
Abstract
Bladder cancer progression is closely linked to metabolic changes within the tumor microenvironment (TME), particularly elevated lactate levels. Lipocalin 2 (LCN2), a protein involved in iron regulation, has been implicated in tumor progression in several cancers, but its regulatory mechanisms and role in bladder cancer remain unclear. Bladder cancer cell lines were treated with exogenous lactate or subjected to inhibition of endogenous lactate production to evaluate changes in LCN2 expression. The role of Signal Transducer and Activator of Transcription 3 (STAT3) was assessed using genetic knockdown and pharmacological inhibitors. Functional assays including migration, epithelial-to-mesenchymal transition (EMT), sphere formation, and stemness marker analysis were performed to determine the biological effects of LCN2. Chemoresistance to gemcitabine was examined with and without LCN2 silencing. Clinically, LCN2 expression was significantly associated with the pathological relevance of bladder tumors. Lactate significantly induced LCN2 expression in bladder cancer cells through activation of STAT3. Disruption of STAT3 signaling reduced LCN2 levels. High LCN2 expression correlated with advanced tumor grade, poor prognosis, and shorter overall survival. Functionally, LCN2 promoted EMT, enhanced cell migration, and increased stem cell-like properties, as evidenced by elevated stemness markers and sphere-forming ability. Moreover, lactate-induced LCN2 expression conferred resistance to gemcitabine treatment, while LCN2 knockdown restored chemosensitivity. This study identifies tumor-derived lactate as a key inducer of LCN2 expression via STAT3 activation in bladder cancer. LCN2 contributes to tumor aggressiveness, cancer stemness, and chemoresistance. Targeting the STAT3-LCN2 signaling axis may offer a promising therapeutic strategy to suppress bladder cancer progression and overcome treatment resistance within a lactate-rich TME.
Similar content being viewed by others


Background
Bladder cancer (BLCA) is the ninth most common malignancy worldwide, and has the sixth highest incidence rate among male malignancies, associated with high recurrence rates and a strong tendency for metastasis1. Pathologically, BLCA is classified into non-muscle-invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC). Once BLCA progresses to an invasive stage, it becomes largely incurable, with a poor prognosis. The 5-year survival rate of metastatic bladder cancer is only 5%, posing a significant clinical challenge2.
Cancer stem cells (CSCs) represent a small but significant subpopulation of tumor cells within the tumor mass that contribute to tumor recurrence, metastasis, and poorer prognosis in BLCA. Targeting CSCs or understanding the underlying mechanisms driving their formation may provide novel therapeutic avenues for bladder cancer treatment3,4. The continuous interactions between tumor cells and diverse extracellular components within the tumor microenvironment (TME) play a crucial role in driving tumor progression5. For example, collagen matrices, the most abundant structural protein in the human extracellular matrix (ECM), have been shown to drive local invasion and distant lung metastasis6 while also modulating immunotherapy efficacy in bladder cancer7. The bladder TME is dynamic and constantly evolving, with ongoing crosstalk between bladder tumor cells and their surrounding TME shaping therapeutic vulnerabilities8. This interplay highlights the importance of deciphering the mechanisms that promote bladder cancer malignancy to develop more effective and targeted treatment strategies. Understanding these interactions could lead to novel therapeutic approaches aimed at disrupting key tumor-promoting pathways within the TME, ultimately improving patient outcomes.
Multiple metabolic abnormalities contribute to bladder tumorigenesis9. The Warburg effect elaborates the unique metabolic adaptations of tumors. Cancer cells preferentially utilize glucose as their primary metabolic fuel and convert it into lactate, even in the presence of sufficient oxygen10. Bladder cancer cells exhibit excessive alterations in glucose and pyruvate metabolism, correlating with disease progression and a highly proliferative state11, suggesting a metabolic shift within the tumor microenvironment (TME), where cancer cells reprogram their metabolism to sustain their malignancy. Tumors are increasingly recognized as metabolic diseases, relying on metabolic reprogramming to accelerate cell proliferation, promote metastasis, and enhance resistance to chemotherapy. This metabolic flexibility allows cancer cells to adapt to harsh microenvironmental conditions, evade immune surveillance, and sustain uncontrolled growth12. Tumor cells catalyze the conversion of glucose-derived pyruvate into lactate by the action of lactate dehydrogenase A (LDHA)13. Notably, high lactate levels (10–30 mM) have been reported in the serum of cancer patients compared to healthy tissue and serum (1.5-3 mM)14,15. Initially regarded as a mere “metabolic waste” product of glycolysis, lactate has garnered increasing attention for its crucial role in tumor biology. Beyond contributing to the acidification of the tumor microenvironment (lactate-rich TME), excessive lactate production actively promotes tumor malignancy by driving cancer progression, immune evasion, and therapeutic resistance16,17.
Lipocalin-2 (LCN2), also known as neutrophil gelatinase-associated lipocalin (NGAL), is a member of the lipocalin superfamily with diverse pathological roles that vary depending on its tissue distribution18. LCN2 has been implicated in lung and liver fibrosis19,20, obesity-related insulin resistance21, and chronic kidney disease22. In cancer, LCN2 expression is dysregulated across multiple tumor types, often contributing to tumor progression. While its oncogenic role has been well documented in several malignancies23, its function remains controversial, as some studies suggest a tumor-suppressive role in colon and pancreatic cancers24,25. In bladder cancer, a clinical study reported elevated LCN2 expression in urine and serum samples, correlating with an aggressive phenotype and poor prognosis26, which implies the clinical significance of LCN2 as a diagnostic biomarker for bladder tumor. However, its precise role in bladder cancer progression, particularly within the metabolically altered lactate-rich tumor microenvironment (lactate-rich TME), remains poorly understood, necessitating further investigation into its regulatory mechanisms and pathological significance.
In this study, we identified tumor-derived lactate as a key regulator of LCN2 expression via STAT3 activation. Both exogenous lactate treatment and inhibition of endogenous lactate production confirmed this mechanism. STAT3 inhibition significantly suppressed lactate-induced LCN2 expression. Clinically, high LCN2 expression correlated with poor prognosis and aggressive tumor characteristics in bladder cancer. The key lactate-producing enzyme, LDHA, and STAT3 expression were positively associated with LCN2 in clinical tumor tissues, reinforcing the link between lactate metabolism and LCN2 expression. Functionally, LCN2 promoted bladder cancer cell migration, epithelial-mesenchymal transition (EMT), and cancer stem-like properties, while its knockdown impaired these processes. Additionally, LCN2 contributed to lactate-driven chemoresistance, as its silencing reduced gemcitabine resistance. These findings establish the STAT3-LCN2 axis as a key driver of bladder cancer progression and therapy resistance, presenting a potential therapeutic target.
Methods
Cell culture, transfection and infection
Human bladder cancer cell lines 5637 (obtained from the Bioresource Collection and Research Center, Taiwan) and MC-SV-HUC T-2 (MC-T2; obtained from the American Type Culture Collection, USA) were cultured in RPMI-1640 medium (Corning, Cat#50-020-PC, NY, USA) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Cat#10437028, Waltham, USA) and 100 µg/ml streptomycin and 100 U/ml penicillin (Corning, Cat. 30-002-CI, NY, USA). Cells were maintained at 37 °C in a humidified incubator with a 5% CO₂ atmosphere. For STAT3, LCN2 and LDHA knockdown, lentivirus-based shRNAs constructs were obtained from the RNAi Core Lab of Sinica in Taiwan. LCN2 shRNA clone: TRCN0000060290; STAT3 shRNA clone: TRCN0000329887 and LDHA shRNA clone: TRCN0000026537. These shRNA constructs were transfected into HEK293T cells using calcium phosphate transfection method to generate lentiviral particles containing the targeted shRNA knockdown sequence. These lentiviral particles were collected from the medium of transfected 293T cells and then transferred to 5637 and MC-T2 cells (40–50% confluence) with polybrene (Santa Cruz, CAS#28728-55-4). The process of selecting stable knockdown cells was conducted by incubating a medium containing 4 µg/ml of puromycin (Sigma-Aldrich Inc.) for two weeks.
Western blot analysis
Cells lysates were harvest by RIPA buffer containing protease inhibitors and phosphatase inhibitors. Protein concentration will be determined using a Bio-Rad DC protein assay kit. 25–30 µg of total protein was loaded onto 10 or 8% SDS-polyacrylamide gel electrophoresis and transfer to a PVDF membrane. The protein was identified by incubating the membrane with primary antibodies and followed by HRP-conjugated secondary antibodies and enhanced chemiluminescence solution.
Antibodies and reagents
LCN2 (proteintech, 26991-1-AP); b-Actin (Santa Cruz, sc-47778); STAT3 (Cell Signaling #9139); p-STAT3 (Tyr705, Cell Signaling #9138); Vimentin (Cell Signaling #5741); N-cadherin (Cell Signaling #13116S); E-cadherin (Cell Signaling #3195) and LDHA (Cell Signaling #3582) antibodies were used in this study. Secondary antibodies for western blot analysis: anti-rabbit and anti-mouse HRP-conjugated secondary antibodies were purchased from Cell signaling. Secondary antibody for immunofluorescence analysis: rabbit anti-mouse IgG-iFluor 594 secondary antibody was from CROYEZ (Cat: C04035). Chemotherapy drug: Gemcitabine (S1714); STAT3 inhibitor-Napabucasin (S7977) were purchased from Selleckchem; Lactate (L6402, Sigma-Aldrich); Oxamate (ThermoFisher) and Sodium L-lactate (L7022, Sigma-Aldrich).
In vitro 3D sphere formation assay
To evaluate the cancer stem-like properties of bladder cancer cells, the cells were cultured in low-adhesion plates to allow tumor sphere formation. Briefly, single-cell suspensions were seeded at a density of 2000–3000 cells per 6-cm ultra-low-adhesion culture dish in RPMI-1640 complete medium supplemented with 10% FBS and 1% penicillin/streptomycin. Under these non-adherent conditions, cells grew into tumor spheres over a period of 10–14 days. At the end of the experiment, the tumor spheres were collected by gentle centrifugation, washed in PBS, and subjected to RNA extraction. The expression of stemness-related genes was then assessed using real-time PCR analysis.
Quantitative reverse transcription real-time polymerase chain reaction (qRT-PCR)
Total RNA from tumor spheres were isolated using the Aurum Total RNA Mini Kit (Cat. #732–6820, BIO-RAD) and reverse-transcribed with the Reverse Transcription Supermix Kit (Cat. #1708841, BIO-RAD). The cDNA products were used to assess the mRNA expression of stemness-related genes. The following primers were used: GAPDH: Forward:5′-TGCACC. ACCAACTGCTTAGC-3′, Reverse: 5′-GGCATGGACTGTGGTCATGAG-3′; LCN2: Forward:5′-GTGAGCACCAACTACAACCAGC-3′, Reverse: 5′-GTTCCGAAGTCAGCTC. CTTGGTT-3′; Oct-4: Forward:5′-TATGCACAACGAGAGGATTTTG-3′, Reverse: 5′- TGTGTCCCAGGCTTCTTTATTT-3′; Nanog: Forward:5′-CAGAAGGCCTCAGCACCTAC-3′, Reverse: 5′- CTGTTCCAGGCCTGATTGTT-3′; SOX2 Forward:5′-AGGGCTGGACTGC. GAACTG-3′, Reverse: 5′- TTTGCACCCCTCCCAATTC-3′; SOX5: Forward:5′-CAGCCAGAGTTAGCACAATAGG-3′, Reverse:5′-CTGTTGTTCCCGTCGGAGTT-3′; SOX9: Forward:5′- AGCTCACCAGACCCTGAGAA-3′, Reverse: 5′-TCCCAGCAATCGT. TACCTTC-3′. Quantitative real-time PCR was performed using iTaq Universal SYBR Green Supermix (BIO-RAD, Cat. #172–5121) and the Roche LightCycler96 machine. The cDNA expression levels of each target gene were normalized to GAPDH.
Trans-well migration assays
Cell migration was assessed using a trans-well migration assay with 24-well plates and 8-µm pore size trans-well chambers (Corning). Bladder cancer cells were pre-treated with 10 mM lactic acid for 24 h before the assay. A total of 5 × 104 cells were resuspended in serum-free medium and seeded into the upper chamber, while 400 µl of culture medium containing 10% FBS was added to the lower chamber to create a nutrient gradient. After 14–16 h of incubation, non-migrated cells in the upper chamber were removed with a cotton applicator. Migrated cells were washed with PBS, fixed with methanol, and stained with 0.1% crystal violet solution. Cell migration was quantified by counting cells in the lower chamber beneath the membrane.
Cell viability assay
To assess the effect of lactic acid on chemoresistance, bladder cancer cell viability was measured using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. A total of 12,000 cells/well were cultured n a 96-well plate and pretreated with 10 mM lactic acid before exposure to gemcitabine at concentrations of 0, 7.8, 15.625, 31.25, 125, 500, 1000, and 2000 nM. After 24 h of incubation, 20 µl of MTT solution (4 mg/ml) in 100 µl culture medium was added to each well and incubated at 37 °C for 2 h. The supernatant was then aspirated, and 100 µl of DMSO was added to dissolve the formazan crystals. Relative cell viability was determined by measuring absorbance at 570 nm using a microplate reader.
Scratch-wound assay
The wound scratch assay was conducted to assess the migratory ability of bladder cancer cells in vitro. Cells were cultured using a Culture-Insert (IB-80209, ibidi) to create a thin, uniform scratch (wound) in the central area of a six-well plate. Detached and damaged cells were carefully removed with PBS, and the remaining cells were culture in growth medium. Wound closure was monitored by a light microscopy and the images of the scratches were captured at the indicated time points (0, 3, 6 and 9 h). The extent of wound closure was quantified by measuring the remaining wound area relative to the initial wound width, using ImageJ software for analysis. All experiments were performed in triplicate to ensure reproducibility.
Immunofluorescence assay
Cells were cultured on 35-mm diameter glass coverslips in a 24 well-plate. Following treatment with 10 mM lactic acid, cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Following fixation, cells were washed with PBS and permeabilized using 0.1% Triton X-100 for 20 min, followed by an additional PBS wash. Slides were blocked with 2% BSA for 30 min at room temperature to minimize nonspecific antibody binding. Cells were then incubated with an anti-STAT3 antibody (1:100 dilution) in a humidified chamber overnight at 4 °C. After primary antibody incubation, cells were washed and incubated with rabbit anti-mouse IgG-iFluor 594 secondary antibody (1:200; CROYEZ) for 1 h at room temperature in the dark. To visualize cell nuclei, DAPI staining was performed by applying fluoroshield mounting medium with DAPI (abcam, AB104139).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 10 software. Results are expressed as mean ± SEM from at least three independent experiments. Student’s t-test and analysis of variance were used to compare two and multiple groups, respectively. Significant differences are indicated by *P < 0.05, **P < 0.01 and ***P < 0.001.
Results
High LCN2 expression is associated with poor prognostic features in bladder tumors
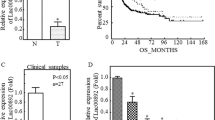
To establish clinical relevance of LCN2 in bladder tumor, Gene Expression Profiling Interactive Analysis (GEPIA2) was first performed. GEPIA2 is a powerful tool for analyzing gene expression in cancer using TCGA (The Cancer Genome Atlas) and GTEx (Genotype-Tissue Expression) datasets27. Figure 1a indicated LCN2 was highly expressed in tumor tissues compared with the normal bladder tissue (N:28 v.s T = 404, p < 0.05) by GEPIA2 analysis. Moreover, the Human Protein Atlas (HPA) is a valuable tool for studying protein localization and expression in human tissues and cells28. We evaluated LCN2 expressions between normal and different grade of bladder tumors from publicly available HPA webpage. Representative immunohistochemical images and anti-LCN2 staining results revealed that compared to normal bladder tissue, bladder tumors expressed high LCN2 levels (Fig. 1b). Notably, moderate LCN2 staining intensity was found in high grade bladder tumors, whereas weak LCN2 staining was in low grade bladder tumors, suggesting LCN2 levels positive correlated with bladder tumor grades. Another independent database [GSE83586, Ta (n = 19), T1 (n = 44), T2 (n = 241) and T3 (n = 2)] confirmed similar results that LCN2 levels were higher in advanced tumors (T2/T3) compared to early-stage tumors (Ta/T1) (Fig. 1c, p = 0.034). Besides, LCN2 expression was associated with poorer survival outcomes in bladder cancer patients, as confirmed by analyses from two independent online databases (GEPIA2 and KM Plotter). GEPIA2 and KM Plotter analyses respectively indicated high LCN2 expression exhibited shorter overall survival in cancer patients (Fig. 1d, p = 0.015 and Fig. 1e, p = 0.015). Collectively, these data revealed the clinical significance of LCN2 expression in bladder tumors.

Lactate regulates LCN2 expression in bladder cancer cells and its association with clinical outcomes. (a) The expression of LCN2 in normal bladder tissues (n = 28, GTEx-BLCA) and bladder cancer tissues (n = 404, TCGA-BLCA cohort), with a significance threshold of p < 0.05 (b) Representative images at magnification x100 were obtained and LCN2 protein expression in normal tissue and bladder tumor tissues were analyzed through the Human Protein Atlas database. Normal urinary bladder tissue (Male, age 55). Low grade urothelial carcinoma (Female, age 73; weak LCN2 staining). High grade urothelial carcinoma (Male, age 81; moderate LCN2 staining). (c) Correlation of LCN2 gene expression with clinical tumor staging of bladder cancer in GSE83586 cohort. Box plots represent LCN2 expression levels across different tumor stages: Ta (n = 19), T1 (n = 44), T2 (n = 241), and T3 (n = 2). Statistical significance and expression trends are indicated (p = 0.0334), highlighting potential associations between LCN2 expression and tumor progression. (d) Kaplan-Meier survival analysis of bladder cancer patients based on LCN2 gene expression using the GEPIA2 online application (http://gepia2.cancer-pku.cn/#index). The follow-up period for overall survival was over 150 months. The blue and red lines represent the survival curve of patients with low (n = 201) and high (n = 201) LCN2 expression, respectively, under a 95% confidence interval for each survival curve. p-value = 0.015 (e) Kaplan-Meier Plotter database (http://kmplot.com/analysis/) was used to assess the correlation between LCN2 expression and overall survival in patients with bladder cancer. The analysis showed that patients with high LCN2 expression (n = 204) had a significantly lower survival rate than those with low LCN2 expression (n = 200) (HR = 1.45, 95% CI, p = 0.015). With a follow-up period of 60 months, suggesting that higher LCN2 expression is associated with a reduced survival probability. (f) 5637 bladder cancer cells were treated with different lactic acid concentrations (10 and 20 mM) and sodium lactate (50 and 100 mM) for 24 h, respectively. Cell lysates were collected and subjected to western blot analysis using anti-LCN2 antibody. b-actin was used as a loading control. (g) The correlation between LCN2 and LDHA expression of the TCGA database were generated by cBioPortal website. (h) 5637 were treated with oxamate (50 and 100 mM for 24 h) to inhibit LDHA activity. Cell lysates were harvested and subjected to western blot analysis to assess LCN2 expression. (i) Cells were stably transduced with either scrambled shRNA control lentivirus or shRNA lentivirus targeting LDHA. Western blotting was performed to examine LDHA and LCN2 protein levels in 5637 cells.
Tumor-derived lactate regulates LCN2 expression
To establish the association between tumor-derived lactate-rich TME and LCN2 expression, different concentrations of exogenous lactate were first applied in bladder cancer treatment to simulate varying pathology severity. 5637 bladder cancer cells were cultured in medium with 10 and 20 mM lactic acid (LA) for 24 h, respectively. The results in Fig. 1f revealed lactic acid treatment dose-dependently increased LCN2 protein expression in 5637 cells. In addition, lactic acid can dissociate into lactate and hydrogen ions (H+), the excessive presence of these two molecules mimics the pathological phenomena of extracellular acidic tumor microenvironment found in tumors of cancer patients. Accordingly, to clarify the effect of lactic acid-induced LCN2 was by lactate or by hydrogen proton, sodium lactate was employed to treat bladder tumor cells. Figure 1f showed sodium lactate treatment phenocopies the effect of lactic acid, inducing LCN2 expression in a dose-dependent manner, thereby demonstrating that lactate itself is essential for LCN2 upregulation. Consistent findings were observed in MC-SV-HUC T-2 (MC-T2) bladder cancer cells, as shown in Supplementary Fig. 1a-b.
Tumor cells preferentially utilize glucose as their primary metabolic fuel, leading to excessive lactate production via aerobic glycolysis, and lactate dehydrogenase A (LDHA) is the key enzyme responsible for catalyzing the conversion of pyruvate to lactate. Accordingly, to establish a mechanistic link where LDHA activity in bladder cancer influences tumor-derived lactate levels, subsequently affects LCN2 expression, TCGA cohort (N = 411) analysis was performed to evaluate the clinical association of LCN2 expression and LDHA expression levels in bladder tumors. Figure 1g showed LCN2 positively and significantly correlated with LDHA expression levels in bladder tumors (p = 0.0005279). Besides, to investigate the effect of tumor-derived lactate on LCN2 expression, we used oxamate, a lactate dehydrogenase inhibitor (LDHi), to block endogenous lactate production in tumor cells29. The results showed an obvious reduction in LCN2 expression in 5637 and MC-T2 bladder cancer cells when LDH activity was inhibited by oxamate (Fig. 1h and Supplementary Fig. 1c). To exclude the possibility that oxamate-mediated reduction of LCN2 was caused by off-target effects and to validate bladder cancer cells with high LDHA levels show elevated LCN2 expression (Fig. 1g), we next examined LCN2 expression in LDHA-silenced cells. LDHA suppression in 5637 and MC-T2 cells were achieved using shRNA knockdown and LCN2 expression was markedly reduced in LDHA-silenced bladder cancer cells (Fig. 1i and Supplementary Fig. 1d). These results reinforced the idea that tumor-derived lactate regulates LCN2 levels. Collectively, these results demonstrated that both exogenous lactate treatment and the inhibition of endogenous lactate production established a link between tumor-derived lactate and LCN2 expression and depicted the regulatory role of lactate metabolism in controlling LCN2 expression in bladder cancer cells.
Tumor-derived lactate modulates STAT3 activity
The expression of LCN2 was shown to be upregulated in response to metabolic changes, such as those induced by lactic acid accumulation in the tumor microenvironment (Fig. 1). Subsequently, the regulatory mechanisms underlying LCN2 expression in bladder cancer cells following lactic acid treatment was investigated. The mRNA levels of LCN2 following lactic acid treatment were assessed by qPCR analysis. The results revealed LCN2 mRNA levels were significantly and dose-dependently up-regulated in both 5637 and MC-T2 cells treated with lactic acid, compared to control cells without lactic acid treatment (Fig. 2a), suggesting lactate-mediated LCN2 expression was through transcriptional regulation. Next, “Network Explore” analysis using Ingenuity Pathway Analysis (IPA) to identify potential signaling networks associated with LCN2 in bladder tumor was conducted. Among the molecules identified, STAT3 emerged as a candidate (Fig. 2b). Furthermore, comparison between the LCN2 high-expression and low-expression groups from the TCGA BLCA cohort revealed distinct differential gene expression profiles. Notably, several immune-regulating pathways, as well as the JAK-STAT signaling pathway, were significantly altered in the LCN2 high-expression group (Fig. 2c). Furthermore, a clinical relevance was evaluated by TCGA cohort analysis, our data revealed that LCN2 mRNA levels positively correlated with STAT3 expression in clinical bladder tumor tissues (Fig. 2d, p = 3.43− 9), highlighting the potential mechanism of STAT3 in regulating LCN2 expressions. Based on these findings, we investigated the regulatory interaction between STAT3 and LCN2 in bladder tumors, particularly within this pathological context of lactate-rich TME. To address this, STAT3 activity was examined in lactic acid and sodium lactate treatment bladder cancer cells. Figure 2and f showed lactic acid and sodium lactate treatment respectively not only induced LCN2 expression, but also increased STAT3 activity (pY705 status) in 5637 cells. In contrast, suppression of LDHA activity using oxamate or knockdown of LDHA expression with shRNA in 5637 cells effectively abrogated both pY705 STAT3 phosphorylation and LCN2 expression (Fig. 2g h). These findings suggest that tumor-derived lactate-induced LCN2 expression is mediated through the STAT3 signaling pathway.

Lactate activates STAT3 in bladder cancer cells. (a) Quantitative PCR (qPCR) was performed to assess LCN2 mRNA levels in 5637 and MC-T2 cells treated with 10 mM and 20 mM for 24 h. Results are expressed as the ratio of LCN2 to GAPDH mRNA levels. Data represent the mean ± SEM from three independent experiments. Statistical significance was determined by an unpaired two-tailed t-test (*p < 0.05, **p < 0.01). (b) Exploring the potential mechanistic network between lactate, LCN2, and STAT3 in bladder tumors using Ingenuity Pathway Analysis (IPA). Gene Ontology (GO) analysis of the TCGA BLCA cohort (N = 411) was performed using the ShinyGO platform, comparing significantly differentially expressed genes between LCN2-high and LCN2-low expression groups (cutoff: median LCN2 expression). (d) A positive correlation of LCN2 and STAT3 mRNA expression in bladder tumors using cBioPortal website. (e) and (f) Western blot analysis of phosphorylated STAT3 (p-Y705 STAT3), total STAT3 and LCN2 expressions of 5637 cells under 10 mM and 20 mM lactic acid in (d), and 50 mM and 100 mM sodium lactate treatment in (e). (g) and (h) p-Y705 STAT3, STAT3 and LCN2 expression levels were examined in oxamate (50 and 100 mM) treatment and LDHA-silenced 5637 cells by western blot analysis.
STAT3 mediates lactic acid-induced LCN2 expression
To further validate the role of STAT3 in promoting LCN2 expression, immunofluorescence analysis was performed to assess STAT3 nuclear translocation in response to lactic acid treatment (5637 cells in Fig. 3a, and MC-T2 cells in Fig. 3b). The results revealed that lactic acid significantly increased nuclear STAT3 expression compared to untreated control cells, indicating that lactic acid promotes STAT3 activation. To further confirm that STAT3 mediates lactate-driven LCN2 expression, two complementary approaches were employed to inhibit STAT3 function in lactic acid-treated bladder cancer cells: shRNA-mediated STAT3 knockdown and treatment with BBI608, a selective STAT3 inhibitor. Western blot analysis confirmed effective silencing of STAT3 expression by STAT3 shRNA in in 5637 and MC-T2 cells, which was accompanied by impaired STAT3 activity (Fig. 3c). Notably, the steady-state protein levels of LCN2 were also affected by STAT3 reduction. Lower LCN2 expression was observed in STAT3-silenced cells compared to control cells. Subsequently, the stable STAT3 knockdown cells were employed to assess the role of STAT3 in regulating LCN2 expression under lactic acid treatment. As shown in Fig. 3d, lactic acid treatment increased both STAT3 activity and LCN2 expression. However, STAT3 depletion markedly abrogated lactic acid-induced LCN2 expression, indicating that STAT3 is required for lactic acid-mediated LCN2 regulation. Additionally, to validate the results observed in STAT3 knockdown cells and to access the clinical potential of targeting the STAT3-LCN2 signaling axis as a therapeutic strategy in bladder cancer, a Phase Ib/II clinical trial STAT3 inhibitor, BBI608 (Napabucasin), was employed in this study30. In the presence of BBI608, lactic acid-induced STAT3-Y705 phosphorylation was markedly inhibited. Furthermore, LCN2 expression was reduced in BBI608-treated cells compared to those treated with lactic acid alone (Fig. 3e). Collectively, together with the findings that oxamate treatment and LDHA silencing both impaired STAT3 activity and LCN2 expression (Fig. 2g,h), these results emphasized the role of STAT3 as a key regulator of LCN2 expression in lactate-rich TME.

Impaired STAT3 function attenuates lactate-induced LCN2 expression. (a) and (b) Nuclear expression of STAT3 under lactic acid treatment by immunofluorescence analysis. Cells were treated with 10 mM lactic acid for 24 h. Following treatment, cells were washed with PBS, fixed, permeabilized, and stained with anti-STAT3 antibody, followed by iFluor 594 secondary antibody. The nuclear stain is DAPI (blue). Quantification of nuclear STAT3 in the absence (control) and presence of lactic acid treatment in 5637 and MC-T2 cells was performed. Scale bar: 20 μm. *p < 0.05, **p < 0.01 (c) 5637 and MC-T2 bladder cancer cells were stably transduced with either scrambled shRNA control lentivirus or shRNA lentivirus against STAT3 (STAT3 KD). STAT3 and LCN2 expression levels were confirmed by western blot analysis. (d) Examining the induction efficiency of lactic acid treatment on LCN2 expression in STAT3-silenced 5637 cells. Scrambled control and STAT3 knockdown (STAT3 KD) cells were treated with 10 mM lactic acid for 24 h. Cell lysates were harvested for western blot analysis. (e) Inhibition of STAT3 activity by BBI608 inhibitor suppresses lactic acid-induced LCN2 expression. Cell lysates of control and 10 mM lactic acid-treated 5637 and MC-T2 cells were treated with or without 1µM STAT3 inhibitor (BBI608) for 24 h. Western blot analysis was performed to assess STAT3 activity (p-Y705) and LCN2 expression.
Attenuation of LCN2 expression suppresses lactic acid-induced migration in bladder cancer cells
We next explored the pathological role of lactate-rich TME and LCN2 in promoting malignant features in bladder cancer, with a particular focus on cell migration ability. As shown in Fig. 4a, lactic acid treatment significantly enhanced the migration ability of 5637 bladder cancer cells, as demonstrated by the transwell assay (p < 0.01). Moreover, lactic acid treatment facilitated the epithelial-to-mesenchymal transition (EMT) by upregulating vimentin and N-cadherin expression while downregulating E-cadherin expression, thereby promoting cancer cell migration (Fig. 4b). Furthermore, silencing LCN2 expression in 5637 cells by shRNA (LCN2 KD) attenuated the mesenchymal phenotype by reducing the expressions of vimentin and N-cadherin while increasing epithelial marker E-cadherin (Fig. 4c). In addition, LCN2 silencing obviously impaired the migration ability of bladder cancer cells compared to scrambled control cells (Fig. 4d). Collectively, these findings suggested that LCN2 regulates cell migration in bladder cancer. Furthermore, lactic acid treatment increased LCN2 expression (Fig. 1), consequently increase cell migration ability (Fig. 4a), whereas silencing LCN2 expression reduced cell migration (Fig. 4d). To further investigate the role of LCN2 in lactic acid-enhanced cancer cell migration, a scratch assay was performed to compare wound healing properties between scramble control and LCN2 knockdown (LCN2 KD) 5637 bladder cancer cells in the presence of lactic acid treatment. Cell migration into the wound area was monitored and compared among the groups to assess the impact of lactic acid and LCN2 silencing on cell motility. The data in Fig. 4e demonstrated lactic acid treatment contributed to progressive wound closure in bladder cancer cells. By 9 h after the scratch, quantification of the wounded area revealed that lactic acid-treated cells covered approximately 79% of the cell-free area, compared to 61% in control cells without lactic acid treatment (p < 0.05). This indicates that 5637 cells exposed to lactic acid migrated faster than untreated control cells. Furthermore, LCN2-silenced 5637 cells were employed to investigate the role of LCN2 in lactic acid-enhanced wound closure. The results showed that silencing LCN2 expression significantly impaired lactic acid-induced wound closure, reducing the covered area from 79% to 46% (p < 0.01). These findings suggested that LCN2 plays a pathological role in driving the malignant progression of bladder tumors within a lactate-rich TME.

Lactate-induced LCN2 expression enhances bladder cancer cells migration. (a) 5637 cells were cultured in the absence and presence of 10 mM lactic acid for 24 h. Cells were then collected and subjected to trans-well assay to evaluate cell migration ability. Representative images of migrated 5637 cells, stained with crystal violet, were captured by an inverted microscope (100x magnification; Scale bar: 200 μm) are shown here. The accompanying histogram quantifies the number of migrated cells per field, showing a significant increase in migration following lactic acid treatment (p < 0.01). (b) Cell lysates from (a) were collected to examine vimentin, N-cadherin and E-cadherin expressions. b-actin was used as a loading control. (c) Examination of vimentin, N-cadherin and E-cadherin expression in scrambled shRNA control or LCN2-silenced 5637 cells (LCN2 KD). (d) Scrambled shRNA control and LCN2-silenced 5637 cells were subjected to a trans-well assay to examine the role of LCN2 in regulating cell migration. Similar experiments were performed as shown in (a). Migrated cells were stained with crystal violet and subsequently quantified (p < 0.01). (e) A scratch assay was performed to evaluate the wound healing ability of control, lactic acid-treated control, and LCN2-silenced 5637 cells (LA + LCN2 KD) over a 9 h incubation period. Cells were pre-treated with 10 mM lactic acid for 24 h, while untreated cells served as the control. Cells migration was photographed at 0, 3, 6, 9 h using an inverted microscope at a 40x magnification (Scale bar: 100 μm). The percentage of wound closure was quantified relative to 0 h time point. Results expressed as mean ± SEM (*p < 0.05, **p < 0.01).
Lactic acid-driven LCN2 confers stem cell-like characteristics to cancer cells
Cancer metastasis, acquired chemoresistance, and tumor recurrence are major clinical challenges in cancer treatment, contributing to poor survival outcomes for bladder cancer patients. The presence of cancer stem-like cells (CSCs) is considered a key driver of these malignant features in cancer patients. Therefore, we next investigated whether lactic acid-driven LCN2 promotes CSCs formation. CSCs represent a small population within the tumor mass, and they can be repopulated over time by the conversion of non-CSCs to CSCs. 3D cultures more accurately mimic the in vivo tumor environment, they are more likely to promote cancer stem-like cells phenotypes. To examine the association between LCN2 expression and CSCs, we first cultured bladder cancer cells under 3D sphere-forming conditions to enrich the CSCs population. Figure 5a presented representative images of 5637 and MC-T2 cells grown under 2D and 3D culture conditions. Additionally, cancer stemness-related genes (Nanog, Oct-4, SOX2 and SOX5) were analyzed to confirm the stemness properties of 3D sphere-forming cells. As shown in Fig. 5b, these stemness-related genes were elevated in 3D sphere-forming cells compared to 2D culture cells. Subsequently, cell lysates from both 2D and 3D cultures were collected to analyze LCN2 expression and STAT3 activity. Figure 5c showed that STAT3 activity, along with LCN2 expression, was increased in 3D sphere-forming cells compared to 2D culture cells in both 5637 and MC-T2 cells. Together with the findings of STAT3 is a key regulator of LCN2 expression (Figs. 2, 3), these results suggested a potential association between the STAT3-LCN2 signaling axis and CSCs. To further investigate the roles of STAT3 and LCN2 in maintaining CSCs, scrambled control, as well as STAT3- and LCN2-silenced 5637 and MC-T2 cells, were cultured under 3D condition. The silencing efficiency of STAT3 and LCN2 in 5637 and MC-T2 cells were confirmed by western blotting analysis and were shown in Figs. 3c and 5d, respectively. Subsequently, a qPCR assay was conducted to evaluate CSC properties of STAT3- and LCN2-silenced spheres by analyzing the expressions of cancer stemness-related genes. As shown in Fig. 5e, the mRNA expression levels of Nanog, HOXA4, Oct-4 and SOX5 were significantly down-regulated in STAT3- and LCN2-silenced spheres compared to control spheres in 5637 cancer cells. Moreover, similar reduction results were observed in MC-T2 cells, where silencing STAT3 and LCN2 expression in spheres attenuated cancer stemness (Fig. 5f), further supporting the role of STAT3 and LCN2 in regulating CSCs. Furthermore, since lactic acid treatment activates STAT3-LCN2 signaling axis in bladder cancer cells (Fig. 2), we next to evaluate the effect of lactic acid treatment in sphere formation. Representative images of spheres in Fig. 5g (left panel) showed lactic acid treatment increased both the sphere size and number in 5637 cells. In addition, qPCR analysis confirmed that lactic acid treatment promotes CSC formation by upregulating the expression of cancer stemness-related genes (Nanog, HOXA4, Oct-4, and SOX2), thereby enhancing cancer stemness in 5637 sphere cells (Fig. 5g, right panel). However, silencing STAT3 and LCN2 expression in lactic acid-treated 5637 sphere cells obviously suppressed lactic acid-induced CSCs formation (Fig. 5g, left panel) and significantly impaired the ability of lactic acid to enhance cancer stemness (Fig. 5g, right panel).

LCN2 regulated lactic acid-mediated cancer stem-like cells. (a) 5637 and MC-T2 cells were cultured in 2D and 3D sphere conditions. Representative images of 2D cells and 3D spheres are shown (200x magnification, scale bar: 100 μm). (b) qPCR analysis of stemness-related markers (Nanog, Oct-4, SOX2 and SOX5) in 2D cells and 3D spheres. Relative gene expression was normalized to GAPDH level and expressed as mean ± SEM (*p < 0.05, **p < 0.01). (c) Cell lysates of 2D and 3D spheres were collected, LCN2 expression and STAT3 activity (pY705) were examined by western blot analysis. (d) Western blot results confirmed the efficient knockdown of LCN2 expression in LCN2-silenced cells compared to scrambled control cells. β-actin was used as a loading control to ensure equal protein loading. (e) Quantitative PCR (qPCR) analysis was performed to evaluate the expression levels of stemness-related markers, including Nanog, HOXA4, Oct-4, and SOX5, in sphere-forming scrambled control, STAT3-silenced, and LCN2-silenced 5637 cells. Gene expression levels were normalized to GAPDH and are presented as mean ± SEM from at least three independent experiments. Statistical significance was determined using an unpaired two-tailed t-test (*p < 0.05, **p < 0.01). (f) Stemness-related markers (Nanog, HOXA4, Oct-4, and SOX2) expression was examined in sphere-forming scrambled control, STAT3-silenced, and LCN2-silenced MC-T2 cells by qPCR analysis (*p < 0.05, **p < 0.01). (g) Representative images of 3D spheres formed by scrambled control, and lactic acid-treated scrambled control, STAT3-silenced, and LCN2-silenced 5637 cells (LA 10 mM) were captured at 100x magnification (scale bar: 100 μm) to assess sphere formation efficiency among the different treatment groups (left panel). Additionally, qPCR analysis was performed to evaluate the expression levels of stemness-related markers (Nanog, HOXA4, Oct-4, and SOX2) in sphere-forming cells. Gene expression was normalized to GAPDH and presented as mean ± SEM (*p < 0.05, **p < 0.01). (h) Evaluation of the role of lactic acid and LCN2 in chemoresistance to gemcitabine. Scrambled control and LCN2-silenced 5637 cells were pre-treated with 10 mM lactic acid before exposure to gemcitabine (0, 7.8, 15.625, 31.25, 125, 500, 1000, 2000 nM). Cell viability was measured using the MTT assay, and the IC50 values were calculated.
Gemcitabine is a widely used chemotherapeutic agent for bladder cancer, but resistance to it remains a significant clinical challenge. The effect of lactic acid in conferring cancer stemness to cancer cells through LCN2 was elucidated. Many bladder cancer cells, particularly those exhibiting cancer stem cell-like (CSC) properties, show reduced sensitivity to gemcitabine, which leads to relapse and poor outcomes. Therefore, we next examined the involvement of LCN2 in modulating the response of bladder cancer cells to gemcitabine treatment. As shown in Fig. 5h, the 50% maximal inhibitory concentration (IC50) value of gemcitabine in 5637 cells is 34.67 nM. However, under lactic acid treatment, the IC50 value of gemcitabine increased to 61.85 nM, indicating cancer cells acquired resistance to chemotherapy. Importantly, LCN2 silencing mitigated this resistance effect, the IC50 was reduced from 61.85 to 46.69 nM, emphasizing the pivotal ability of LCN2 in lactate-rich TME in modulating the chemotherapeutic response in bladder tumor.
Discussion
This study explores the molecular mechanisms underlying the induction of LCN2 by tumor-derived lactate through STAT3 signaling. Clinically, high LCN2 expression was linked to poor prognosis, higher tumor grades, and worse survival outcomes. Additionally, LDHA and STAT3 expression positively correlated with LCN2 mRNA levels in bladder tumor tissues, further reinforcing the connection between lactate metabolism and LCN2 regulation. Tumor-derived lactate upregulates LCN2 expression in bladder cancer cells through STAT3 activation. Both exogenous lactate treatment and inhibition of endogenous lactate production confirmed this regulatory mechanism. Disrupting STAT3 activity markedly suppressed lactate-induced LCN2 expression, highlighting STAT3 as a critical mediator of this process. Functionally, LCN2 was shown to play an essential role in lactate-driven bladder cancer cell migration and the acquisition of cancer stem-like properties, as evidenced by increased expression of stemness markers and enhanced sphere formation. Importantly, lactic acid treatment induced chemoresistance to gemcitabine. Furthermore, LCN2 silencing mitigated this resistance, indicating its role in promoting tumor cell survival under chemotherapeutic pressure. The diagram of Fig. 6 summarizes the relationship between the Warburg effect, lactate accumulation, and STAT3-mediated LCN2 expression, ultimately driving tumor growth and malignancy.

Lactate-rich tumor microenvironment promotes LCN2 expression and tumor progression. The Warburg effect leads to a lactate-rich tumor microenvironment (TME) due to aerobic glycolysis, where LDHA-driven lactate production activates STAT3 signaling. This activation induces LCN2 expression in the lactate-rich TME. The increased production of LCN2 plays a crucial role in tumor progression, promoting an aggressive cancer phenotype.
The tumor microenvironment drives cancer progression, with metabolic reprogramming as a key feature. Analysis of bladder cancer cells with high LDHA levels showed correspondingly high LCN2 expression. This supports the idea that tumor cells relying on glycolysis (high LDHA activity) also expressed high LCN2 levels. Tumor-derived lactate upregulates LCN2 expression in a dose-dependent manner, confirmed by exogenous lactate treatment and LDHA inhibition. Sodium lactate produced similar effects, indicating lactate, rather than pH alterations, is primarily responsible for inducing LCN2 expression. LDHA silencing significantly reduced LCN2 expression, confirming that LCN2 regulation is LDHA-dependent and not just a side effect of oxamate treatment. Aberrant immune modulation determines the therapeutic efficiency in bladder cancer. Regulatory T cells, myeloid-derived suppressor cells (MDSCs) and type 2 immunity (TH2 cells and ILC2s) are key regulators of anti-tumor responses in bladder cancer31,32. Beyond promoting tumor progression33, LCN2 plays diverse roles in inflammation and immune response34, including regulates the Treg/Th17 balance35 and neutrophil function36. LCN2 is also implicated in MDSCs regulation, particularly in polymorphonuclear MDSCs (PMN-MDSCs). Pathological conditions like stroke elevate LCN2 expression in PMN-MDSCs, leading to an immunosuppressive tumor microenvironment that supports cancer progression37. Dendritic cells-secreted LCN2 regulates the balance between TH1 and TH2 and may have therapeutic potential in immune modulation38. Collectively, together with these studies, it implies that regulation of LCN2 by lactate accumulation in the TME may also modulate immune evasion in bladder tumors. However, the potential immunological role of LCN2 in a lactate-rich TME needs to be carefully addressed in future studies.
Beyond our study, accumulating studies demonstrated the critical role of lactate in the tumor microenvironment beyond metabolism. Lactate acts as a key signaling molecule in metabolic remodeling, lactylation, immunosuppression, drug resistance, epigenetics, and metastasis, all linked to poor cancer prognosis16,39. STAT3 is a key transcription factor frequently hyperactivated in cancer, tumor-derived lactate modulates its activation as well. However, the precise mechanism by which lactate activates STAT3 remains unclear. Recently, lactate had been shown to promote Treg generation by lactylating Lys72 in MOESIN, enhancing its interaction with TGF-β receptor I and SMAD3 signaling40. Based on this, investigating whether lactate modulates the JAK/STAT signaling pathway is a promising direction for future research41.
STAT3 activity associates with cancer stem-like cells42. Our study revealed that STAT3-LCN2 signaling axis was upregulated in 3D sphere-forming bladder cells, and tumor-derived lactate increased LCN2 expression through STAT3 to confer bladder cancer cells with stemness prosperities. Silencing LCN2 and STAT3 significantly reduced CSC markers expression and sphere formation capacity, supporting STAT3-LCN2 axis is essential for maintaining cancer stem-like properties. Moreover, STAT3 and LCN2 likely promote the expression of genes associated with stemness, and their knockdown leads to a loss of these traits, potentially making the cells less tumorigenic or more differentiated. Due to the widespread role of STAT3 in cancer progression, STAT3 is considered a promising therapeutic target43. A Phase Ib/II clinical trial STAT3 inhibitor BBI608 (Napabucasin) obviously reduced LCN2 expression, stemness-related genes expression and 3D spheres formation in this study, highlighting the clinical significance of STAT3 as a therapeutic target in bladder cancer.
Gemcitabine is a widely used chemotherapy drug for treating various human cancers, including bladder cancer. However, the emergence of resistance in cancer cells presents a significant challenge to its effectiveness. In addition to inducing DNA damage, gemcitabine increases reactive oxygen species (ROS) levels, leading to mitochondrial damage and apoptosis44. Nevertheless, cancer cells can adapt by upregulating ROS-scavenging systems such as glutathione, superoxide dismutase, and catalase, which neutralize ROS and protect against oxidative stress-induced apoptosis45. The balance of redox homeostasis plays a crucial role in determining the cytotoxic efficacy of gemcitabine46. Low ROS level and efficient drug efflux ability by upregulating the expression of drug efflux transporters are two characteristics of CSCs contributing to higher drug resistance47,48. LCN2 is involved in lactate-mediated stemness properties of bladder cancer. LCN2 is a versatile innate immune protein that exerts antimicrobial effects by sequestering bacterial siderophores, regulates iron homeostasis, and enhances cellular resilience against oxidative stress49. Lactate enables bladder cancer cells to resist gemcitabine treatment, potentially through the role of LCN2 in oxidative stress and the maintenance of stemness. High LCN2 levels enhance oxidative stress resistance, protecting cancer cells from oxidative stress induced by chemotherapy.
Conclusions
Our findings identified a regulatory mechanism between tumor-derived lactate and LCN2 expression in bladder cancer. LCN2 expression was clinically correlated with aggressive bladder tumor features. Lactate-rich TMEs drive LCN2 expression via STAT3 activation, which in turn promotes bladder cancer cell migration, CSC maintenance, and therapy resistance. Targeting this pathway may provide a promising therapeutic strategy to mitigate bladder cancer aggressiveness and overcome chemoresistance, particularly in lactate-rich TME.
Data availability
The data presented in this study are available upon reasonable request from the corresponding author.
Abbreviations
- BLCA:
-
Bladder cancer
- NMIBC:
-
Non-muscle-invasive bladder cancer
- MIBC:
-
Muscle-invasive bladder cancer
- LCN2:
-
Lipocalin 2
- STAT3:
-
Signal Transducer and Activator of Transcription 3
- TME:
-
Tumor microenvironment
- EMT:
-
Epithelial-to-mesenchymal transition
- LA:
-
Lactic acid
- CSCs:
-
Cancer stem cells
- ECM:
-
extracellular matrix
- LDHA:
-
Lactate dehydrogenase A
- NGAL:
-
neutrophil gelatinase-associated lipocalin
- GEPIA2:
-
Gene Expression Profiling Interactive Analysis
- TCGA:
-
The Cancer Genome Atlas
- GTEx:
-
Genotype-Tissue Expression
- HPA:
-
Human Protein Atlas
- MC-T2:
-
MC-SV-HUC T-2
- IPA:
-
Ingenuity Pathway Analysis
- PMN-MDSCs:
-
polymorphonuclear MDSCs
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74 (3), 229–263 (2024).
Saginala, K. et al. Epidemiology of bladder cancer. Med. Sci. 8 (1), 15 (2020).
Abugomaa, A. et al. Emerging roles of cancer stem cells in bladder cancer Progression, Tumorigenesis, and resistance to chemotherapy: A potential therapeutic target for bladder cancer. Cells, 9(1). (2020).
Hamid, A. et al. The expression of stem cells markers and its effects on the propensity for recurrence and metastasis in bladder cancer: A systematic review. PLoS One. 18 (5), e0269214 (2023).
de Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell. 41 (3), 374–403 (2023).
Lee, Y. C. et al. Collagen-rich airway smooth muscle cells are a metastatic niche for tumor colonization in the lung. Nat. Commun. 10 (1), 2131 (2019).
Zhang, X. et al. High expression of COL6A1 predicts poor prognosis and response to immunotherapy in bladder cancer. Cell. Cycle. 22 (5), 610–618 (2023).
Lee, Y. C. et al. The dynamic roles of the bladder tumour microenvironment. Nat. Rev. Urol. 19 (9), 515–533 (2022).
Massari, F. et al. Metabolic phenotype of bladder cancer. Cancer Treat. Rev. 45, 46–57 (2016).
Warburg, O., Wind, F., Negelein, E. & THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol., 8(6): 519–530. (1927).
Conde, V. R. et al. The progression from a lower to a higher invasive stage of bladder cancer is associated with severe alterations in glucose and pyruvate metabolism. Exp. Cell. Res. 335 (1), 91–98 (2015).
Hsu, P. P. & Sabatini, D. M. Cancer cell metabolism: Warburg and beyond. Cell 134 (5), 703–707 (2008).
Sharma, D., Singh, M. & Rani, R. Role of LDH in tumor glycolysis: regulation of LDHA by small molecules for cancer therapeutics. Sem. Cancer Biol. 87, 184–195 (2022).
Brizel, D. M. et al. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 51 (2), 349–353 (2001).
Walenta, S. et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 60 (4), 916–921 (2000).
Chen, S. et al. The emerging role of lactate in tumor microenvironment and its clinical relevance. Cancer Lett. 590, 216837 (2024).
de la Cruz-López, K. G. et al. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 9, 1143 (2019).
Flower, D. R., North, A. C. T. & Attwood, T. K. Mouse oncogene protein 24p3 is a member of the Lipocalin protein family. Biochem. Biophys. Res. Commun. 180 (1), 69–74 (1991).
Galaris, A. et al. Increased lipocalin-2 expression in pulmonary inflammation and fibrosis. Front. Med. (Lausanne). 10, 1195501 (2023).
Chen, J. et al. Hepatic Lipocalin 2 promotes liver fibrosis and portal hypertension. Sci. Rep. 10 (1), 15558 (2020).
Yan, Q. W. et al. The adipokine Lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes 56 (10), 2533–2540 (2007).
Viau, A. et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J. Clin. Invest. 120 (11), 4065–4076 (2010).
Candido, S. et al. Roles of neutrophil gelatinase-associated Lipocalin (NGAL) in human cancer. Oncotarget 5 (6), 1576–1594 (2014).
Lee, H. J. et al. Ectopic expression of neutrophil gelatinase-associated Lipocalin suppresses the invasion and liver metastasis of colon cancer cells. Int. J. Cancer. 118 (10), 2490–2497 (2006).
Tong, Z. et al. Neutrophil gelatinase–associated lipocalin: a novel suppressor of invasion and angiogenesis in pancreatic cancer. Cancer Res. 68 (15), 6100–6108 (2008).
Candido, S. et al. Diagnostic value of neutrophil gelatinase-associated lipocalin/matrix metalloproteinase-9 pathway in transitional cell carcinoma of the bladder. Tumour Biol. 37 (7), 9855–9863 (2016).
Tang, Z. et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45 (W1), W98–W102 (2017).
Thul, P. J. & Lindskog, C. The human protein atlas: A Spatial map of the human proteome. Protein Sci. 27 (1), 233–244 (2018).
Zhao, Z. et al. Oxamate-mediated Inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt–mTOR signaling pathway. Cancer Lett. 358 (1), 17–26 (2015).
Becerra, C. et al. A phase Ib/II study of cancer stemness inhibitor Napabucasin (BB608) combined with weekly Paclitaxel in advanced non-small cell lung cancer. J. Clin. Oncol. 34 (15_suppl), 9093–9093 (2016).
Schneider, A. K., Chevalier, M. F. & Derré, L. The multifaceted immune regulation of bladder cancer. Nat. Reviews Urol. 16 (10), 613–630 (2019).
Crispen, P. L. & Kusmartsev, S. Mechanisms of immune evasion in bladder cancer. Cancer Immunol. Immunother. 69 (1), 3–14 (2020).
Živalj, M., Van Ginderachter, J. A. & Stijlemans, B. Lipocalin-2: A nurturer of tumor progression and a novel candidate for targeted cancer therapy. Cancers (Basel), 15(21). (2023).
Abella, V. et al. The potential of lipocalin-2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers 20 (8), 565–571 (2015).
Xu, L. & Shi, M. Lipocalin 2 (LCN2) knockdown regulates Treg/Th17 balance to improve asthma in mice. J. Asthma Allergy. 16, 1323–1332 (2023).
Janas, R. M. et al. Neutrophil gelatinase-associated Lipocalin in blood in children with inflammatory bowel disease. J. Gastroenterol. Hepatol. 29 (11), 1883–1889 (2014).
Huang, T. et al. Stroke exacerbates cancer progression by upregulating LCN2 in PMN-MDSC. Front. Immunol. 11, 299 (2020).
Floderer, M., Prchal-Murphy, M. & Vizzardelli, C. Dendritic cell-secreted lipocalin2 induces CD8 + T-cell apoptosis, contributes to T-cell priming and leads to a TH1 phenotype. PLoS One. 9 (7), e101881 (2014).
Jin, M. et al. Tumor-Derived lactate creates a favorable niche for tumor via supplying energy source for tumor and modulating the tumor microenvironment. Front. Cell. Dev. Biol. 10, 808859 (2022).
Gu, J. et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell. Rep. 39 (12), 110986 (2022).
Hu, X. et al. The JAK/STAT signaling pathway: from bench to clinic. Signal. Transduct. Target. Therapy. 6 (1), 402 (2021).
Galoczova, M., Coates, P. & Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell. Mol. Biol. Lett. 23 (1), 12 (2018).
Hu, Y., Dong, Z. & Liu, K. Unraveling the complexity of STAT3 in cancer: molecular Understanding and drug discovery. J. Exp. Clin. Cancer Res. 43 (1), 23 (2024).
Maleki, F., Handali, S. & Rezaei, M. The role of mitochondrial dysfunction in the cytotoxic synergistic effect of gemcitabine and arsenic on breast cancer. PLoS One. 20 (1), e0312424 (2025).
Ozben, T. Oxidative stress and apoptosis: impact on cancer therapy. J. Pharm. Sci. 96 (9), 2181–2196 (2007).
Ju, H. Q. et al. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol. Cancer Ther. 14 (3), 788–798 (2015).
Mengistu, B. A. et al. Comprehensive review of drug resistance in mammalian cancer stem cells: implications for cancer therapy. Cancer Cell. Int. 24 (1), 406 (2024).
Murayama, T. & Gotoh, N. Drug resistance mechanisms of cancer stem-like cells and their therapeutic potential as drug targets. Cancer Drug Resist. 2 (3), 457–470 (2019).
Saha, P. et al. Ectopic expression of innate immune Protein, Lipocalin-2, in Lactococcus lactis protects against gut and environmental stressors. Inflamm. Bowel Dis. 23 (7), 1120–1132 (2017).
Acknowledgements
We sincerely appreciate all members of the Lee Laboratory for their valuable suggestions and insightful discussions. Their expertise and collaborative spirit have significantly contributed to the progress of this research. We also extend our gratitude to our collaborators and colleagues for their constructive feedback.
Funding
We acknowledge the National Science and Technology Council, Taiwan (110-2320-B-038-072-MY2 and 113-2314-B-038-034-MY3) for providing the necessary resources and funding that made this work possible.
Author information
Authors and Affiliations
Contributions
YCL and KHL conceived and designed the study. SYY, SHH and MCH performed the experiments. SYY conducted data analysis and interpretation. YCL and HYL wrote the original draft, while CYW and WJW and reviewed and edited the manuscript. HYL and KXY provided methodology assistance. YCL supervised the project and acquired funding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, SY., Li, HY., Wang, CY. et al. Tumor-derived lactate fuels the STAT3-LCN2 pathway to promote bladder cancer malignancy and chemoresistance. Sci Rep 15, 41610 (2025). https://doi.org/10.1038/s41598-025-25534-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-25534-y

{kind=link}