while Bonferroni correction was retained where applicable for virus-category comparisons. A p value < 0.05 was considered statistically significant. Statistical analyses were conducted using SAS version 9.4 (SAS Institute, Cary, NC, USA).
Ethics statement
NHIRD served as the primary data source for this study, standing as Taiwan’s most exhaustive healthcare research database. Diagnoses and procedures within NHIRD are classified following ICD-9-CM. The database is also connected to the National Death Registry and employs encryption of national identification numbers to safeguard individual privacy. Personal data within NHIRD is encrypted to maintain patient confidentiality. This study adhered to the Declaration of Helsinki and received approval from the Research Ethics Committee of the Tri-Service General Hospital, National Defense Medical Center (TSGHIRB: No: E202516027), and the requirement for informed consent was waived due to the use of de-identified administrative data.
Results
Basic characteristics of patients in the study
Table 1 shows the demographic characteristics of the study population and their association with viral infection. A total of 32,280 individuals were included, of whom 6456 had viral infections and 25,824 did not. The mean age was approximately 40 years, with 57.67% being male and 42.33% female. No significant differences were found between the groups in terms of sex, age, age groups, monthly insured premium, season, or level of care. However, significant differences were observed in the Charlson Comorbidity Index (CCI), location, and urbanization level. Patients with viral infections had a higher mean CCI score compared to those without (p = 0.001). Furthermore, variations in infection rates were also noted across different regions and urbanization levels (p = 0.043 and p = 0.046, respectively).
Characteristics of patient endpoints in the study
Unless otherwise noted, demographic characteristics in this section correspond to the end of follow-up (censoring date) for each participant (i.e., incident cardiomyopathy, death, disenrollment, or study end—whichever occurred first). Baseline characteristics used for propensity-score matching and covariate adjustment are reported in Sect. “Basic characteristics of patients in the study” and the Methods. Table 2 presents the demographic characteristics and clinical features of individuals with and without viral infections. The mean age of the study population was 49.04 ± 20.98 years. The male-to-female ratio was 57.67 to 42.33%, with no significant difference between groups. Significant differences between the two groups were observed in the presence of cardiomyopathy, age group distribution, CCI, and level of care (all p < 0.05). Patients with viral infections were more likely to have cardiomyopathy and had a slightly higher CCI. Furthermore, the distribution across age groups and the level of care differed significantly between the two groups.
Years from viral infection to cardiomyopathy
The mean time after diagnosis from the index date to the diagnosis of cardiomyopathy was 10.23 (SD = 9.72) years earlier than patients without viral infection (10.34 [SD = 9.86] years). Patients with viral infection developed cardiomyopathy at a mean age of 5.31 (SD = 4.29) years earlier than patients without viral infection (5.97 [SD = 4.83] years) (Tables S3-1, S3-2).
Use Cox regression to analyze the influencing factors of cardiomyopathy in viral infection
Figure 2 summarizes the results of the multivariable Cox proportional hazards regression analysis. Compared to individuals without viral infection, those with viral infections had a significantly higher risk (adjusted hazard ratio [aHR] = 2.915, 95% CI 1.177–4.828, p < 0.001). Male sex was associated with a lower risk compared to females (aHR = 0.626, 95% CI 0.429–0.869, p < 0.001). Increasing age was associated with a higher risk, with individuals aged 50–64 years (aHR = 1.134, p = 0.002) and those aged 65 years and older (aHR = 1.267, p < 0.001) showing elevated risks compared to the 20–49 years age group. Higher CCI scores were significantly associated with an increased risk (aHR = 1.349, 95% CI 1.020–1.634, p = 0.040). Regarding urbanization level, individuals residing in areas with the highest urbanization (level 1) had a significantly higher risk compared to those living in the least urbanized areas (aHR = 1.883, 95% CI 1.382–2.418, p < 0.001). In terms of level of care, patients treated at hospital centers (aHR = 1.691, p < 0.001) and regional hospitals (aHR = 1.108, p = 0.034) had higher risks compared to those treated at local hospitals.

Adjusted hazard ratios (aHRs) for the risk of cardiomyopathy by covariates. Each point represents the aHR with its corresponding 95% confidence interval (CI), derived from multivariate Cox regression. Variables include demographic, socioeconomic, seasonal, and healthcare-level factors. An aHR above 1 indicates increased risk, while values below 1 suggest reduced risk relative to the reference category.
Viral infection using Cox regression to stratify cardiomyopathy factors by the variables listed in the table
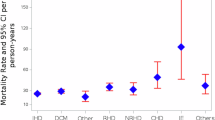
Figure 3 presents the stratified analysis of the association between viral infection and the outcome of interest. In the overall cohort, patients with viral infection had a significantly higher risk compared to those without (aHR = 2.915, 95% CI 1.177–4.828, p < 0.001). In the sex-stratified analysis, both males (aHR = 2.775, 95% CI 1.121–4.597, p < 0.001) and females (aHR = 3.097, 95% CI 1.250–5.129, p < 0.001) with viral infection showed significantly increased risks. Across age groups, viral infection was associated with a higher risk in individuals aged 20–49 years (aHR = 2.438, p < 0.001), 50–64 years (aHR = 2.865, p < 0.001), and ≥ 65 years (aHR = 3.271, p < 0.001). When stratified by insured premium, individuals in all premium categories (< 18,000, 18,000–34,999, and ≥ 35,000 NT$) exhibited significantly elevated risks associated with viral infection (all p < 0.01). Regarding seasons, viral infection was associated with increased risks during spring, summer, autumn, and winter, with the highest aHR observed in winter (aHR = 3.284, p < 0.001). In terms of urbanization level, patients residing in the highest urbanization areas (level 1) had the greatest risk elevation (aHR = 3.333, p < 0.001), though increased risks were noted across all urbanization levels. Finally, regarding the level of care, patients treated at hospital centers, regional hospitals, and local hospitals all showed significantly increased risks associated with viral infection (all p < 0.001).

Factors of cardiomyopathy stratified by variables listed in the table by using Cox regression and Bonferroni correction for multiple comparisons.
Factors of cardiomyopathy across various subgroups of viral infection using Cox regression and applying Bonferroni correction for multiple comparisons
Table S4 summarizing the distribution of viral infections at the index encounter. Cases are grouped as Enterovirus, Viral hepatitis, Viral pneumonia (including influenza), and other viruses, with the corresponding ICD-9-CM code sets listed in the table. Table S4 also reports group counts (n), cohort percentage, person-years (PYs), events, and incidence rates per 100,000 PYs. As requested, Influenza is included under Viral pneumonia.
Figure 4 demonstrates the association between different types of viral infections and the outcome of interest. Overall, patients with viral infections had a significantly increased risk compared to those without (aHR = 2.915, 95% CI 1.177–4.828, p < 0.001). Among specific viral infections, enterovirus infection was associated with a significantly elevated risk (aHR = 2.479, 95% CI 1.001–4.105, p = 0.049). Viral hepatitis showed the highest associated risk (aHR = 3.435, 95% CI 1.386–5.689, p < 0.001), followed by viral pneumonia (aHR = 3.091, 95% CI 1.249–5.121, p < 0.001) and influenza (aHR = 3.002, 95% CI 1.211–4.978, p < 0.001). In contrast, infection with other viruses did not reach statistical significance (aHR = 2.195, 95% CI 0.887–3.630, p = 0.124).

Factors of cardiomyopathy among different viral infection subgroups by using Cox regression and Bonferroni correction for multiple comparisons.
Kaplan–Meier cumulative incidence curve of cardiomyopathy in viral infection
Figure 5 Kaplan–Meier curves for incident cardiomyopathy comparing individuals with viral infection versus matched controls. Shaded areas denote 95% confidence intervals; tables show numbers at risk at 0, 1, 3, 5, and 10 years. Log-rank p < 0.001. Note: KM treats death as non-informative censoring; competing-risk results (Fine–Gray sub distribution models) are provided in Fig. 5/Table S5 and show a similar pattern.
We visualized time-to-event differences using Kaplan–Meier (KM) curves comparing individuals with a first recorded viral infection to matched unexposed controls. Time zero was the index date (first qualifying encounter for the exposed; matched index year for controls). Participants were censored at cardiomyopathy diagnosis, death, disenrollment, or end of follow-up, whichever came first. Curves display pointwise 95% confidence bands and numbers at risk at pre-specified intervals.
The cumulative incidence of cardiomyopathy was higher in the viral-infection group throughout follow-up. Separation of the curves emerged early and persisted over time. A log-rank test indicated a significant difference between groups. These unadjusted curves complement, but do not replace, the adjusted estimates from the Cox models; corresponding adjusted hazard ratios (aHRs) are reported in the main results and were consistent with the KM pattern.
Because standard KM treats death as a non-informative censoring event, we provide Fine–Gray competing-risk analyses in Table S5 to account for death as a competing event; the direction and magnitude of the association were similar. For readability, we truncated the x-axis at the 95th percentile of observed follow-up; complete curves are shown in Fig. 5.

Kaplan–Meier for cumulative risk of cardiomyopathy aged 20 and over stratified by viral infection with log-rank test.
Discussion
In this nationwide, propensity-matched cohort, clinically recorded viral infection was associated with an increased long-term risk of incident cardiomyopathy compared with matched unexposed controls (aHR 2.915, 95% CI 1.177–4.828; p < 0.001). Virus-category analyses suggested non-uniform risk: associations were strongest for viral hepatitis (aHR 3.435, 1.386–5.689) and viral pneumonia/Influenza (aHR 3.091, 1.249–5.121; aHR 3.002, 1.211–4.978, respectively), with enterovirus showing a smaller yet significant elevation (aHR 2.479, 1.001–4.105). “Other viruses” did not reach significance (aHR 2.195, 0.887–3.630). These effect sizes were observed alongside higher absolute incidence rates among infected groups (e.g., 115.90 per 100,000 PYs for viral hepatitis; overall infected 98.41 vs. unexposed 45.69). The magnitude and consistency of these associations across several common virus categories support a generalizable elevation in long-term cardiomyopathy risk following infection. While causality cannot be inferred from observational data, the pattern that hepatotropic and respiratory pathogens carry larger relative hazards is clinically coherent and actionable for risk-adapted follow-up. Notably, influenza grouped under viral pneumonia showed an aHR ≈3.0, reinforcing that vaccine-preventable infections may hold downstream implications for structural heart disease beyond the acute phase.
Principal findings
Individuals with a clinically recorded viral infection experienced a higher long-term incidence of cardiomyopathy than matched unexposed controls. The association was consistent across multiple robustness checks—including competing-risk adjustment for death, proportional-hazards diagnostics, alternative exposure/lag definitions, and subgroup analyses—and persisted across age, sex, and comorbidity strata1,2,3,4,13,14. While our design estimates association rather than causation, the directionality, persistence, and internal consistency of the signal align with contemporary evidence linking post-viral myocardial injury, immune dysregulation, and fibrotic remodelling to subsequent structural heart disease15,16,17,18.
Context with prior evidence
Viral pathogens are established precipitants of myocarditis; a proportion of patients demonstrate progression from post-inflammatory injury to dilated or arrhythmogenic phenotypes, particularly when inflammation is protracted or recurrent15,16,17,18,19. Large population-based analyses during and after the COVID-19 era have further demonstrated an excess of long-term cardiovascular events—including dysrhythmias, heart failure, and myocarditis—after infection, supporting the plausibility of downstream structural disease and latent remodelling20,21,22,23. Our findings extend this literature by focusing on incident cardiomyopathy as a defined outcome in routine care with long follow-up, leveraging a near-universal coverage health system and rigorous covariate balance, which improve generalizability and reduce selection bias relative to single-centre cohorts1,4,7,24.
Biological underpinnings
Mechanistically, cardiotropic and systemic viruses can trigger direct cytopathic injury, antigen-mimicry–mediated autoimmunity, and maladaptive host responses (e.g., interferon signalling, inflammasome activation), all of which may culminate in extracellular-matrix deposition, impaired energetics, and adverse remodelling16,17,18,25,26,27. Persistent low-grade inflammation and microvascular dysfunction—as described after several respiratory and hepatotropic infections—offer convergent explanations for the delayed manifestation of ventricular dysfunction and arrhythmogenic substrate observed in longitudinal cohorts21,22,26,28. These pathways are not mutually exclusive and likely interact with host factors such as age, metabolic disease, genetics, and prior myocardial reserve25,27,28,29.
Heterogeneity by pathogen and host. Although our primary objective was to estimate an overall association, exploratory analyses suggested heterogeneity across pathogen groups and clinical subgroups. Signals appearing stronger for selected respiratory and hepatotropic viruses are concordant with prior descriptions of cardiotropic potential, systemic inflammatory burden, and organ-crosstalk (e.g., cardio-hepatic, cardio-pulmonary axes)15,16,17,18,26,30. Host factors also matter older age, multimorbidity, and higher baseline healthcare utilization—proxies for frailty and physiological vulnerability—were associated with larger relative or absolute risk differences in some strata, echoing prior precision-risk findings after viral illness20,23,28,31. These patterns, while hypothesis-generating, highlight targets for pathogen-resolved surveillance and tailored prevention19,26,30,31,32.
Clinical implications
Given the ubiquity of viral infections, even modest relative risk elevations can translate into meaningful population burden. Our results support pragmatic, risk-adapted post-infection surveillance—especially for older adults, individuals with cardiometabolic disease, and those with severe or recurrent infections1,20,23,33. In practice, this could include symptom-prompted natriuretic peptide testing, echocardiography (including global longitudinal strain when available), and ambulatory rhythm monitoring in selected patients, coupled with timely initiation of guideline-directed medical therapy when overt dysfunction is detected15,18,34. From a systems perspective, vaccination and infection-control policies—endorsed across contemporary guidance—may yield downstream cardiac benefits that extend beyond preventing acute illness and should be viewed as part of comprehensive cardiovascular risk management12,20,21,33,34,35.
Public-health and health-system perspective
Health systems could pilot longitudinal “infection-to-cardiac-care” pathways: electronic prompts after a documented infection for patients at elevated baseline risk; streamlined access to follow-up evaluation; and integration with primary-care and cardiology services7,24,33,36. Such programs warrant evaluation for benefit, cost-effectiveness, and equity, as infection burden and access to follow-up care are socially patterned and may widen disparities if unaddressed31,36,37. These estimates justify targeted post-infection surveillance, particularly after viral hepatitis and respiratory viral illnesses: symptom-prompted natriuretic peptides, echocardiography (including strain where available), and rhythm monitoring in higher-risk patients (older age, cardiometabolic comorbidities, severe/recurrent infections). From a population perspective, the observed doubling of incidence relative to unexposed cohorts underscores potential secondary cardiac benefits of vaccination and infection-control strategies.
Clinical protocol for dyspnoea after viral infection
To support implementation in publicly funded systems, we propose a stepwise pathway for patients with new or persistent dyspnoea after a clinically recorded viral infection (Table S6) (e.g., symptoms ≥ 4 weeks or exertional limitation):
Step 1—Primary-care screening (all patients)
History/physical; vitals and SpO2 (rest and post-walk if feasible).
12-lead ECG and chest radiograph.
Natriuretic peptide (BNP or NT-proBNP) using locally validated non-acute thresholds to help rule out heart-failure–related dyspnea in primary care.
Basic labs as available (CBC, renal/electrolytes, thyroid), and cardiometabolic risk review.
Step 2—Risk-tiering and next actions
Low risk (normal vitals/SpO2, normal ECG/CXR, natriuretic peptide not suggestive of HF, no red flags; mild outpatient infection; age < 65; no major cardiometabolic comorbidity): education, optimization of risk factors, and re-assessment at 4–6 weeks.
Intermediate risk (any single abnormal screen—e.g., borderline natriuretic peptide or ECG changes—or persistent dyspnea affecting function): arrange transthoracic echocardiography within 4–12 weeks; consider ambulatory rhythm monitoring for palpitations/syncope; manage contributory conditions (anemia, thyroid, COPD/asthma).
High risk/red flags (resting SpO2 < 94% or exertional desaturation, new pathologic ECG changes, markedly elevated natriuretic peptide, rising troponin or ischemic pain, syncope, signs of heart failure, or rapidly progressive symptoms): urgent cardiology referral; same-week echocardiography; cardiac MRI considered only when echo or biomarkers suggest myocardial involvement and MRI access exists.
Where imaging capacity is limited, prioritize natriuretic peptides + ECG/CXR as gatekeepers for echocardiography; use point-of-care ultrasound if available; defer cardiac MRI to cases with persistent diagnostic uncertainty after echo/biomarkers. This staged approach focuses specialist resources on the small subset most likely to benefit, while remaining scalable and generalizable. (We emphasize that our cohort demonstrates an association rather than causation; the pathway aims to guide pragmatic follow-up, not mandate universal advanced imaging.)
Methodological considerations
Several features strengthen inference. First, we used a large, population-based database with stable coverage and transparent governance, enabling reproducible identification of exposures, outcomes, and covariates7,24. Second, propensity-score matching balanced demographics, comorbidities, medications, and utilization patterns, mitigating confounding by indication and healthcare-seeking behaviour9,38,39. Third, we complemented Cox models with diagnostics for the proportional-hazards assumption and sensitivity analyses (lag/washout windows) to reduce reverse causation and diagnostic-work-up bias10,40,41. Fourth, we accounted for the competing risk of death using sub distribution hazards models, consistent with contemporary recommendations when competing events are non-negligible10,11,41. Finally, outcome ascertainment used claims-based algorithms supported by recent validation literature in cardiovascular populations, acknowledging that performance varies by setting and phenotype12,35,37.
This observational design has constraints limitations. First, misclassification of exposure and outcome is possible because ascertainment relied on diagnosis codes rather than virologic confirmation, imaging, or adjudication. Although we applied validated definitions and robustness checks, nondifferential misclassification would likely bias estimates toward the null12,35,37. Second, the KM curves show persistent separation, and competing-risk analyses yielded directionally concordant results. Outcome phenotyping was limited by code-based ascertainment (ICD-9-CM 425.xx), precluding reliable separation of dilated, hypertrophic, and other cardiomyopathy subtypes in this database, which we now state prominently in the abstract, and methods. Third, residual confounding cannot be excluded despite extensive matching and adjustment (e.g., frailty, physical activity, genetic susceptibility, socio-economic gradients)9,38,39, but sensitivity analyses (proportional-hazards diagnostics, washout/lag windows, inpatient vs outpatient strata) did not materially alter the conclusions.
Fourth, we lacked granular clinical data—cardiac MRI, biomarker panels, strain, and ejection-fraction trajectories-precluding mechanistic inference and phenotype-specific risk estimates16,17,18,26,34. Fifth, infection severity and treatment were approximated by care-setting and utilization proxies; dose–response inferences should therefore be cautious20,21,22. Sixth, coding practices and pathogen epidemiology in Taiwan may differ from other regions, which may limit external generalizability; harmonized definitions and external replication are needed7,24,36. Finally, Generalizability to PLWH. Because individuals with HIV were excluded to avoid etiologic and measurement heterogeneity, findings may not generalize to PLWH; HIV-specific analyses would require clinical biomarkers (CD4, viral load), antiretroviral regimen data, and adjudicated outcomes.
Future directions
Linking claims to electronic health records, imaging repositories, and biospecimens will enable refined phenotyping and causal inference. Priorities include: (i) pathogen-resolved cohorts with standardized cardiac follow-up after infection; (ii) integration of cardiac MRI and omics/immune profiling to map trajectories from inflammation to fibrosis; (iii) evaluation of host susceptibility (genetics, immune signatures) and environmental modifiers; and (iv) trials testing whether risk-adapted surveillance or early rhythm/heart-failure management reduces transition to overt cardiomyopathy15,16,17,18,20,21,22,23,26,28,30,34. At the population level, modelling the absolute burden preventable through vaccination and infection-control strategies may inform policy and resource allocation, particularly for high-risk groups33,35,36,37.
Conclusions
In a large, real-world population, viral infection was associated with a sustained elevation in the long-term risk of incident cardiomyopathy. Although causal attribution cannot be established, convergence with contemporary biological and epidemiologic evidence supports post-infection cardiovascular vigilance and prevention strategies, while motivating pathogen-specific, mechanism-oriented research to refine risk stratification and interventions.
Data availability
The data that supports the findings of this study are available from the corresponding author upon reasonable request.
References
Arbelo, E. et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 44(37), 3503–3626. https://doi.org/10.1093/eurheartj/ehad194 (2023).
Tschöpe, C. et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 18(3), 169–193. https://doi.org/10.1038/s41569-020-00435-x (2021).
Fairweather, D. L. et al. COVID-19, myocarditis and pericarditis. Circ. Res. 133(9), e891–e909. https://doi.org/10.1161/CIRCRESAHA.123.321878 (2023).
Xie, Y., Xu, E., Bowe, B. & Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 28(3), 583–590. https://doi.org/10.1038/s41591-022-01689-3 (2022).
Ying, C. et al. Viral myocarditis. Int. J. Mol. Sci. 25(10), 8127. https://doi.org/10.3390/ijms25108127 (2024).
U.S. FDA. Real-world data: Assessing electronic health records and medical claims data to support regulatory decision-making for drug and biological products (final guidance). 2024-07-25.
Wang, T. H. et al. A national health insurance research database. Int. J. Med. Inform. 178, 105208. https://doi.org/10.1016/j.ijmedinf.2023.105208 (2023).
Huang, J. Y. et al. The risk of endometrial cancer and uterine sarcoma… using Taiwan’s LGTD2000. eClinicalMedicine 56, 101804 (2023).
Medaglio, D. et al. Research and scholarly methods: Propensity scores. J. Orthop. Traumatol. 23(1), 7. https://doi.org/10.1186/s10195-022-00636-8 (2022).
Sjölander, A. Why test for proportional hazards—or any other model assumption?. Am. J. Epidemiol. 193(6), 926–928. https://doi.org/10.1093/aje/kwae013 (2024).
Mansournia, M. A. et al. A practical guide to handling competing events in etiologic time-to-event studies. Stat. Med. 41(17), 3299–3326. https://doi.org/10.1002/sim.9392 (2022).
Bates, B. A. et al. Validation of ICD-10 diagnosis codes for identification of acute heart failure hospitalizations. Circ. Cardiovasc. Q. Outcomes 16(4), e009078. https://doi.org/10.1161/CIRCOUTCOMES.122.009078 (2023).
Gluckman, T. J. et al. 2022 ACC expert consensus decision pathway on cardiovascular sequelae of COVID-19 in adults. J. Am. Coll. Cardiol. 79(17), 1717–1756. https://doi.org/10.1016/j.jacc.2022.02.003 (2022).
Vrsalovic, M. & Vrsalovic, P. A. Cardiac complications in patients with COVID-19: A systematic review and meta-analysis of longitudinal studies. Eur. J. Intern. Med. 83, 34–47. https://doi.org/10.1016/j.ejim.2020.11.044 (2021).
Caforio, A. L. P. et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: A position statement. Eur. Heart J. 44(17), 1510–1532. https://doi.org/10.1093/eurheartj/ehad061 (2023).
Ferreira, V. M. et al. Cardiovascular magnetic resonance in myocarditis: Expert recommendations update. J. Am. Coll. Cardiol. 79(20), 2077–2095. https://doi.org/10.1016/j.jacc.2022.03.350 (2022).
Mevorach, D. et al. Myocarditis after BNT162b2 mRNA vaccine in Israel. N. Engl. J. Med. 385(23), 2140–2149. https://doi.org/10.1056/NEJMoa2109730 (2021).
Witberg, G. et al. Myocarditis after Covid-19 vaccination in a large health care organization. N. Engl. J. Med. 385(23), 2132–2139. https://doi.org/10.1056/NEJMoa2110737 (2021).
Buckley, B. J. R. et al. Prevalence and clinical outcomes of post-COVID syndrome: A systematic review and meta-analysis. Eur. Heart J. Q. Care Clin. Outcomes 8(7), 700–712. https://doi.org/10.1093/ehjqcco/qcac058 (2022).
Xie, Y., Xu, E. & Al-Aly, Z. Risks of mental health outcomes in people with COVID-19: Cohort study. BMJ 376, e068993. https://doi.org/10.1136/bmj-2021-068993 (2022).
Kotecha, T. et al. Patterns of myocardial injury in recovered COVID-19 patients assessed by CMR. J. Am. Coll. Cardiol. Img. 14(11), 2337–2352. https://doi.org/10.1016/j.jcmg.2021.04.025 (2021).
Puntmann, V. O. et al. Longitudinal CMR in patients after COVID-19. Eur. Heart J. Cardiovasc. Imaging 23(6), 714–725. https://doi.org/10.1093/ehjci/jeab271 (2022).
Ssentongo, P. et al. Cardiovascular disease burden among long-COVID patients: Systematic review and meta-analysis. Open Heart. 10, e002196. https://doi.org/10.1136/openhrt-2022-002196 (2023).
Huang, J. Y. et al. Methods using Taiwan’s LGTD2000: A nationwide cohort resource. eClinicalMedicine 56, 101804. https://doi.org/10.1016/j.eclinm.2022.101804 (2023).
Bozkurt, B., Kamat, I. & Hotez, P. J. Myocarditis with COVID-19 mRNA vaccines: Mechanisms and management. Nat. Rev. Cardiol. 18(12), 779–780. https://doi.org/10.1038/s41569-021-00662-w (2021).
Heymans, S. & Cooper, L. T. Jr. Myocarditis after COVID-19 mRNA vaccination: Clinical observations and potential mechanisms. Nat. Rev. Cardiol. 19(2), 75–77. https://doi.org/10.1038/s41569-021-00662-w (2022).
Tschöpe, C. & Van Linthout, S. New insights into immune-mediated myocardial injury and fibrosis. Cardiovasc. Res. 117(9), e95–e97. https://doi.org/10.1093/cvr/cvab171 (2021).
Chen, C. et al. Global prevalence of post-COVID symptoms: A meta-analysis. Nat. Commun. 13, 2057. https://doi.org/10.1038/s41467-022-29513-z (2022).
Thavendiranathan, P. et al. Use of myocardial strain imaging (GLS) for early detection of subclinical LV dysfunction. J. Am. Soc. Echocardiogr. 34(7), 763–778. https://doi.org/10.1016/j.echo.2021.03.010 (2021).
Aimo, A. et al. Biomarkers for myocarditis and inflammatory cardiomyopathy. Heart Fail. Rev. 27(3), 625–643. https://doi.org/10.1007/s10741-021-10096-5 (2022).
Yelin, D. et al. Patterns of somatic and autonomic symptoms in long COVID and cardiovascular risk. Clin. Microbiol. Infect. 29(5), 741–748. https://doi.org/10.1016/j.cmi.2022.12.014 (2023).
Sudre, C. H. et al. Attributes and predictors of long COVID. Nat. Med. 27(4), 626–631. https://doi.org/10.1038/s41591-021-01292-y (2021).
Mesnier, J. et al. Hospital admissions for heart disease during COVID-19 and vaccination eras: Nationwide analysis. Lancet Reg. Health Eur. 16, 100358. https://doi.org/10.1016/j.lanepe.2022.100358 (2022).
Vaduganathan, M. et al. Initiation and intensification of GDMT for heart failure—clinical implications. Lancet 398(10305), 1834–1847. https://doi.org/10.1016/S0140-6736(21)01947-0 (2021).
Quan, H. et al. Updating and validating the Charlson comorbidity index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am. J. Epidemiol. 173(6), 676–682 (2011).
Marmot, M. et al. Build back fairer: The COVID-19 Marmot review—health equity implications for CVD care pathways. J. Epidemiol. Comm. Health 75(3), 242–244. https://doi.org/10.1136/jech-2020-215091 (2021).
Patel, A. P. et al. Social determinants, COVID-19, and cardiovascular outcomes. Circulation 143(9), 873–875. https://doi.org/10.1161/CIRCULATIONAHA.120.050354 (2021).
Austin, P. C. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav. Res. 46(3), 399–424 (2011).
Franklin, J. M. & Schneeweiss, S. When and how can real-world data analyses substitute for randomized trials?. Clin. Pharmacol. Ther. 111(1), 24–31. https://doi.org/10.1002/cpt.2379 (2022).
Grambauer, N., Schumacher, M. & Beyersmann, J. Proportional hazards for flexible hazard models: Checking assumptions in practice. Stat. Med. 40(10), 2471–2486. https://doi.org/10.1002/sim.8901 (2021).
Austin, P. C., Lee, D. S. & Fine, J. P. Introduction to the analysis of survival data in the presence of competing risks. Circulation 141(6), 601–603. https://doi.org/10.1161/CIRCULATIONAHA.119.043866 (2020).
Acknowledgements
We appreciate the support from the Tri-Service General Hospital Research Foundation and the Medical Affairs Bureau, Ministry of Defense, Taiwan, ROC. We also appreciate the database provided by the Health and Welfare Data Science Center, Ministry of Health and Welfare (HWDC, MOHW).
Funding
This study was funded by the Tri-Service General Hospital, grant number: TSGH-A-114010, TSGH-B-114022, TSGH-D-114196.
Author information
Authors and Affiliations
Contributions
P.-C.Y., Y.-C.L., T.-H.W., Y.-C.H., R.-J. C., L.-Y. F., C.-H.C., C.-S.T., and W.-C.C.: conception and design, analysis and interpretation of the data, critical review, and approval of the final version submitted for publication. P.-C.Y., S.-H.H., R.-J. C., B.-L.W., H.-T.H., Y.-C.L., C.-S.T., L.-Y. F., and Y.-C.H.: statistical analysis, critical review, and approval of the final version submitted for publication. P.-C.Y., S.-H.H., B.-L.W., H.-T.H., T.-H.W., Y.-C.H., R.-J. C., C.-H.C., Y.-C.L., C.-S.T., L.-Y. F., and W.-C.C.: drafting of the paper, critical review, and approval of the final version submitted for publication. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Ethics approval and consent to participate
This study was conducted according to the Code of Ethics of the World Medical Association (Declaration of Helsinki). The Institutional Review Board of the Tri-Service General Hospital approved this study (TSGHIRB: No: E202516027) and waived the need for individual consent since all the identification data were encrypted in the NHIRD.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, PC., Hsin, HT., Chung, RJ. et al. Increased cardiomyopathy risk after viral infection evidence from a nationwide cohort in Taiwan. Sci Rep 15, 41561 (2025). https://doi.org/10.1038/s41598-025-25454-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-25454-x
