
Abstract
Fuchs Endothelial Corneal Dystrophy (FECD) is an age-related disorder that affects about 4% of the population over the age of 40 years and is genetically associated with CTG repeat expansion in Transcription factor-4 (TCF4) gene. Although both genetic variants as well as environmental exposures like ultraviolent-A light (UVA) have been reported to cause FECD, there are no pharmacological treatments due to the lack of understanding of disease pathogenesis. To characterize the corneal endothelial (CE) cells in FECD individuals with or without CTG repeat expansions in TCF4, we performed RNA-sequencing (RNAseq) of three normal CE cell lines and seven FECD cell lines, including 3 generated from donors with TCF4 repeat expansions (FECD-R) and 4 from non-expanded donors (FECD-NR). Analysis of transcriptomic profiles in all 7 FECD cell lines compared to normal, revealed 214 differentially expressed genes or DEGs with 193 upregulated and 21 downregulated genes. Ingenuity Pathway Analysis (IPA) detected hepatic fibrosis and endothelial-mesenchymal transition pathways as top canonical pathways, which is consistent with extracellular matrix (ECM) deposition in the form of guttae seen in FECD. We identified and further validated transcriptional regulator genes like MAFB, TFAP2B, and POU6F2 to be differentially downregulated in FECD. Furthermore, 48 common DEGs were detected in both FECD-R and FECD-NR, and their upstream regulators included β-estradiol, TGF-β1, Aryl hydrocarbon receptor (AHR), and transcription regulators like CEBPA, CEBPB and SMARCA4. There were 29 genes identified to be differentially regulated only in FECD-R, compared to normal cells. In addition, other canonical pathways like tryptophan and melatonin degradation, Wnt signaling, AHR signaling, mitochondrial dysfunction, and estrogen receptor signaling were also highly enriched in FECD. Our findings support the previously proposed underlying mechanisms of disease progression, including role of transcriptional regulation of endothelial cells, mitochondrial dysfunction, Wnt- and estrogen receptor signaling, as well as the phenotypic clinical FECD hallmark of ECM deposits. Further investigation focusing on differentially expressed genes related to these pathways may be beneficial for elucidating disease-causing mechanisms and developing novel therapies for FECD.
Similar content being viewed by others

Introduction
Corneal diseases like Fuchs Endothelial Corneal Dystrophy (FECD) represent one of the leading causes of vision loss and blindness worldwide1. FECD is a bilateral, genetically heterogeneous degenerative disease2 of corneal endothelial cells (CEnCs) occurring in approximately 4% of the U.S. population over 40 years of age, with higher incidence in women3,4,5,6,7. Among the prominent characteristics of FECD are the formation of corneal guttae and extracellular matrix (ECM) deposition8,9,10. Guttae and subsequent thickening or remodeling of Descemet’s membrane (DM) cause light scatter and account for glare and visual complaints in FECD patients. In FECD, CEnCs exhibit stress-induced antioxidant imbalance11, oxidation of DNA bases12, heightened reactive oxygen species (ROS) production13, mitochondrial damage14, and sustained ER stress15. However, the mechanism of guttae formation or ECM deposition is not clearly understood. We have previously established that physiological stressor, ultraviolet-A (UVA) light, causes DNA damage and leads to FECD phenotype in both in vitro and in vivo disease models16,17. Recently we demonstrated that the endo-degenerative phenotype in FECD is indeed driven by estrogen metabolite-dependent corneal endothelial (CE) cell loss that is exacerbated in the absence of NAD(P)H quinone oxidoreductase 1 (NQO1); thus, shedding light on the mechanism accounting for the higher incidence of FECD in females18.
Most cases of FECD are caused by expansion of the intronic CTG trinucleotide repeat within the TCF4 (Transcription factor 4) gene, making it the largest contributing locus for FECD19,20,21,22. TCF4 expansions of greater than 40 CTG repeats confer significant risk for developing FECD23. FECD can also be caused by a CTG expansion within the 3′-untranslated region (3′-UTR) of the DMPK (Myotonic dystrophy protein kinase) gene, implicating mutant expanded CUG RNA as the root cause for repeat-associated FECD24,25,26. FECD cases with (FECD-R) or without (FECD-NR) repeat expansions are indistinguishable by clinical observation and require genetic analysis like TCF4 CTG18.1 polymorphism genotyping for classification. To date, several non-mutually exclusive mechanisms have been proposed that drive and/or exacerbate the onset of disease. These mechanisms include dysregulation of TCF4 expression27; RNA-mediated toxicity28,29; repeat-associated non-AUG dependent (RAN) translation30; and somatic instability of CTG repeat length31,32. However, the relative contribution of these proposed mechanisms in disease pathogenesis is currently unknown.
Currently, corneal transplantation is the only treatment for FECD, accounting for the vast majority of the 32,000 endothelial keratoplasties performed in the U.S. annually33. There is a significant unmet need to investigate and understand mechanisms involved in the pathogenesis of FECD to help develop targeted pharmacotherapeutic interventions. RNA sequencing (RNAseq) has become an indispensable tool for transcriptome-wide analysis of differential gene expression to infer biological meaning in further studies and to understand the disease pathogenesis34. This technique is also suitable for analyzing many aspects of RNA biology and has introduced the use of RNA-based biomolecules as useful diagnostic, prognostic, and therapeutic means in various diseases35. This implies that RNAseq analysis of FECD related samples could be valuable in identifying causative genes as well as pathways.
Although there had been four previous studies employing RNAseq technique, all these studies have been conducted using corneal endothelium from patients with FECD and normal controls36,37,38,39. Here, instead of using RNA from the corneal endothelium of FECD patients for RNAseq analysis, we utilized the FECD and normal cell lines that were generated by collecting DM and endothelium from corneas of cadaveric donors and FECD patients undergoing endothelial keratoplasty, followed by simian virus-40 T-antigen cell immortalization of primary CEnCs. To identify differentially expressed genes (DEGs) and pathways that may be potentially related to the pathogenesis of FECD, we performed RNAseq using RNA from 3 normal cell lines, 3 FECD cell lines derived from donors with TCF4 repeat expansions (FECD-R) and 4 FECD cell lines derived from donors without TCF4 repeat expansions (FECD-NR). Transcriptomic data from the three groups, normal, FECD-R and FECD-NR, revealed various differentially expressed genes that are (i) shared between FECD-R and FECD-NR or (ii) uniquely altered in FECD-R but not FECD-NR. Pathways related to hepatic fibrosis and epithelial–mesenchymal transition (EMT) were found to be shared by FECD-R and FECD-NR, thus supporting extracellular matrix (ECM) depositions (guttae) seen in FECD. These results will help understand the underlying mechanism of pathogenesis in FECD as well as other trinucleotide repeat diseases, thus providing potential targets for therapeutic design.
Results
Identification of differentially expressed genes and pathways in FECD compared to normal corneal endothelial cell lines
To elucidate the differences in gene expression profiles of CEnCs between FECD and normal controls, we compared the CE cell lines derived from FECD (n = 7) subjects (irrespective of their TCF4 CTG repeat expansion status) undergoing endothelial keratoplasty with normal cadaveric donors (n = 3) by RNA sequencing (RNAseq) (Fig. 1A). Genetic analysis from blood samples and CEnCs identified 3 FECD samples with a single allelic CTG repeat expansion in the TCF4 gene (FECD-R), 4 FECD samples without the CTG repeat expansion (FECD-NR), and 3 normal control samples without the CTG repeat expansion (Fig. 1B). Since FECD is a genetically heterogenous disease, the FECD-R and FECD-NR cell lines were tested for mutation in specific genetic variants in other FECD-associated genes namely LOXHD1, SLC4A11, AGBL1, KANK4 and ZEB1 (Supplementary Fig. 1A-B). The selection of alleles tested was based on that allele being predominant in the familial cases of FECD and/or having been reported most commonly in FECD cases in literature (Supplementary Fig. 1A). All the 7 FECD cell lines were found to be wild type for all the alleles tested, except for ZEB1 where one FECD-NR cell line (SVF2-61F) was heterozygous for rs35753967 (Supplementary Fig. 1B). Multi-dimensional scaling (MDS) plot demonstrated clustering of FECD and normal CEnCs (Supplementary Fig. 2). To classify changes as significant, we used the cutoff of < 0.05 for False Discovery Rate (FDR). Volcano plot allows a global overview of individual gene expression changes and is useful in visualizing pattern of changes and identifying ‘outlier’ genes by combining highly significant changes in gene expression with higher fold changes. Evaluation of volcano plot and heatmap [log2 RPKM (reads per kilobase of transcript per million reads mapped), FDR = 0.05) for RNAseq analyses identified a total of 214 statistically significant DEGs between FECD and normal cells with 193 downregulated (log2 fold change (log2FC) < = 1, FDR < 0.05) and 21 upregulated (log2FC > 1, FDR < 0.05) genes in FECD cell lines (Fig. 2A-B, Supplementary Tables 1–3). While the MDS plot (Supplementary Fig. 2) illustrates the qualitative relative similarity between FECD and normal cells for the top 300 most varied genes in our cohort, the heatmap (Fig. 2B) is generated using only the 214 differentially expressed genes (DEGs) between the two cohorts. This explains the differences between the clustering of samples in the MDS plot and the heatmap. Therefore, although the one healthy control cell line (SVN2-67F) clustered oddly in the MDS plot, the evaluation of heatmap (Fig. 2B) confirmed that its gene expression pattern is more similar to the other normal than the FECD samples. The top 20 differentially overexpressed genes identified in FECD cells include transcriptional regulator genes like ZNF711 (Fig. 2C). To elucidate the potential impact of changes in the expression of individual genes between FECD and normal cells on physiological processes, we applied Ingenuity Pathway Analysis (IPA) to our RNAseq data set. Intriguingly, the top canonical pathway was hepatic fibrosis pathway (Fig. 2D). Involvement of hepatic fibrosis genes in FECD is consistent with the accumulation of extracellular matrix (ECM) proteins with thickening of DM and guttae formation9. Genes of the fibrosis pathway that showed statistically significant decrease in expression include EGFR, COL12A1, COL9A3, BAMBI, COL4A3, COL8A2, CD14, COL21A1, COL26A1 and FGFR2 (Fig. 2E). The pathway analysis also showed the role of Epithelial–mesenchymal transition (EMT) related genes in FECD (Fig. 2D-E), including FGF5, TCF4, EGFR, WNT7B, FGF10 and FGFR2, some of which have already been explored and studied in the disease pathogenesis15,40,41. Subsequently, we performed an IPA upstream regulator analysis and identified β-estradiol as the top upstream regulator in our RNAseq data set, which included 50 target molecules out of the 214 DEGs (Fig. 2F). Additional upstream regulators included SMARCA4, estrogen receptor 2, and ARNT2 as predicted inhibitors and mir155-5p as a predicted activator (Fig. 2F). PANTHER analysis of 214 DEGs identified in FECD cells demonstrated a variety of molecular functions with the largest representation involving a binding function, and statistically significant enrichment of extracellular matrix (ECM) structural constituents conferring tensile strength, heparin binding, glycosaminoglycan binding, cation channel activity and calcium ion binding (Supplementary Fig. 3A-B).

Experimental scheme and sample cohorts. (A) Descemet’s membrane and endothelium were collected from donor corneas (n = 3) and patients with FECD undergoing endothelial keratoplasty (n = 7). Primary CEnCs were immortalized using the simian virus-40 T-antigen cell immortalization kit and the number of repeats were estimated in CTG18.1 in the TCF4 gene of FECD cells. Cell lines from normal (SVN1-67F, SVN2-67F and SVN3-65M), FECD with repeat expansions (FECD-R; SVF1-73F, SVF5-54F, and SVF6-61M) and FECD without repeat expansions (FECD-NR; SVF2-61F, SVF3-76M, SVF4-61F, and SVF7-74F) were used for RNA sequencing followed by data analysis. (B) Table representing age, sex, passage number, and TCF4 trinucleotide CTG repeats status of normal and FECD cell lines used in this study.

RNAseq analysis of differential gene expression and pathways in FECD versus normal control cell lines. (A) Volcano plot for FECD (FECD-R and FECD-NR) versus normal control (FDR < 0.05) showing 193 downregulated (log2FC < = 1) and 21 upregulated (log2FC > 1) DEGs. (B) Heatmap (log2 RPKM, FDR = 0.05) comparing the differential expression level of total 214 genes in FECD and control cohorts, verifies that the gene expression pattern of FECD cases is distinct from that of normal controls. Samples are indicated at the bottom. Red stripes represent high expression levels, while blue stripes represent low expression levels. (C) Top 20 downregulated and upregulated genes in FECD versus control cohorts. (D) Top 20 ingenuity canonical pathways with -log(p-value) for each pathway shown in the second column. The two pathways involved in FECD pathogenesis are boxed in red. (E) Differentially expressed genes in the Hepatic Fibrosis and Epithelial Mesenchymal Transition (EMT) pathways in FECD vs. normal cohorts. (F) Top 18 upstream regulators that were identified as significantly inhibited (z-score <-0.2) or activated (z-score > = 0.2) in FECD as compared to normal. The activation z-scores and p-values for each upstream regulator are shown in the last two columns.
Differentially expressed transcriptional regulator genes in FECD cells
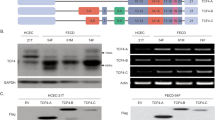
The primary regulators of gene expression are transcription factors that play a crucial role in maintaining cellular sustainability and finely regulating complex internal metabolic networks42. Two-thirds of the FECD cases have been found to be associated with the CTG repeat expansions within transcription factor 4 (TCF4)20,43, a basic helix-loop-helix transcription factor and an important regulator of neurodevelopment44,45 and epithelial-mesenchymal transition46. To advance our understanding of the role of additional transcription factors in FECD, we inspected differentially expressed transcriptional regulators in our dataset of 214 DEGs in FECD. Ten out of the 214 DEGs were identified as transcription regulators (Fig. 3A-B). While ZNF711 was the only transcription regulator that was identified as upregulated in FECD, the remaining 9 genes including TCF4, MAFB, TFAP2B, POU6F2, SOX21, ARX, PPARGC1A, ZNF423, and LMX1B were all found to be downregulated in FECD as compared to normal cells (Fig. 3A-B). Interestingly, small Maf (MAF BZIP transcription factor) genes like MAFB have been previously shown to heterodimerize with activated Nrf247,48,49 and bind to antioxidant response elements (ARE) in the DNA to facilitate the transcription of a myriad of genes encoding antioxidants and protect against oxidative stress, an underlying cause of FECD50,51. Additionally, transcription factors TFAP2B and POU6F2 are shown to play a role in corneal development and maintenance52,53,54. We therefore validated the downregulation of MAFB, TFAP2B and POU6F2 proteins in cell lysates from 2 normal cell lines, SVN1-67F and SVN3-65M; and 4 FECD cell lines, SVF1-73F, SVF5-54F, SVF2-61F and SVF3-76M by western blotting (Fig. 3C). The densitometry analysis of western blots demonstrated MAFB/β-actin and POU6F2/β-actin protein levels were markedly downregulated by 4-fold (p < 0.01) and 2-fold (p < 0.05) respectively, in FECD compared to normal cell lines (Fig. 3C). Lifelong exposure of the cornea to oxidative stress can play a role in the pathogenesis of FECD11. We have previously demonstrated that UVA light, an environmental stressor of the human cornea, causes macromolecular damage via the production of ROS, leading to FECD phenotype in a mouse model16,17,50. A recent study revealed a distinct regional variation in FECD progression in humans, with relative sparing of the superior cornea suggesting that UVA light could be an etiologic factor in FECD pathogenesis55. Therefore, we evaluated the effect of UVA on the expression of MAFB, TFAP2B and POU6F2 proteins. Interestingly, UVA-irradiation decreased the levels of MAFB and TFAP2B but did not affect POU6F2 in SVN-67F cells (Fig. 3C).

Differential expression of transcriptional regulators in FECD compared to normal cell lines. (A) List of 9 downregulated and 1 upregulated transcription regulator genes in FECD as compared to normal controls. The log2 fold change values and gene functions for each of 10 differentially regulated genes are shown in the last two columns. The three interesting candidate genes shown to be involved in FECD related pathways or corneal endothelial cell development are boxed in red. (B) Expression profiles of 9 downregulated and 1 upregulated transcription regulator genes for normal and FECD cell lines (FDR < 0.05). (C) Western blot assay showing downregulation of MAFB, TFAP2B and POU6F2 in FECD as compared to control cell lines. UVA-irradiated normal cells showed downregulation of MAFB and TFAP2B, but not POU6F2 at 24-hours post UVA (5 J/cm2). Densitometry was analyzed using Image J software and relative protein expression levels were determined by normalizing band intensities to that of β-actin. * Indicates p < 0.05 applying two-tailed unpaired t-test with Welch’s correction.
Differentially expressed genes and altered pathways in FECD-R and FECD-NR cell lines
We next investigated whether we could further identify common DEGs between FECD CE cell lines with and without CTG repeat expansions in TCF4, compared to normal cell lines. We identified 77 DEGs (60 downregulated and 17 upregulated) between FECD with the CTG repeat expansion in TCF4 (FECD-R) and normal (Fig. 4A; Supplementary Fig. 4); and 484 DEGs (338 downregulated and 146 upregulated) between FECD without the CTG repeat expansion in TCF4 (FECD-NR) and normal cell lines (Fig. 4B; Supplementary Fig. 4). Overlapping the DEGs between Normal vs. FECD-R and Normal vs. FECD-NR, revealed forty-eight DEGs (45 downregulated and 3 upregulated) to be common among both the data sets (Fig. 4C-D). The 48 common gene list retained the transcription factors MAFB, and TAFB2P as downregulated genes and identified FAM196B, STEAP1 and ALDH1A3 as the three upregulated genes (Fig. 4D). Among the top canonical pathways revealed by IPA of the 48 common genes included some known FECD-associated signaling pathways like hepatic fibrosis, Wnt signaling, EMT pathway, mitochondrial dysfunction, NF-kB signaling and estrogen receptor signaling (Fig. 4E). Additionally, there were some interesting new pathways like serotonin and melatonin degradation, PTEN signaling, Aryl hydrocarbon signaling, and signaling by Rho GTPases, that need further investigation with respect to FECD pathogenesis. Furthermore, we performed an IPA upstream regulator analysis and identified three transcriptional regulators CEBPA, CEBPB and SMARCA4, along with β-estradiol, TGF-β1, ESR1 and AHR (aryl hydrocarbon receptor) (Fig. 4F). PANTHER analysis of 48 common DEGs demonstrated a diversity of molecular functions with the largest representation involving a binding function, and statistically significant catalytic and transporter activity (Supplementary Fig. 5).

Analysis of differential gene expression and pathways in FECD-R, FECD-NR, and normal control cell lines. (A) Heatmap (log2 RPKM, FDR = 0.05) comparing the differential expression level of total 77 genes (60 down- and 17 up-regulated) in FECD-R and normal control cohorts. (B) Heatmap (log2 RPKM, FDR = 0.05) comparing the differential expression level of total 484 genes (338 down- and 146 up-regulated) in FECD-NR and normal control cohorts. Samples are indicated at the bottom. Red stripes represent high expression levels, while blue stripes represent low expression levels. (C) Venn diagram showing the overlap for gene expression of between Normal vs. FECD-R and Normal vs. FECD-NR. Numbers indicate the number of differentially expressed genes identified in each individual region. (D) Significant expression level changes in 48 common DEGs genes in FECD-R and FECD-NR cohorts. (E) Top 20 ingenuity canonical pathways with -log(p-value) for each pathway shown in the second column. (F) Top 10 upstream regulators that were identified as significantly inhibited (z-score <-0.2) or activated (z-score > = 0.2) as common between Normal vs. FECD-R and Normal vs. FECD-NR. The molecule type and p-values for each upstream regulator are shown in the last two columns.
Furthermore, we also investigated differences in gene expression profiles that were unique to FECD-R cell lines. We identified 29 (14 downregulated and 15 upregulated) DEGs in Normal vs. FECD-R but absent in Normal vs. FECD-NR (Fig. 4C; Table 1). The downregulated genes in FECD-R include MAPK4, NPY, ZBED2, TAC1, PRSS21, L1CAM, CDH11, TMTC1, APOD, DMKN and BOC, while some of the upregulated genes in FECD-R are NMT2, DRAM1, SLIT2, FGF5, ARL4C, DLC1, IL31RA, TNFGSF1B and IL1RL1. It will be interesting to further investigate if any of these genes or their altered pathways play a role in TCF4 repeat expansion, seen in FECD.
Discussion
Identifying the drivers of late-onset diseases like FECD is important for understanding disease progression and developing therapeutics. Transcriptome analysis of corneal endothelium has been previously reported in vitro55 and ex vivo38,56,57. However, many of the studies were performed with low sample size, pooled samples from multiple corneas, limited statistical analysis, or lack of TCF4 repeat expansion status. Cell lines derived from FECD patient tissues serve as an in vitro model to not only study disease causing pathways but also screen therapeutic compounds. Therefore, it is essential to characterize them to identify known and unknown pathways implicated in FECD that are still preserved in the immortalized cell lines obtained from patient tissues. RNA-seq is a great tool for transcriptome profiling and allows examining the expression of all genes at once58. The present study was designed to utilize RNA-sequencing technique and evaluate the gene expression changes in the corneal endothelial cell-lines generated from FECD tissues with or without CTG repeat expansions in TCF4 and thereby highlight novel disease-causing molecular pathways/mechanisms of FECD. In this study, we identified a total of 214 genes that met our criteria as DEGs between FECD and normal cells with 193 downregulated (log2FC < = 1, FDR < 0.05) and 21 upregulated (log2FC > 1, FDR < 0.05) in FECD cell lines. 48 common DEGs were detected in both FECD-R and FECD-NR, compared to normal cells. Moreover, we also identified 29 DEGs (14 downregulated and 15 upregulated) that were specifically altered in FECD-R, but not in FECD-NR compared to normal cell lines. The molecular signatures identified for FECD-R would be useful for developing biomarkers to characterize FECD with and without TCF4 repeat expansions.
CEnCs secrete and deposit extracellular matrix (ECM) proteins that form a specialized basement membrane called Descemet’s membrane (DM). A normal DM contains collagen type VIII, collagen type IV (chains α1–α2), and fibronectin on its stromal side and entactin, laminin, perlecan, and collagen type IV (chains α3–α6) on its endothelial side59,60. The main feature of late-onset FECD is its irregular and thickened DM61,62 due to abnormal accumulation of ECM components including collagens, transforming growth factor-β induced (TGFβ-I) and clusterin9,63,64; resulting in guttae formation. ECM accumulation and tissue remodeling during fibrosis is often a result of continued endothelial-mesenchymal transition (EMT), a repair-associated process whereby endothelial cells lose their cell-cell adhesion, develop migratory and invasive properties leading to tissue fibrosis40. Among the top canonical pathways identified by IPA analysis of our RNAseq data confirmed the involvement of hepatic fibrosis and EMT pathways, consistent with the abnormal ECM regulation and EMT in FECD pathogenesis, and with previously reported RNA measurements using tissues from late-stage FECD9. Hepatic fibrosis genes showing significant downregulation in FECD cells included EGFR, CD14, FGFR2 and collagens like COL8A2, COL4A3, and COL12A1. Mutations in COL8A2 gene have consistently shown association with early-onset FECD, associated with thickening of Descemet membrane and subsequent increase in central corneal thickness (CCT)65,66. Corneal endothelial expression of COL4A3 due to loss of ZEB1 has been shown to result in an abnormal endothelial proliferation, corneal thickening, and iridocorneal adhesions observed in individuals affected with posterior polymorphous corneal dystrophy type 3 (PPCD3) and in Zeb1-heterozygous and -null mice67. Collagens XII have been shown to be critical for corneal function and their absence results in the delayed maturation of corneal endothelium and abnormal corneal compaction68. Among the EMT-related genes in our dataset, while FGF5 showed a significant increase, TCF4, EGFR, WNT7B, FGF10 and FGFR2 genes markedly decreased in FECD, as compared to normal cells. Gao et al., performed a genome-wide association study in 2016, and identified WNT7B as a novel locus for CCT in Latinos69. Interestingly, FGF10 (Fibroblast Growth Factor 10) has been recently shown to play a significant role in the repair of the corneal endothelium, primarily by promoting wound healing, inhibiting inflammatory responses, and enhancing the function of endothelial cells70.
Two transcription factors have been implicated in the pathogenesis of FECD: TCF4 (Transcription factor-4) and TCF8 (Transcription factor-8). An intronic CTG repeat in TCF4 gene is the most common mutation in FECD is that leads to its altered expression20,21,23,71. In the United States, about 70% of the FECD cases are caused by an intronic trinucleotide repeat expansion in TCF420,21,22,23. Additionally, mutations in TCF8 have been linked to late-onset FECD72. TCF8 encodes the ZEB1 protein and is upregulated by the E2-2 protein encoded by TCF472,73. Although TCF4 and TCF8 have similar biological functions, their specific role in the pathogenesis of FECD is unclear. Interestingly, we identified a heterozygous ZEB1 variant, c.1721 A > G; p.K553R (rs35753967), in SVF2-61 F, a FECD-NR cell line. This ZEB1 variant has been previously reported in 3 non-familial FECD cases74 (Supplementary Fig. 1A-B), however, functional studies are needed to determine its role in FECD pathogenesis. Since in this study our criterion of grouping the cell lines was the presence or absence of TCF4 repeat expansion, we do not think that our current transcriptomic analyses would be impacted even if ZEB1 mutation identified in SVF2-61 F cells is found to be pathogenic. To further explore the altered expression of additional transcription factors in FECD, we utilized our RNA-seq dataset and identified MAFB (MAF BZIP transcription factor B) to have a lower expression in FECD, compared to normal cells. Interestingly, small Maf proteins including Mafb have been known to heterodimerize with another transcription factor Nrf247,48,49,75. Suppression or loss of Nrf2 in FECD causes mitochondrial dysfunction, DNA damage, excessive lipid peroxidation and ultimately corneal endothelial cell death50,51. Additionally, Nrf2 is deficient in FECD and leads to loss of its transcriptional target NQO1, rendering ocular tissue to be highly susceptible to oxidative stress18,76,77,78. Furthermore, our dataset identified decreased expression of TFAP2B (transcription factor AP-2 beta) and POU6F2 (POU class 6 homeobox 2) genes in FECD, which was further validated by western blots. Both TFAP2B and POU6F2 are shown to be CEnC-specific transcription factors52,53. While TFAP2B transcriptionally regulates corneal endothelial-related genes52, POU6F2 expression is found to be upregulated during human CEnC differentiation, suggesting it to be pivotal in terminal differentiation of corneal endothelium from neural crest53. King et al. used the mouse genetic reference panel and identified Pou6f2 as one of the transcription factors that modulates corneal thickness during development and provides a potential link with the risk for glaucoma54. Further research on these corneal endothelium transcription factors may increase our knowledge on the corneal endothelial differentiation process and homeostatic conservation transcription and may also be important in regenerative medicine using cell-based therapy with CEnCs and pluripotent stem cells.
FECD has been shown to have greater incidence in females16. Our lab has recently demonstrated the role of estrogen genotoxicity in preferential development of FECD in females18. Specifically, we showed that injecting wild-type male mice with estrogen (E2, β-estradiol) increases CEnC loss, followed by increased production of estrogen metabolites and mitochondrial DNA damage18. Overwhelmingly, the IPA upstream regulator analysis of 214 DEGs identified β-estradiol, the primary female sex hormone, as the top upstream regulator with predicted inhibition (activation Z-score < -2.0), thus confirming our previous findings. ESR2 (Estrogen receptor-2), SMARCA4 (SWI/SNF Related, Matrix Associated, Actin Dependent Regulator of Chromatin, subfamily A, Member 4) and ARNT2 (aryl hydrocarbon receptor nuclear translocator 2) genes are also identified as the upstream regulators with predicted inhibition. Rosenbloom et al. utilized chromatin immunoprecipitation sequence (ChIP-seq) and determined that TCF4 rs613872 single nucleotide polymorphism (SNP) is present within the binding site for the SMARCA4 transcription factor79. It has been hypothesized that variation at rs613872 SNP affects the spatiotemporal expression of TCF4 through SMARCA4 and hence its targets. ARNT2 is a transcription factor that is known to dimerize with Aryl hydrocarbon receptor (AhR) and then translocate to nucleus where it binds with specific xenobiotic responsive elements to upregulate CYP-family of enzymes80. Interestingly, CYP1B1 is the key enzyme in the metabolism of estrogen, that is shown to be upregulated in FECD tissues16. Additionally, the upstream regulators with predicted activation included miR-155-5p. miR-155 (microRNA-155) are demonstrated to play a significant role in regulating the function of CEnCs, primarily by impacting their cell cycle and barrier function through the modulation of the E2F2 protein, potentially contributing to conditions like high myopia where endothelial damage is observed81.
We identified 6-fold more DEGs in normal vs. FECD-NR, compared to normal vs. FECD-R, suggesting that the FECD in patients with non-expanded TCF4 repeats (FECD-NR) may involve a multitude of genes and pathways that converge into disease-causing mechanism leading to FECD. We further investigated the 48 DEGs found in the cell lines with the previously published RNA-seq data set taken from FECD post-keratoplasty tissues38, comparing control vs. FECD tissues taken from pre-symptomatic (Pre-S), late stage FECD with TCF4 repeat expansions (FECD-REP), or FECD without TCF4 repeat expansions (FECD-NR). Notably, we found 3 out of 48 genes, namely ALDH1A3, MAOB and MAFB from our dataset to be also dysregulated in at least 2 of the 3 tissue groups from that study38. While ALDH1A3 was upregulated, the other 2 genes, MAOB and MAFB, were found to be downregulated in both immortalized FECD cell lines and ex vivo FECD tissues, indicating the consistent findings between both data sets.
The canonical pathway analysis for the 48 DEGs detected in both FECD-R and FECD-NR indicated that these common genes were significantly enriched in some of the pathways like fibrosis, Wnt signaling, EMT signaling, mitochondrial dysfunction82, and estrogen receptor signaling18 that are known to be involved in FECD pathogenesis. Additionally, some novel and interesting pathways like tryptophan degradation, serotonin/melatonin degradation, and AhR signaling, were also discovered that require further investigations with respect to FECD pathogenesis. Previous studies have shown that mitochondrial biogenesis, and autophagy are tightly regulated by melatonin, an evolutionarily conserved metabolite of tryptophan and serotonin83. Although the expression of melatonin receptors (MT1 and MT2) has been detected in corneal endothelium84,85, melatonin has been shown to stimulate antioxidative responses and DNA repair via receptor independent pathways86. Melatonin, along with its metabolites, has been shown to protect against UV-radiation87, and act as ligands for AhR88. Interestingly, in our dataset of the upstream regulators for these 48 common DEGs, we found AhR and TGF-β (transforming growth factor-β), the most potent fibrogenic cytokine. Recently, AhR is shown to be activated by environmental89 factors like smoking, leading to oxidative stress and corneal endothelial dysfunction in mice90,91. Additionally, TGF-β family genes are known to be involved in regulating cell differentiation, proliferation and extracellular matrix production in ocular tissue92. In fact, TGF-β2 null mice exhibit an absence of endothelium and fusion of the lens with the cornea93. Overall, our data supports the notion that these pathways including melatonin, AhR, and TGF-β signaling may be important for FECD pathogenesis, irrespective of their TCF4 repeat expansion status. Future studies are required to investigate (a) the non-receptor mediated, mitochondrial function of melatonin in the eye, especially in response to AhR activation, and (b) the role of TGF-β related genes in corneal endothelial dysfunction, seen in FECD. Use of immortalized cell lines offers advantages, including increased proliferative capacity and consistency and reproducibility of the results due to their homogenous population. However, cell lines pose limitations of reduced physiological relevance compared to primary cells due to the genetic manipulation and induction of replication that is not physiologic in post-mitotic cells such as corneal endothelium. Therefore, it will be important to further validate findings with patient specimen data and animal models. Although FECD more commonly affects females than males at a ratio 4 − 3:1, different genetic variants have variable sex-specific association. For example, COL8A2 mutations affect both men and women equally66, while LAMC1 carries greater risk in women94. A study by Zarouchlioti et al. showed TCF4 mutation carries a greater risk in men in white European individuals95; however, there is a recent study demonstrating no sex bias for the prevalence of TCF4 repeat expansion in a multiethnic population96. The goal of this study was to investigate the common causative pathways of FECD based on the most common genetic association (presence of TCF4 repeat expansions), irrespective of sex-differences. The current study is not powered for sex-related differences and future studies are needed to investigate the differential expression of genes in male vs. female FECD samples.
In conclusion, we have identified several differentially expressed genes, related canonical pathways enriched in these genes, and their upstream regulators, in FECD cell lines with or without TCF4 repeat expansions. These results shed light on the genes/pathways/mechanisms that are shared between FECD caused or not caused due to TCF4 repeat expansions. Our data also provided transcriptomic evidence that FECD caused by expanded TCF4 repeats possesses a set of distinct functional properties. However, further studies for the functional characterization of the genes identified in this study are necessary to discern the molecular mechanisms underlying FECD pathogenesis. The generation of stable corneal endothelial cell lines overexpressing or lacking genes of interest could facilitate the development of therapeutic strategies for FECD, thus reducing the need for corneal transplantation. Identification of signature pathways that trigger disease onset in individuals harboring the expanded mutation in FECD may also provide pointers for studying the pathogenesis of other oxidative stress and repeat expansion disorders.
Methods
Generation of Simian virus-40 T-antigen immortalized normal and FECD cell lines
All experiments were performed in accordance with relevant guidelines and regulations. The protocol for generating CE cell lines from FECD patient tissues was approved by the Massachusetts Eye and Ear Institutional Review Board. Written and informed consent was obtained from patients and donors97. The generation of the normal and FECD cell lines by simian virus (SV)-40 (SV-40) immortalization in our lab has been described previously97. Cell lines were prepared from corneal endothelial cells extracted from single corneas of cadaveric donors harvested at Eversight or SightLife eye banks or of FECD patients undergoing routine Descemet’s membrane endothelial keratoplasty (DMEK) at the Massachusetts Eye and Ear Infirmary (Boston, Massachusetts) and Price Vision Group (Indianapolis, Indiana). All FECD patients were at the end-stage of FECD requiring corneal transplantation, with Krachmer grade 5 (≥ 5 mm central confluent guttae without stromal edema) or 6 (≥ 5 mm central confluent guttae with stromal edema) assessed by slit lamp microscopy4,98. Specimens were incubated in complete Chen’s medium (OptiMEM-I; Invitrogen) containing 8% fetal bovine serum (HyClone), 5 ng/ml epidermal growth factor (Millipore), 100 mg/ml bovine pituitary extract (Invitrogen), 200 mg/l calcium chloride (Sigma-Aldrich), 0.08% chondroitin sulfate (Sigma-Aldrich), 50 mg/ml gentamicin (Invitrogen), and 1:100 diluted antibiotic and antimycotic solution (Sigma-Aldrich) for 24 to 48 h. Chen’s medium was removed from specimens and incubated at 37° C, 5% CO2, with 5 ml prewarmed 0.02% ethylenediaminetetraacetic acid (EDTA) solution (Sigma-Aldrich) for up to 45 min to disrupt cell-to-cell junctions. CEnCs were centrifuged for 3 min at 3000 rpm at room temperature to pellet DM and cells, which were resuspended in prewarmed Chen’s media with gentle mechanical trituration and cells and DM were plated in a 24-well tissue culture plate precoated with undiluted FNC Coating (AthenaES). CEnCs were incubated for approximately 48 h at 37° C, 5% CO2, before changing the media. Subculturing of CECs was performed using 0.05% trypsin (Invitrogen) for 5 min at 37° C, 5% CO2. Primary CECs were immortalized SV-40 T-antigen cell immortalization kit (Alstem Cell Advancements). At passage 1, CECs were divided into 2 wells and infected with 2 µl/ml lentivirus (SV-40 T antigen) with 1 µl/ml TransPlus reagent (Alstem Cell Advancements). After 24 h, viral supernatant was removed, and CECs were grown in Chen’s media for 72 h. Transduced CECs underwent puromycin selection (2 µg/ml) until passage 5, after which they were grown in Chen’s media without puromycin. Using this method, we generated 3 normal cell lines (SVN1-67F, SVN2-67F and SVN3-65M) and 7 FECD cells lines (SVF1-73F, SVF5-54F, SVF6-61M, SVF2-61F, SVF3-76M, SVF4-61F, and SVF7-74F) that were further analyzed for TCF4 CTG repeat expansion status.
TCF4 CTG18.1 polymorphism genotyping
The number of repeats in CTG18.1 in the TCF4 gene in the blood samples of patients and donors from which DM and endothelium were collected to generate cell lines, were estimated by short tandem repeat (STR) and triplet-primed PCR (TP-PCR) based fragment analysis as previously established99. For CTG repeat size estimation, genomic DNA was isolated from the peripheral blood mononuclear cells (PBMCs) obtained from same FECD cases. The fragment analysis was performed on data generated by an ABI3730 genetic analyzer in the Genomics Core Facility of Mass Eye and Ear. The literature on FECD suggests that a repeat length of 40 or more CTG repeats is accepted as an expanded allele associated with FECD20. Accordingly, the FECD patients and their respective immortalized cell lines were stratified as either those with mono- or bi-allelic CTG repeats greater than 40 (expanded or FECD-R) or those with bi-allelic CTG repeats less than 40 (non-expanded or FECD-NR) in TCF4. Genetic analysis in blood samples (normal and FECD) as well as newly generated cell lines revealed an expansion of the CTG repeat sequence in the TCF4 gene in 3 out 7 samples (SVF1-73F with 74 repeats, SVF5-54F with 73 repeats, and SVF6-61M with 80 repeats). FECD cell lines retained the TCF4 repeat expansion until at least passage 7.
Genotyping for mutations in other FECD-associated genes
The FECD cell lines (FECD-R and FECD-NR) were further tested for specific FECD-associated genetic variants in LOXHD1, SLC4A11, AGBL1, and KANK4 genes. PCR was performed using primers containing universal M13 tags to target the genetic variants listed in Supplementary Fig. 1. The primer sequences used are provided in Supplementary Fig. 6. PCR products were purified using ExoSAP-IT™ Express PCR Product Cleanup Reagent (Thermo Fisher, Grand Island, NY). Sanger sequencing reactions were then prepared using the purified PCR products and the BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher, Grand Island, NY). Bi-directional sequencing was carried out on the Applied Biosystems 3730xl DNA Analyzer (Thermo Fisher, Grand Island, NY).
RNA isolation and sequencing
Total RNA was extracted from 10 immortalized human corneal endothelial cell lines (3 normal and 7 FECD cell lines; Fig. 1B), with early passage numbers (p5-p7) (Fig. 1B), using RNeasy Micro kit (Qiagen) and sent to the Genomics Core Facility at the Massachusetts General Hospital (Boston, MA) for bulk RNA sequencing. Illumina sequencing libraries were constructed from total RNA using polyA selection followed by NEBNext Ultra II Directional RNA Library Prep protocol (New England Biolabs). Sequencing was performed on Illumina HiSeq 2500 instrument (Illumina) resulting in approximately 30 million of 50 bp reads per sample.
Analysis of differentially expressed gene
Sequencing reads (50 bp single-end reads) were mapped in a splice-aware fashion to the human reference transcriptome (hg19/GRCh37.75 assembly) using STAR100. Read counts over transcripts were calculated using HTSeq101 based on the Ensembl annotation for hg19/GRCh37.75 assembly. For differential expression analysis, we used the EdgeR method102 and classified genes as differentially expressed based on the cutoffs of 2-fold change in expression value and false discovery rates (FDR) below 0.05.
Ingenuity pathway analysis (IPA) and Panther analysis
The datasets of DEGs identified by RNA-seq after comparing (1) the 3 normal cell lines vs. 7 FECD cell lines; (2) the common genes between the 3 normal cell lines vs. 3 FECD cell lines with expanded CTG repeats and 3 normal cell lines vs. 4 FECD cell lines with normal CTG repeats; were uploaded to the server and analyzed through the use of IPA (Ingenuity Pathway Analysis, QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis)103. The canonical pathway and upstream regulator revealed by IPA were further analyzed as gene networks. Additionally, the datasets were uploaded to an online website for Protein Annotation Through Evolutionary Relationship (Panther, version 16.0, http://www.pantherdb.org/) analysis following instructions by Mi et al.104. The gene was grouped to different categories by molecular function and further analyzed by overrepresentation (enrichment) test by using PANTHER Go-Slim molecular function annotation dataset.
Validation of differential gene expression at protein level using Western blot
Whole cell lysates were prepared by lysis in RIPA buffer containing HALT protease and phosphatase inhibitors (#78440, Life Technologies) for 30 min on ice. Protein concentration was determined using the BCA assay kit (#23225, Life Technologies). Whole cell lysates were resolved by loading onto a 4–12% Bis-Tris gel for SDS-PAGE and blotted onto a polyvinylidene difluoride membrane (#IPVH00010, Millipore) which was blocked in 5% nonfat milk (#1706404, Bio-Rad) or 5% bovine serum albumin (#BP1600, Fisher Scientific) in tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h and incubated overnight with primary antibody at 4 °C. The primary antibodies used were MAFB (1:1000, #ab243902, Abcam, USA), TFAP2B (1: 1000, #sc-376709, Santa Cruz Biotechnology, USA), POU6F2 (1: 1000, #2509, Cell Signaling, USA), β-actin (1: 5000, #A1978, Sigma, USA), and secondary antibodies were horseradish peroxidase-conjugated mouse anti-rabbit IgG (1:1000, #sc-2357), anti-mouse IgG (1:1000, #sc-516102), and mouse anti-goat (1:1000, sc-2354), from Santa Cruz Biotechnology Inc., USA. The membrane was developed with SuperSignal West Pico or Femto (#34577 or # 34096, Life Technologies, USA) plus chemiluminescent substrate. Densitometry was analyzed with ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD), and protein content was normalized relative to β-actin protein content. Experiments were repeated a minimum of three times. Results were averaged and SEM values were calculated.
In vitro treatment with UVA light
The procedure for in vitro treatment with ultraviolet-A light has been previously described17. In short, 0.4 × 10^5 cells were seeded in modified Chen’s medium (phenol red-free OptiMEM, Life Technologies) and 8% charcoal-stripped fetal bovine serum (FBS, Gemini Bio-products) on FNC-coated 6-well plates overnight. For UVA irradiation, fresh growth medium was added, and cells were exposed to two 19.5-inch UVA tubes (Analytik Jena US LLC) emitting 365 nm light to deliver a fluence of 5 J/cm2 (6 min), followed by a 24 h recovery period in the same medium.
Statistical analysis
Statistical analysis was performed using a student t-test or either a one-way or two-way analysis of variance followed by a Welch’s correction. A p-value < 0.05 was considered statistically significant.
Data availability
The data generated and/or analyzed during the current study are available in the NCBI- Gene Expression Omnibus (GEO) repository, Accession Number GSE285203 (private until Dec 2025).For reviewer access only: [GEO Accession viewer](https:/www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE285203)Token information: ojmhawkwtfmpnuv.
References
Barrientez, B. et al. Corneal injury: clinical and molecular aspects. Exp. Eye Res. 186, 107709. https://doi.org/10.1016/j.exer.2019.107709 (2019).
Zhu, A. Y., Eberhart, C. G. & Jun, A. S. Fuchs endothelial corneal dystrophy: a neurodegenerative disorder? JAMA Ophthalmol. 132, 377–378. https://doi.org/10.1001/jamaophthalmol.2013.7993 (2014).
Zhang, C. et al. Immunohistochemistry and electron microscopy of early-onset Fuchs corneal dystrophy in three cases with the same L450W COL8A2 mutation. Trans. Am. Ophthalmol. Soc. 104, 85–97 (2006).
Krachmer, J. H., Purcell, J. J. Jr., Young, C. W. & Bucher, K. D. Corneal endothelial dystrophy. A study of 64 families. Arch. Ophthalmol. 96, 2036–2039. https://doi.org/10.1001/archopht.1978.03910060424004 (1978).
Rosenblum, P., Stark, W. J., Maumenee, I. H., Hirst, L. W. & Maumenee, A. E. Hereditary fuchs’ dystrophy. Am. J. Ophthalmol. 90, 455–462. https://doi.org/10.1016/s0002-9394(14)75011-1 (1980).
Bahn, C. F. et al. Classification of corneal endothelial disorders based on neural crest origin. Ophthalmology 91, 558–563. https://doi.org/10.1016/s0161-6420(84)34249-x (1984).
Babacic, H., Mehta, A., Merkel, O. & Schoser, B. CRISPR-cas gene-editing as plausible treatment of neuromuscular and nucleotide-repeat-expansion diseases: A systematic review. PLoS One. 14, e0212198. https://doi.org/10.1371/journal.pone.0212198 (2019).
Goyer, B. et al. Extracellular matrix and integrin expression profiles in Fuchs endothelial corneal dystrophy cells and tissue model. Tissue Eng. Part. A. 24, 607–615. https://doi.org/10.1089/ten.TEA.2017.0128 (2018).
Weller, J. M. et al. Extracellular matrix alterations in late-onset fuchs’ corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 55, 3700–3708. https://doi.org/10.1167/iovs.14-14154 (2014).
Poulsen, E. T. et al. Proteomics of fuchs’ endothelial corneal dystrophy support that the extracellular matrix of descemet’s membrane is disordered. J. Proteome Res. 13, 4659–4667. https://doi.org/10.1021/pr500252r (2014).
Jurkunas, U. V., Bitar, M. S., Funaki, T. & Azizi, B. Evidence of oxidative stress in the pathogenesis of Fuchs endothelial corneal dystrophy. Am. J. Pathol. 177, 2278–2289. https://doi.org/10.2353/ajpath.2010.100279 (2010).
Halilovic, A. et al. Menadione-Induced DNA damage leads to mitochondrial dysfunction and fragmentation during rosette formation in Fuchs endothelial corneal dystrophy. Antioxid. Redox Signal. 24, 1072–1083. https://doi.org/10.1089/ars.2015.6532 (2016).
Azizi, B. et al. p53-regulated increase in oxidative-stress–induced apoptosis in Fuchs endothelial corneal dystrophy: a native tissue model. Invest. Ophthalmol. Vis. Sci. 52, 9291–9297. https://doi.org/10.1167/iovs.11-8312 (2011).
Schmedt, T. et al. Telomerase immortalization of human corneal endothelial cells yields functional hexagonal monolayers. PLoS One. 7, e51427. https://doi.org/10.1371/journal.pone.0051427 (2012).
Okumura, N. et al. Involvement of ZEB1 and Snail1 in excessive production of extracellular matrix in Fuchs endothelial corneal dystrophy. Lab. Invest. 95, 1291–1304. https://doi.org/10.1038/labinvest.2015.111 (2015).
Liu, C. et al. Ultraviolet A light induces DNA damage and estrogen-DNA adducts in Fuchs endothelial corneal dystrophy causing females to be more affected. Proc. Natl. Acad. Sci. U S A. 117, 573–583. https://doi.org/10.1073/pnas.1912546116 (2020).
Ashraf, S. et al. Modulation of ATM enhances DNA repair in G2/M phase of cell cycle and averts senescence in Fuchs endothelial corneal dystrophy. Commun. Biol. 7, 1482. https://doi.org/10.1038/s42003-024-07179-1 (2024).
Kumar, V. et al. Estrogen genotoxicity causes Preferential development of Fuchs endothelial corneal dystrophy in females. Redox Biol. 69, 102986. https://doi.org/10.1016/j.redox.2023.102986 (2024).
Mootha, V. V. et al. TCF4 triplet repeat expansion and nuclear RNA foci in fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 56, 2003–2011. https://doi.org/10.1167/iovs.14-16222 (2015).
Wieben, E. D. et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 7, e49083. https://doi.org/10.1371/journal.pone.0049083 (2012).
Soliman, A. Z., Xing, C., Radwan, S. H., Gong, X. & Mootha, V. V. Correlation of severity of Fuchs endothelial corneal dystrophy with triplet repeat expansion in TCF4. JAMA Ophthalmol. 133, 1386–1391. https://doi.org/10.1001/jamaophthalmol.2015.3430 (2015).
Xing, C. et al. Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with fuchs’ corneal dystrophy in Chinese implies common causal variant. Invest. Ophthalmol. Vis. Sci. 55, 7073–7078. https://doi.org/10.1167/iovs.14-15390 (2014).
Mootha, V. V., Gong, X., Ku, H. C. & Xing, C. Association and Familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 55, 33–42. https://doi.org/10.1167/iovs.13-12611 (2014).
Gattey, D., Zhu, A. Y., Stagner, A., Terry, M. A. & Jun, A. S. Fuchs endothelial corneal dystrophy in patients with myotonic dystrophy: a case series. Cornea 33, 96–98. https://doi.org/10.1097/ICO.0000000000000018 (2014).
Mootha, V. V. et al. Fuchs’ endothelial corneal dystrophy and RNA foci in patients with myotonic dystrophy. Invest. Ophthalmol. Vis. Sci. 58, 4579–4585. https://doi.org/10.1167/iovs.17-22350 (2017).
Winkler, N. S. et al. Fuchs’ endothelial corneal dystrophy in patients with myotonic dystrophy, type 1. Invest. Ophthalmol. Vis. Sci. 59, 3053–3057. https://doi.org/10.1167/iovs.17-23160 (2018).
Okumura, N. et al. Effect of trinucleotide repeat expansion on the expression of TCF4 mRNA in fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 60, 779–786. https://doi.org/10.1167/iovs.18-25760 (2019).
Du, J. et al. RNA toxicity and missplicing in the common eye disease Fuchs endothelial corneal dystrophy. J. Biol. Chem. 290, 5979–5990. https://doi.org/10.1074/jbc.M114.621607 (2015).
Wieben, E. D. et al. Trinucleotide repeat expansion in the transcription factor 4 (TCF4) gene leads to widespread mRNA splicing changes in fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 58, 343–352. https://doi.org/10.1167/iovs.16-20900 (2017).
Soragni, E. et al. Repeat-Associated Non-ATG (RAN) translation in fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 59, 1888–1896. https://doi.org/10.1167/iovs.17-23265 (2018).
Morales, F. et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum. Mol. Genet. 21, 3558–3567. https://doi.org/10.1093/hmg/dds185 (2012).
Zarouchlioti, C. et al. Tissue-specific TCF4 triplet repeat instability revealed by optical genome mapping. EBioMedicine 108, 105328. https://doi.org/10.1016/j.ebiom.2024.105328 (2024).
Eye Banking Statistical Report. Eye Bank Association of America (2016).
Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nat. Rev. Genet. 20, 631–656. https://doi.org/10.1038/s41576-019-0150-2 (2019).
Byron, S. A., Van Keuren-Jensen, K. R., Engelthaler, D. M., Carpten, J. D. & Craig, D. W. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat. Rev. Genet. 17, 257–271. https://doi.org/10.1038/nrg.2016.10 (2016).
Wieben, E. D. et al. Gene expression and missplicing in the corneal endothelium of patients with a TCF4 trinucleotide repeat expansion without fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 60, 3636–3643. https://doi.org/10.1167/iovs.19-27689 (2019).
Nikitina, A. S. et al. Dataset on transcriptome profiling of corneal endothelium from patients with Fuchs endothelial corneal dystrophy. Data Brief. 25, 104047. https://doi.org/10.1016/j.dib.2019.104047 (2019).
Chu, Y. et al. Analyzing pre-symptomatic tissue to gain insights into the molecular and mechanistic origins of late-onset degenerative trinucleotide repeat disease. Nucleic Acids Res. 48, 6740–6758. https://doi.org/10.1093/nar/gkaa422 (2020).
Nakagawa, T. et al. RNA-Seq-based transcriptome analysis of corneal endothelial cells derived from patients with Fuchs endothelial corneal dystrophy. Sci. Rep. 13, 8647. https://doi.org/10.1038/s41598-023-35468-y (2023).
Katikireddy, K. R. et al. NQO1 downregulation potentiates menadione-induced endothelial-mesenchymal transition during rosette formation in Fuchs endothelial corneal dystrophy. Free Radic Biol. Med. 116, 19–30. https://doi.org/10.1016/j.freeradbiomed.2017.12.036 (2018).
White, T. L., Deshpande, N., Kumar, V., Gauthier, A. G. & Jurkunas, U. V. Cell cycle re-entry and arrest in G2/M phase induces senescence and fibrosis in Fuchs endothelial corneal dystrophy. Free Radic Biol. Med. 164, 34–43. https://doi.org/10.1016/j.freeradbiomed.2020.12.445 (2021).
Oksuz, O. et al. Transcription factors interact with RNA to regulate genes. Mol. Cell. 83, 2449–2463. https://doi.org/10.1016/j.molcel.2023.06.012 (2023). e2413.
Fautsch, M. P. et al. TCF4-mediated Fuchs endothelial corneal dystrophy: insights into a common trinucleotide repeat-associated disease. Prog Retin Eye Res. 81, 100883. https://doi.org/10.1016/j.preteyeres.2020.100883 (2021).
Forrest, M. P. et al. The psychiatric risk gene transcription factor 4 (TCF4) regulates neurodevelopmental pathways associated with Schizophrenia, Autism, and intellectual disability. Schizophr Bull. 44, 1100–1110. https://doi.org/10.1093/schbul/sbx164 (2018).
Mesman, S., Bakker, R. & Smidt, M. P. Tcf4 is required for correct brain development during embryogenesis. Mol. Cell. Neurosci. 106, 103502. https://doi.org/10.1016/j.mcn.2020.103502 (2020).
Sanchez-Tillo, E. et al. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. U S A. 108, 19204–19209. https://doi.org/10.1073/pnas.1108977108 (2011).
Li, W. et al. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim. Biophys. Acta. 1783, 1847–1856. https://doi.org/10.1016/j.bbamcr.2008.05.024 (2008).
Katsuoka, F., Motohashi, H., Engel, J. D. & Yamamoto, M. Nrf2 transcriptionally activates the MafG gene through an antioxidant response element. J. Biol. Chem. 280, 4483–4490. https://doi.org/10.1074/jbc.M411451200 (2005).
Motohashi, H., Katsuoka, F., Engel, J. D. & Yamamoto, M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. U S A. 101, 6379–6384. https://doi.org/10.1073/pnas.0305902101 (2004).
Liu, C., Vojnovic, D., Kochevar, I. E., Jurkunas, U. V. & UV-A Irradiation Activates Nrf2-Regulated antioxidant defense and induces p53/Caspase3-Dependent apoptosis in corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 57, 2319–2327. https://doi.org/10.1167/iovs.16-19097 (2016).
Lovatt, M., Kocaba, V., Neo, H., Soh, D. J., Mehta, J. S. & Y. Q. & Nrf2: A unifying transcription factor in the pathogenesis of fuchs’ endothelial corneal dystrophy. Redox Biol. 37, 101763. https://doi.org/10.1016/j.redox.2020.101763 (2020).
Hara, S. et al. Transcription factor TFAP2B up-regulates human corneal endothelial cell-specific genes during corneal development and maintenance. J. Biol. Chem. 294, 2460–2469. https://doi.org/10.1074/jbc.RA118.005527 (2019).
Yoshihara, M. et al. Restricted presence of POU6F2 in human corneal endothelial cells uncovered by extension of the Promoter-level expression atlas. EBioMedicine 25, 175–186. https://doi.org/10.1016/j.ebiom.2017.10.024 (2017).
King, R. et al. Genomic locus modulating corneal thickness in the mouse identifies POU6F2 as a potential risk of developing glaucoma. PLoS Genet. 14, e1007145. https://doi.org/10.1371/journal.pgen.1007145 (2018).
Serbecic, N., Lahdou, I., Scheuerle, A., Hoftberger, R. & Aboul-Enein, F. Function of the Tryptophan metabolite, L-kynurenine, in human corneal endothelial cells. Mol. Vis. 15, 1312–1324 (2009).
Jun, A. S. et al. Microarray analysis of gene expression in human donor Corneas. Arch. Ophthalmol. 119, 1629–1634. https://doi.org/10.1001/archopht.119.11.1629 (2001).
Frausto, R. F., Wang, C. & Aldave, A. J. Transcriptome analysis of the human corneal endothelium. Invest. Ophthalmol. Vis. Sci. 55, 7821–7830. https://doi.org/10.1167/iovs.14-15021 (2014).
Geraci, F., Saha, I., Bianchini, M. & Editorial RNA-Seq analysis: Methods, applications and challenges. Front. Genet. 11, 220. https://doi.org/10.3389/fgene.2020.00220 (2020).
Ljubimov, A. V. et al. Human corneal basement membrane heterogeneity: topographical differences in the expression of type IV collagen and laminin isoforms. Lab. Invest. 72, 461–473 (1995).
Levy, S. G., Moss, J., Sawada, H., Dopping-Hepenstal, P. J. & McCartney, A. C. The composition of wide-spaced collagen in normal and diseased descemet’s membrane. Curr. Eye Res. 15, 45–52. https://doi.org/10.3109/02713689609017610 (1996).
Shousha, M. A. et al. Use of ultra-high-resolution optical coherence tomography to detect in vivo characteristics of descemet’s membrane in fuchs’ dystrophy. Ophthalmology 117, 1220–1227. https://doi.org/10.1016/j.ophtha.2009.10.027 (2010).
Yuen, H. K. et al. A morphologic study of Fuchs dystrophy and bullous keratopathy. Cornea 24, 319–327. https://doi.org/10.1097/01.ico.0000148288.53323.b2 (2005).
Jurkunas, U. V. et al. Decreased expression of Peroxiredoxins in fuchs’ endothelial dystrophy. Invest. Ophthalmol. Vis. Sci. 49, 2956–2963. https://doi.org/10.1167/iovs.07-1529 (2008).
Jurkunas, U. V., Bitar, M. & Rawe, I. Colocalization of increased transforming growth factor-beta-induced protein (TGFBIp) and clusterin in Fuchs endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 50, 1129–1136. https://doi.org/10.1167/iovs.08-2525 (2009).
Biswas, S. et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum. Mol. Genet. 10, 2415–2423. https://doi.org/10.1093/hmg/10.21.2415 (2001).
Gottsch, J. D. et al. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of Fuchs corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 46, 1934–1939. https://doi.org/10.1167/iovs.04-0937 (2005).
Yellore, V. S. et al. Analysis of the role of ZEB1 in the pathogenesis of posterior polymorphous corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 53, 273–278. https://doi.org/10.1167/iovs.11-8038 (2012).
Hemmavanh, C., Koch, M., Birk, D. E. & Espana, E. M. Abnormal corneal endothelial maturation in collagen XII and XIV null mice. Invest. Ophthalmol. Vis. Sci. 54, 3297–3308. https://doi.org/10.1167/iovs.12-11456 (2013).
Gao, X. et al. Genome-wide association study identifies WNT7B as a novel locus for central corneal thickness in Latinos. Hum. Mol. Genet. 25, 5035–5045. https://doi.org/10.1093/hmg/ddw319 (2016).
Wang, X. et al. Multiple roles of FGF10 in the regulation of corneal endothelial wound healing. Exp. Eye Res. 205, 108517. https://doi.org/10.1016/j.exer.2021.108517 (2021).
Yan, J. et al. Transcription factor 4 promotes increased corneal endothelial cellular migration by altering microtubules in Fuchs endothelial corneal dystrophy. Sci. Rep. 14, 10276. https://doi.org/10.1038/s41598-024-61170-8 (2024).
Riazuddin, S. A. et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45–53. https://doi.org/10.1016/j.ajhg.2009.12.001 (2010).
Vedana, G., Villarreal, G. Jr. & Jun, A. S. Fuchs endothelial corneal dystrophy: current perspectives. Clin. Ophthalmol. 10, 321–330. https://doi.org/10.2147/OPTH.S83467 (2016).
Minear, M. A. et al. Genetic screen of African Americans with Fuchs endothelial corneal dystrophy. Mol. Vis. 19, 2508–2516 (2013).
Malhotra, D. et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734. https://doi.org/10.1093/nar/gkq212 (2010).
Venugopal, R. & Jaiswal, A. K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. U S A. 93, 14960–14965. https://doi.org/10.1073/pnas.93.25.14960 (1996).
Venugopal, R. & Jaiswal, A. K. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 17, 3145–3156. https://doi.org/10.1038/sj.onc.1202237 (1998).
Miyajima, T. et al. Loss of NQO1 generates genotoxic estrogen-DNA adducts in Fuchs endothelial corneal dystrophy. Free Radic Biol. Med. 147, 69–79. https://doi.org/10.1016/j.freeradbiomed.2019.12.014 (2020).
Rosenbloom, K. R. et al. ENCODE whole-genome data in the UCSC genome browser. Nucleic Acids Res. 38, D620–D625. https://doi.org/10.1093/nar/gkp961 (2010).
Dunlap, T. L. et al. Red clover Aryl hydrocarbon receptor (AhR) and Estrogen receptor (ER) agonists enhance genotoxic Estrogen metabolism. Chem. Res. Toxicol. 30, 2084–2092. https://doi.org/10.1021/acs.chemrestox.7b00237 (2017).
Yuan, S., He, G. & Li, L. Hsa-miR-155 regulates the cell cycle and barrier function of corneal endothelial cells through E2F2. Am. J. Transl Res. 13, 1505–1515 (2021).
Kumar, V. & Jurkunas, U. V. Mitochondrial dysfunction and mitophagy in Fuchs endothelial corneal dystrophy. Cells 10. https://doi.org/10.3390/cells10081888 (2021).
Reiter, R. J. et al. Melatonin and pathological cell interactions: mitochondrial glucose processing in cancer cells. Int. J. Mol. Sci. 22. https://doi.org/10.3390/ijms222212494 (2021).
Meyer, P. et al. Melatonin MT-1-receptor immunoreactivity in the human eye. Br. J. Ophthalmol. 86, 1053–1057. https://doi.org/10.1136/bjo.86.9.1053 (2002).
Crooke, A. et al. Effect of melatonin and analogues on corneal wound healing: involvement of Mt2 melatonin receptor. Curr. Eye Res. 40, 56–65. https://doi.org/10.3109/02713683.2014.914540 (2015).
Reiter, R. J., Tan, D. X., Terron, M. P., Flores, L. J. & Czarnocki, Z. Melatonin and its metabolites: new findings regarding their production and their radical scavenging actions. Acta Biochim. Pol. 54, 1–9 (2007).
Fischer, T. W., Kleszczynski, K., Hardkop, L. H., Kruse, N. & Zillikens, D. Melatonin enhances antioxidative enzyme gene expression (CAT, GPx, SOD), prevents their UVR-induced depletion, and protects against the formation of DNA damage (8-hydroxy-2’-deoxyguanosine) in ex vivo human skin. J. Pineal Res. 54, 303–312. https://doi.org/10.1111/jpi.12018 (2013).
Slominski, A. T. et al. Melatonin and its metabolites can serve as agonists on the Aryl hydrocarbon receptor and peroxisome proliferator-activated receptor gamma. Int. J. Mol. Sci. 24. https://doi.org/10.3390/ijms242015496 (2023).
Roohbakhsh, A., Shamsizadeh, A., Hayes, A. W., Reiter, R. J. & Karimi, G. Melatonin as an endogenous regulator of diseases: the role of autophagy. Pharmacol. Res. 133, 265–276. https://doi.org/10.1016/j.phrs.2018.01.022 (2018).
Guerra-Ojeda, S. et al. The role of Aryl hydrocarbon receptor in the endothelium: A systematic review. Int. J. Mol. Sci. 24. https://doi.org/10.3390/ijms241713537 (2023).
Parekh, M. et al. Long-term persistent exposure to cigarette smoke induces AhR driven corneal endothelial dysfunction in mice. Exp. Eye Res. 248, 110089. https://doi.org/10.1016/j.exer.2024.110089 (2024).
Motegi, Y., Usui, T., Ishida, K., Kato, S. & Yamashita, H. Regulation of bovine corneal endothelial cell cycle by transforming growth factor-beta. Acta Ophthalmol. Scand. 81, 517–525. https://doi.org/10.1034/j.1600-0420.2003.00128.x (2003).
Saika, S. et al. TGFbeta2 in corneal morphogenesis during mouse embryonic development. Dev. Biol. 240, 419–432. https://doi.org/10.1006/dbio.2001.0480 (2001).
Afshari, N. A. et al. Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat. Commun. 8, 14898. https://doi.org/10.1038/ncomms14898 (2017).
Zarouchlioti, C. et al. Antisense therapy for a common corneal dystrophy ameliorates TCF4 repeat Expansion-Mediated toxicity. Am. J. Hum. Genet. 102, 528–539. https://doi.org/10.1016/j.ajhg.2018.02.010 (2018).
Zhang, X., Kumar, A., Gong, X., Xing, C. & Mootha, V. V. Prevalence of transcription factor 4 gene triplet repeat expansion associated with fuchs’ endothelial corneal dystrophy in the united States and global populations. Ophthalmol. Sci. 5, 100611. https://doi.org/10.1016/j.xops.2024.100611 (2025).
Ong Tone, S. et al. Increased corneal endothelial cell migration in Fuchs endothelial corneal dystrophy: A live cell imaging study. Ophthalmol. Sci. 1, 100006. https://doi.org/10.1016/j.xops.2021.100006 (2021).
Kocaba, V. et al. Association of the Gutta-Induced microenvironment with corneal endothelial cell behavior and demise in Fuchs endothelial corneal dystrophy. JAMA Ophthalmol. 136, 886–892. https://doi.org/10.1001/jamaophthalmol.2018.2031 (2018).
Vasanth, S. et al. Expansion of CTG18.1 trinucleotide repeat in TCF4 is a potent driver of fuchs’ corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 56, 4531–4536. https://doi.org/10.1167/iovs.14-16122 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. https://doi.org/10.1093/bioinformatics/bts635 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. https://doi.org/10.1093/bioinformatics/btu638 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. EdgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Kramer, A., Green, J., Pollard, J. Jr. & Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. https://doi.org/10.1093/bioinformatics/btt703 (2014).
Mi, H., Muruganujan, A., Ebert, D., Huang, X. & Thomas, P. D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. https://doi.org/10.1093/nar/gky1038 (2019).
Acknowledgements
The work was supported by NIH/NEI R01EY020581 (to U.V.J).
Author information
Authors and Affiliations
Contributions
U.V.J. conceptualized the project and acquired the funding. S.A., S.O.T., S.Z., N.D., M.C., G.M., M.O.P., and F.W.P.J. curated the data. S.A., S.Z., N.D., and M.C. performed the formal analysis of the dataset. S.O.T., G.M. and H.H. designed methodology. S.A. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ashraf, S., Tone, S.O., Zhu, S. et al. Transcriptome analyses of human corneal endothelial cell lines derived from patients with Fuchs endothelial corneal dystrophy. Sci Rep 15, 42140 (2025). https://doi.org/10.1038/s41598-025-26025-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-26025-w
