
Abstract
Persistent pain affects one in five people worldwide, often with severely debilitating consequences. While current treatments can be effective for mild or acute pain, they are largely inadequate for managing moderate to severe chronic pain, underscoring the urgent need for new therapeutics. The somatostatin receptor 4 (SSTR4), expressed in sensory neurons of the peripheral nervous system, has recently emerged as a promising target for non-opioid pain relief. However, the presence of several closely related receptors with similar ligand-binding surfaces complicates the design of receptor-specific agonists. Here, we report the discovery of consomatin Fj1, a potent and selective SSTR4-targeting peptide derived from the venom gene repertoire of marine cone snails. Consomatin Fj1 is a mimetic of the endogenous hormone somatostatin but features a minimized receptor binding motif that provides target selectivity. Peripheral administration of synthetic consomatin Fj1 provides analgesia in mouse models of postoperative and neuropathic pain. Using structure–activity studies, we designed and functionally evaluated several Fj1 analogs, resulting in compounds with further improved potency and selectivity. These findings highlight the therapeutic potential of venom-derived peptides as a novel strategy for targeting the SSTR4 and open new avenues for the development of effective treatments for persistent pain.
Similar content being viewed by others

Introduction
Persistent pain, or chronic pain, poses a significant global challenge, impacting more than one in five individuals and exerting substantial personal, societal, and economic burden1. Current treatments, including acetaminophen (paracetamol), non-steroidal anti-inflammatory drugs (NSAIDs), anticonvulsants, antidepressants, and opioids2, are often effective for mild or acute pain but fall short for moderate to severe persistent pain. Moreover, many of these options carry serious side effects, including the risk of tolerance, dependence, and addiction3. The opioid crisis4, in particular, has highlighted the urgent need for new, non-opioid pain therapeutics.
One promising target for opioid-independent pain relief is somatostatin receptor 4 (SSTR4), a G protein-coupled receptor (GPCR) expressed in peripheral sensory neurons and activated by the peptide hormones somatostatin (SST) and cortistatin. The human somatostatin receptor (SSTR) family includes five subtypes (SSTR1–5), which share moderate sequence similarity and overlapping ligand-binding features5. Although all five receptors respond to the same endogenous ligands and signal through Gαi/o proteins, they have distinct expression patterns and physiological roles. For instance, SSTR2 and SSTR5 are prominently expressed in neuroendocrine tumors, where their activation inhibits cell growth6, and receptor-specific agonists targeting these subtypes are used clinically to treat endocrine disorders and certain cancers7,8,9.
The first indication of the potential role of SSTR4-selective agonists for the treatment of pain came from the SSTR1,4-targeting peptide, TT-232, originally developed for the treatment of cancer but later found to exhibit anti-inflammatory and anti-nociceptive effects10,11,12. These results, as well as additional studies on the small molecule agonist J-215613,14, established the SSTR4 as a novel target for pain relief. Although the molecular mechanisms of SSTR4-mediated antinociception are not fully understood, several studies have suggested that SSTR4 activation in nociceptive neurons of the dorsal root ganglia (DRG) and trigeminal ganglia leads to downstream Gβγ-mediated activation of G protein-coupled inwardly rectifying potassium channels (GIRKs), allowing an outward flux of potassium ions, thereby hyperpolarizing cells involved in pain sensing and/or transmission15. Furthermore, SSTR4 activation can reduce capsaicin-induced transient receptor potential cation channel subfamily V member 1 (TRPV1) currents in DRG neurons through Gαi signaling, further hyperpolarizing the cell15. Thus, SSTR4-selective ligands provide analgesia through the inhibition of peripheral pain signals and do not require central nervous system (CNS) administration or penetration.
Our previous discovery of a venom-derived agonist of SSTR1 and SSTR4, consomatin Ro1, from a marine cone snail16 provided proof of principle that cone snail venoms serve as a source of novel SSTR ligands. While consomatin Ro1 exhibited analgesic effects in vivo, its potency at the SSTR4 was moderate, and it lacked receptor selectivity16.
Here, to investigate whether cone snails evolved additional SSTR4-targeting toxins with improved potency and selectivity profiles, we conducted a computational search of venom gene datasets from 247 cone snail species, identifying 529 SST-like sequences. From this pool, eight candidates were selected, synthesized and profiled, leading to the discovery of consomatin Fj1, a highly potent and selective SSTR4 agonist. In mouse models of postoperative and neuropathic pain, consomatin Fj1 produces robust analgesic effects. Structure–activity relationship studies, supported by molecular dynamics (MD) simulations, revealed further opportunities to optimize this compound for therapeutic development. Together, these findings highlight the potential of venom-derived peptides as a novel approach to treating persistent pain by targeting peripheral pathways.
Methods
Toxin sequence extraction and principal component analysis (PCA)
Consomatin sequences were extracted from the SRA datasets of cone snails from Bioproject PRNJ526781, as described previously17. Briefly, data were downloaded from NCBI and preprocessed using fastp v0.20.1. Consomatin-encoding reads were extracted using tblastn with several previously identified consomatins as queries16. These hits were assembled using Trinity v.2.13.218 as single reads. The generated contigs with more than fivefold coverage were translated in all reading frames and putative toxins were extracted with regular expressions (“[^C]{0,2}C[^C]{0,3}WK[^C]{0,3}C[^RKC]{0,6}”) which allows the “WK” motif to appear at any position in the disulfide loop. We next extracted the length, molecular weight, isoelectric point, hydropathy, aliphaticity, aromaticity, polarity, positive and negative charge, and percentage of unique amino acids (glycine and proline), as well as the count of the 140 most common amino acid 1-to-3-mers across all sequences using an in-house script. Principal components were calculated using sklearn v.1.1.319 in Python 3.9.16 on data scaled to remove the mean and unit variance. The first two principal components were visualized in R v.4.2.2 using ggplot v.3.4.2.
Peptide synthesis and purification
Peptides were either purchased from Genscript or synthesized in-house (to > 90% purity) as described below. Peptides were synthesized by solid-phase peptide synthesis (SPPS) at a 0.1 or 0.05 mmol scale using preloaded Fmoc-protected Tentagel R HMPA resin from Rapp Polymere (Tübingen, Germany) for C-terminal carboxylic acid sequences and TentaGel® S RAM Resins for C-terminal amide sequences. Fmoc-protected amino acids, coupling reagents, and solvents used for synthesis were purchased from Iris Biotech (Marktredwitz, Germany). Synthesis was performed using a Syro I instrument (Biotage, Uppsala, Sweden). The coupling conditions were room temperature (RT) for 2 × 120 min using 5.2 equivalents of amino acids, 4.7 equivalents of N-[(1H-benzotriazol-1-yl)(dimethylamino) methylene]-N-methylmethanaminium hexafluorophosphate N-oxide, 5.2 equivalents of 1-hydroxy-7-azabenzotriazol, and 8 equivalents of N,N-diisopropylethylamine in dimethylformamide (DMF) relative to the resin. Deprotection was performed at RT with 40% piperidine in DMF for 3 min, followed by 20% piperidine in DMF for 15 min. The washing steps were performed with 2 × N-methyl-2-pyrrolidone, 1 × dichloromethane (DCM), and 1 × DMF. After completion of peptide assembly, the peptidyl resin was washed with 3 × DCM and dried. The peptide was released from approximately 0.025 mmol of resin using a mixture (2 mL) of 95% trifluoroacetic acid (TFA), 2.5% triethylsilyl, and 2.5% water over 2.5 h. For methionine-containing sequences, the 2 mL mixture contained 87.5% TFA, 2.5% triethylsilyl, 2.5% water, 2.5% thioanisole, and 5% ethanethiol, or alternatively 87.5% TFA, 2.5% triethylsilyl, 2.5% water, 2.5% thioanisole, and 5% ethanethiol with approximately 0.4 mg sodium iodide (NaI). Cold diethyl ether (13 mL stored at − 20 °C) was added, and the mixture was further cooled to − 85 °C for 30 min before the peptide was isolated by centrifugation. The peptide was redissolved in a mixture of acetic acid, water, and acetonitrile (ACN) in a volume ratio of 1:10:4 and freeze-dried to remove unwanted carboxylation of Trp. For peptides containing methionine, acetic acid was replaced with half the volume of TFA. The freeze-dried compound was dissolved in a few drops of TFA and 5 mL of ACN, followed by the addition of 5 mL of water. Phosphate buffer (0.1 M) was added to this mixture until a total volume of 18 mL. The pH was adjusted to 6 by using 1 M hydrochloride acid. Dimethyl sulfoxide (2.0 mL) was added (10%, v/v) and the mixture was stirred for 40–72 h at RT to allow disulfide bond formation. The mixture was filtered and directly purified using one of two methods: the peptides were purified using a Luna C18(2) column from Phenomenex (Torrance, USA, 5 μm, 100 Å, 250 × 10 mm) on a Dionex Ultimate 3000 HPLC system (Thermo Fisher, Waltham, USA) or a 50 g Biotage Sfär Bio C18 D column on a Selekt System (Biotage, Sweden). For both systems, a gradient of 5–100% ACN in water/0.1% formic acid (or 0.1% TFA for methionine-containing sequences) was applied. The product was isolated and lyophilized by freeze-drying. The final product was analyzed by liquid chromatography-mass spectrometry using a Dionex Ultimate 3000 ultrahigh-performance liquid chromatography system (Thermo Fisher) connected to an Impact HD mass spectrometer (Bruker, Bremen, Germany). Information on peptide sequences, purity and integrity as determined by LC and MS is provided the supplemental information under peptide synthesis.
PRESTO-Tango beta arrestin recruitment assay
Synthetic peptides were screened at the five human SSTRs using the PRESTO-Tango β-arrestin recruitment assay as previously described20. HTLA cells (a kind gift from Prof. Hans Bräuner-Osborne) maintained in DMEM (Gibco) supplemented with 10% FBS (Biowest, Nuaillé, France), 100 U/mL penicillin/100 mg/mL streptomycin (Gibco), 100 µg/mL hygromycin B (ThermoFisher Scientific), and 2 µg/mL puromycin (Gibco) (growth medium) were maintained in a water jacketed 5% CO2 incubator and passaged every 2–3 days using trypsin/EDTA (Gibco). 1E6 cells were seeded into 6 well plates. The following day, the medium was changed, and the cells were transfected with 2 µg of DNA from the relevant receptor construct (obtained from Addgene) and 20 µL PolyFect (Qiagen) into 100 µL DMEM and incubated for 10 min. Growth medium (400 µL) was added and the mixture was added to the cells. 14–18 h later, 4E4 cells per well in 40 µL of DMEM supplemented with 1% dialyzed FBS (assay medium) were seeded into white clear bottom 386 well plates (Corning). The following day, the medium was changed to 40 µL new assay medium and peptides were reconstituted and added at 5× final concentration in HBSS supplemented with 20 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, and 0.1% BSA (stimulation buffer). The following day, the medium was discarded, and 20 µL of a 20× dilution of BrightGlo (Promega) in stimulation buffer was added. The plate was incubated in the dark at RT for 20 min, and luminescence was read at 1000 ms integration time on a SpectraMax iD5 (Molecular Devices).
G protein dissociation assay
To determine downstream receptor signaling using native receptor sequences, we used a bioluminescence resonance energy transfer (BRET)-based assay that measures GPCR activation by monitoring the association of labeled Gβγ subunits and a G protein-coupled receptor kinase (GRK) fragment, following dissociation of Gβγ subunits from the Gα subunit as previously described21. HEK293 cells (ATCC) maintained in DMEM (Gibco) supplemented with 10% FBS (Biowes, Nuaillé, France) and 100 U/mL penicillin/100 mg/mL streptomycin (Gibco) (growth medium) were maintained in a water jacketed 5% CO2 incubator and passaged every 2–3 days using trypsin/EDTA (Gibco). 1E6 cells were seeded into 6 well plates. The following day, the medium was changed, and the cells transfected by adding 0.33 µg of SSTR construct, 0.66 µg of Gα protein, 0.33 µg Venus 156–239-Gβ, 0.33 µg Venus 1–155-Gγ, and 0.33 µg masGRK3ct-Nluc DNA into 27 µL of 150 mM NaCl, and 12 µL of 100 mg/mL linear polyethyleneimine (PEI) at molecular weight 25 kDa (Polysciences, Warrington, PA, USA) into 38 µL of a 150 mM NaCl solution. The two tubes were mixed and incubated for 10–15 min before addition to the cells. The following day, 5E4 cells per well were seeded in 100 µL of growth medium into poly-d-lysine-coated white 96 well OptiPlates (PerkinElmer). The following day, the cells were washed in 80 µL of HBSS supplemented with 20 mM HEPES, 1 mM CaCl2, and 1 mM MgCl2 (assay buffer), and 80 µL of assay buffer supplemented with 0.1% BSA (stimulation buffer) was added. 10 µL of a 1:250 dilution of furimazine (Promega) in stimulation buffer was added, and after 120 s, 10 µL of a 10× solution of the compounds reconstituted in stimulation buffer (HBSS supplemented with 20 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, and 0.1% BSA) was added. 180 s after furimazine addition, the plate was placed in a SpectraMax iD5 and read using a program consisting of 60 s of shaking, followed by 500 ms integration reads with a 485/20 nm filter, followed by a 535/25 nm filter.
Animal ethics
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Arizona and were conform to the guidelines for the use of laboratory animals of the National Institutes of Health (NIH publication no. 80–23, 1966), or in accordance with the guidelines of the Danish Animal Experimentation Inspectorate (permission number 2021-15-0201-01036) in a fully AAALAC-accredited facility, under the supervision of the local animal welfare committee at the University of Copenhagen. Experiments using animals are reported in accordance with ARRIVE (Animal Research: Reporting of In Vivo Experiments).
Paw incision model of postoperative pain
Male or female C57Bl/6j mice (6–8 weeks old, The Jackson Laboratory, Bar Harbor, ME) were housed one to six per box (single housing due to fighting) with standard bedding and ad libitum access to standard chow and tap water. Mice were kept in 12 h/12 h light/dark cycles and left to habituate for 1 week prior to testing. To analyze post-surgery acute incisional hypersensitivity, a plantar incision model was used as described by Brennan et al.22. Briefly, a 0.5 cm long incision, from the heel toward the toes, was made through the skin and fascia of the plantar aspect of the left hind paw, including the underlying muscle. The plantaris muscle was then elevated and longitudinally incised, leaving the origin and insertion intact. After hemostasis with gentle pressure, the skin was closed with two mattress sutures of 5–0 nylon on a curved needle.
Measurement of tactile sensory thresholds for postoperative pain model
Tactile sensory thresholds were assessed by measuring the withdrawal response to probing the plantar surface of the hind paw with a series of calibrated fine filaments (von Frey) following administration of peptide or vehicle (saline: 0.9% sodium chloride (NaCl) in sterile water). Each filament was applied perpendicular to the plantar surface of the paw of mice held in suspended wire mesh cages. The “up and down” method was used to identify the mechanical force required for a paw withdrawal response. The size range of stimuli was between 2.44 (0.4 mN) and 4.56 (39.2 mN). The starting filament was 3.61 (3.9 mN). The filament was placed perpendicular to the skin with a slowly increasing force until it bent; it remained bent for approximately 1 s and was then removed. Data were analyzed using the nonparametric method of Dixon, as described by Chaplan et al.23. The results are expressed as the mean withdrawal threshold that induced a paw withdrawal response in 50% of the animals. Statistical analysis was performed as described in the “Statistical analysis” section below.
Spared nerve injury (SNI) model of neuropathic pain
C57BL/6Nrj male mice, approximately 10 weeks of age (Janvier, France), were housed 1–6 per box (single housing due to fighting) with standard bedding and ad libitum access to standard chow (Altromin 1342, Brogaarden, Denmark) and tap water. Mice were kept in 12 h/12 h light/dark cycles (experiments performed during their inactive phase) and left to habituate to the facility for 1 week prior to testing. Mice were anesthetized with 2% isoflurane gas followed by subcutaneous injection of buprenorphine (0.1 mg/kg, Department of Experimental Medicine, University of Copenhagen (UCPH)), and SNI surgery was performed on the left hind leg as follows. The skin on the lateral surface was incised for approximately 0.5 cm between the hip and knee, followed by cutaneous application of a single drop of a mixture of 10 mg/kg lidocaine and 5 mg/kg bupivacaine (Department of Experimental Medicine, UCPH) before lengthwise division of the biceps femoris muscle, leading to exposure of the three branches of the sciatic nerve. The sural branch was left intact, whereas the peroneal and tibial branches were ligated with a single surgical knot and axotomized distal to the ligation. Wounds were closed with surgical glue, and animals were monitored daily for signs of stress or discomfort, but in all cases, recovered uneventfully. Immediately post-surgery, the mice were administered carprofen (5 mg/kg, Department of Experimental Medicine, UCPH). On day 16 post-surgery, mice were injected i.p. with a volume of 10 µL/g of the indicated doses of Fj1, 30 mg/kg of gabapentin (Sigma Aldrich), or vehicle (phosphate buffered saline).
Measurement of tactile sensory thresholds for SNI model
For the SNI experiments, von Frey filaments ranging from 0.04 to 2 g (g = gram-forces) (0.04, 0.07, 0.16, 0.4, 0.6, 1.0, 1.4, 2.0) were used to determine the mechanical paw withdrawal threshold (PWT). Filaments were applied in ascending order to the frontolateral plantar surface of the hind paws innervated by the sural nerve. Mice were placed in red PVC plastic cylinders (8 (Ø) × 7.5 (h) cm) on a wire mesh and allowed to habituate for a minimum of 20 min prior to the experiment initiation. Each von Frey hair was applied five times with adequate resting periods between each application, and the number of withdrawals was recorded. The withdrawal threshold was determined as the von Frey filament eliciting at least three positive trials out of five applications in two consecutive filaments. A positive trial was defined as a sudden paw withdrawal, flinching, and/or paw licking induced by the filament. The animals were habituated to the experimental room for a minimum of 60 min before the initiation of the experiment, and the experimenter (female) was blinded to the treatment groups. Statistical analysis was performed as described in the Statistical analysis section below.
Structural preparation
Modelling of consomatin Fj1 at the SSTR4 made use of the X-ray crystal structure of consomatin Ro1 (PDB: 7SMU)16, and the cryoEM structure of the SSTR4 in complex with the Gi1 subunit and SST-14 ligand (PDB: 7XMS)24 as modeling templates. Because of the sequence similarity of consomatin ligands and SST-14, specifically the core Trp-Lys receptor-binding motif mimicking the broader conserved Phe-Trp-Lys-Thr residues present within SST ligands, Fj1 was assumed to bind the same orthosteric binding pocket as the endogenous ligands. Modeller (v.10) utility was used to create complexes of ligands bound to the SSTR4 binding pocket, generating 50 models with the top model assessed using the Modeller objective function to seed subsequent simulations. Due to the absence of N-terminal SSTR4 residues present within the cryoEM structure, only residues Met47-Phe322 were included within the final complex, with the Gi1 subunit removed. Non-standard amino acids hydroxyproline (Hyp9) and D-tryptophan (D-Trp4) were included, with parameters assigned from the CHARMM36m force field. The resulting complex was inserted into a model POPC membrane using the CHARMM-GUI server and solvated in a box of CHARMM-modified TIP3P (mTIP3P) water, extending > 10 Å beyond all protein atoms. The resulting bilayer spanned an area of 100 × 100 Å2, with a total of 130 lipids each in the upper and lower leaflets. Sodium and chloride ions were included to neutralize the system and attain an ionic strength of 0.1 M.
Molecular dynamics simulations
Simulations were performed using the GROMACS v2022)25 package with the CHARMM36m force field26,27. Temperature coupling was achieved using velocity rescaling applying a coupling time of 0.1 ps with the protein, membrane and water/ions coupled separately at 310 K. Semi-isotropic pressure coupling was maintained using the Parrinello–Rahman method with a coupling time of 2.0 ps. All simulations were performed with a single non-bonded cut-off of 12 Å, with van der Waals interactions switched at 1.0 Å. The Verlet neighbor searching cut-off scheme was applied with a neighbor-list update frequency of 25 steps (50 fs); the time step used in all the simulations was 2 fs. Periodic boundary conditions were applied using the particle-mesh Ewald method to account for long-range electrostatic interactions. The bond lengths were constrained using the P-LINCS algorithm28. Initial minimization and equilibration of all simulations followed established procedures outlined by the CHARMM-GUI via a steepest descent protocol, followed by short positionally restrained equilibration in the NVT (canonical) and NPT ensemble. The systems were then allowed to progress for 1 μs, with each system simulated in 10 replicas, for a total simulation time of 10 μs per consomatin analog.
Statistical analysis
All statistical analyses, fittings, and plotting of curves were performed using GraphPad Prism 10.
For GPCR assays, experiments were conducted in triplicate, and EC50 and pEC50 ± 95% confidence intervals (CI95) were calculated in Prism. When statistical analysis could not be performed (e.g., for peptides with very low activity that did not reach saturation at the highest concentration tested), assays were performed in duplicate. Some peptides, such as controls, were tested in additional independent experiments (i.e., more than three replicates); the number of replicates for these cases is indicated in parentheses in Figs. 1 and 5, as described in the figure legends.

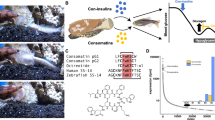
Computational selection and SSTR profiling of consomatins from a large toxin gene dataset. (A) Principal component analysis (PCA) of 529 SST-like toxin sequences was used as the basis for selecting lead compounds for SSTR screening. Percentages refer to the proportion of the total variance in the data explained by the given principal component. A subset of the PCA plot, including vertebrate SS-like sequences and selected consomatins, is shown (right box). (B) Synthesized consomatin sequences selected from the PCA plot. Cleavage sites and post-translational modifications, such as D-Trp, were predicted based on the original somatostatin-like venom peptide, consomatin Ro1. Cysteines forming intramolecular disulfide bonds are shown in yellow and the essential Trp-Lys motif is highlighted in blue and green. Post-translational modifications are indicated by γ = γ-carboxyglutamate, w = D-Trp, O = hydroxyproline, and # = C-terminal amidation. SST-14 is human somatostatin-14. Seven of the listed sequences are from snails of the Asprella clade; Ro1 and Ro2 from C. rolani, Fj1, Fj2, and Fj3 from C. fijisulcatus, and SuSa1 and SuSa2 from C. sulcatus samiae, two are from the snails of the Phasmoconus clade; Oc3 from C. ochroleucus; Pr1 from C. parius, while pG1 was predicted from the transcriptome of C. geographus from the Gastridium clade16. (C) Heatmap of activity values of selected consomatins tested at the five human SSTRs using the PRESTO-Tango β-arrestin recruitment assay. Rows correspond to the sequences shown in (B). Data are represented as EC50 (top line, nanomolar, two significant digits shown) and pEC50 ± 95% confidence intervals (CI95) with the number of independent repeats in parentheses (bottom line, two significant decimals shown). Approximate values (~) or “higher than” (>) and “lower than” (<) are used when we did not obtain full curves within the concentrations tested (see Fig. S1 for representative curves).
For the paw incision model, the effects at different time points were analyzed using two-way ANOVA with Geisser-Greenhouse correction and Dunnett’s post-test for multiple comparisons (all compared to vehicle), while the area under the curve plot was analyzed using one-way ANOVA with Dunnett’s post-test for multiple comparisons (all compared to vehicle). For the SNI model, the effect over time was analyzed as described for the paw incision model (all compared to the vehicle on the ipsilateral side), while the area under the curve was analyzed using a two-way ANOVA with Dunnett’s post-test for multiple comparisons (all compared to the vehicle on the ipsilateral side).
Images and visual representation
All graphs were created using either GraphPad Prism 10 or a custom Python script for heatmaps (https://github.com/Aephir/create_heatmap, v2.0.6 used). All images that are not data graphs or plots were created using Biorender, Adobe Illustrator, and/or Inkscape.
Data analysis
The BRET signals in the G protein dissociation assay were calculated by dividing the signal at 535 nm by that at 485 nm. All concentration–response curves were fitted using a variable slope (four-parameter) equation:
Potency calculations were performed using log10 transformed values (pEC50) before transforming the average to EC50 values.
For the MD data, representative orientations of ligand complexes were determined by clustering all simulation replica trajectories (limited to 100,000 frames) using the GROMOS method with a cut-off between clusters of 0.25 nm. VMD was used along with the tachyon renderer for visualization, and the images were encoded to produce videos with ffmpeg.
Results
Identification of candidate SSTR agonists from large venom gene library
We recently discovered that cone snails have evolved a family of peptide toxins called consomatins, which resemble the hormone SST and related peptides like cortistatin and urotensin17. These toxins originated from an SST-like peptide used in the snail’s own internal signaling and later diversified after being recruited into the venom system17. To identify consomatins that might target vertebrate SSTRs, especially the SSTR4, we analyzed genomic data from 247 cone snail species. We found 529 consomatin sequences across 169 species and used principal component analysis (PCA) to explore their evolutionary relationships. We also included three previously characterized consomatins (Ro1, Ro2, and pG116,29) in our analysis. Since cone snails specialize in different prey (worms, snails, or fish), we compared their toxins to natural SST-like peptides from vertebrates, annelid worms, and mollusks. As seen in our previous study17, toxins from worm-hunting snails grouped with worm hormones, while many fish-hunting snail toxins (including Ro1) clustered with vertebrate peptides (Fig. 1A). No consomatins were found in snail-hunting species. For subsequent experimental testing, we prioritized toxin sequences from fish-hunting species over those from worm hunting species, as the fish SST receptors targeted by these toxins show greater similarity to human receptors than those of worms. Furthermore, we focused on species that are closely related to the species expressing consomatin Ro1, Conus rolani from the Asprella clade. Finally, we selected those sequences that clustered closely with vertebrate hormones in the PCA, as they were more likely to activate human SSTRs. The final list of candidates is shown in Fig. 1B.
Receptor profiling identifies potent and selective agonist of the SSTR4
The predicted mature peptide sequences selected from the PCA plot were synthesized to > 90% purity using standard solid-phase peptide synthesis and verified using reverse-phase high-performance liquid chromatography and mass spectrometry. Post-translational modifications were predicted based on modifications previously observed for the venom peptide consomatin Ro116. Modifications included γ-carboxylation of Glu, hydroxylation of Pro (Hyp), L-to-D epimerization of a Trp positioned within the disulfide loop, and C-terminal amidation based on the presence of a Gly-Arg/Lys motif30. Disulfide bonds were predicted based on the presence of two cysteine residues. We note that the predicted modifications and the proteolytic N- and C-terminal cleavage sites may differ from those found in the native toxins. Future proteomics studies on collected venom are needed to establish the exact chemical identity of the toxins studied here.
Synthetic peptides were screened at the five human SSTRs using the PRESTO-Tango β-arrestin recruitment assay20 (Fig. 1C and Fig. S1). Human somatostatin-14 (SST-14) and the previously characterized peptides, consomatin Ro1 and pG1, were included for comparison. The eight new peptides that were tested showed a range of SSTR activity profiles demonstrating that cone snail venoms are a rich source for the discovery of novel ligands of the human SSTRs (Fig. 1C). For example, Fj2 activates the SSTR2 and SSTR4 with EC50 values of 0.20 µM and 2.0 µM, respectively, Pr1 activates the SSTR1 and SSTR2 with EC50 values of 130 nM and 41 nM, respectively, and Oc3 shows activity at SSTR2, SSTR3, and SSTR4 with EC50 values ranging from 5.0 nM to 4.2 µM. Interestingly, although the sequence of Ro2 is distinct from the previously characterized Ro1, the two peptides that are both derived from the same cone snail species, C. rolani, have very similar activity profiles at the human SSTR1 and SSTR4. Notably, none of the peptides tested showed any significant activity at SSTR5, although it should be noted that the peptides were tested for agonism only; thus, we cannot rule out antagonist activity. Of particular interest to us was Fj1, which activated the SSTR4 with an EC50 value of 22 nM, showing 173-fold selectivity over SSTR1 (EC50 = 3.8 µM) and no measurable activity at the other three receptor subtypes when tested at concentrations up to 100 µM.
Consomatin Fj1 is a somatostatin evolog with a minimized receptor binding motif
Consomatin Fj1 is a ten amino acid long cyclic peptide from Conus fijisulcatus that contains four residues inside a disulfide loop and two residues at both its N- and C-termini (Sequence: Pro1-Val2-Cys3-D-Trp4-Lys5-Phe6-Gly7-Cys8-Hyp9-Leu10, Fig. 2A,B). The minimized disulfide loop of Fj1 closely mirrors that of several SSTR2-selective somatostatin drug analogs, such as octreotide and lanreotide. Incorporation of a shortened disulfide loop and a D-Trp in these analogs significantly prolonged their half-lives compared to the native human hormone9. In contrast to these analogs, which were developed using medicinal chemistry approaches, consomatin Fj1 and other consomatins evolved from an endogenous SST-like signaling peptide following the principles of natural selection. Thus, we refer to these naturally evolved analogs as evologs16. Notably, despite these distinctively different approaches, the final products exhibit a remarkably high degree of similarity (Fig. 2B). One notable difference that distinguishes consomatin Fj1 from other SST analogs and evologs is that the characteristic D-Trp-Lys motif essential for SSTR activation6 resides immediately following the first cysteine residue (Fig. 2B). As discussed in more detail below, these differences partly contribute to the potency and selectivity of the peptide for the SSTR4.

Consomatin Fj1 is a naturally evolved SSTR4-selective somatostatin evolog. (A) Chemical structure of consomatin Fj1, a ten-residue long cyclic peptide predicted from the venom gene-encoding exons of Conus fijisulcatus. Standard amino acid one or three letter abbreviations are used, extended with O/Hyp = 4-hydroxyproline, w/D-Trp = D-tryptophan, and -ol = C-terminal alcohol group in place of a carboxylic acid group. (B) Schematic comparison of the contrasting origins of consomatin Fj1 and the SST drug analog octreotide, both representing minimized SST scaffolds. Fj1 is a minimized SSTR4-selective evolog that originated from an endogenous SS-like signaling gene, while the SSTR2-selective drug analog octreotide was designed using medicinal chemistry approaches. (C) Representative concentration–response curves for G protein (GαoA) dissociation of SST-14 and Fj1 at the five human SSTRs. Fj1 shows nM potency at the human SSTR4, with low potency at SSTR1 and no discernable activity at the other SSTR subtypes at 1 µM. Error bars represent the standard deviation of two technical replicates.
Consomatin Fj1 induces G protein dissociation at the SSTR4
To determine downstream receptor signaling using native receptor sequences, we assessed the activity of Fj1 at the SSTR4 using a BRET-based assay that measures GPCR activation by monitoring the association of labeled Gβγ subunits and a G protein-coupled receptor kinase (GRK) fragment, following dissociation of Gβγ subunits from the Gα subunit21. We tested the activity of Fj1 at the SSTR4 using a range of different Gα proteins in the Gαi family, including Gαi1, Gαi2, Gαi3, as well as the two dominant splice variants of Gαo, and observed similar potencies for each Gα protein (Fig. S2). As the Gαo type is the most highly expressed type in DRGs31, the proposed site for SSTR4-mediated analgesia, we performed subsequent tests with the canonical Gαo sequence (GαoA). Here, we observed similar overall receptor activation profiles as in the PRESTO-Tango screening assay: potent activation of SSTR4 (EC50 = 6.0 nM) with only limited activity at SSTR1 (~ 30% activation at 1 µM) and no activation of the SSTR2,3,5 when tested at up to 1 µM (Fig. 2C).
Peripheral consomatin Fj1 administration provides analgesia in a postoperative pain model
Having established that consomatin Fj1 selectively activates the SSTR4 in the PRESTO-Tango and G protein dissociation assays, we next performed a dose response study for potential analgesic action in a model of postoperative pain, the paw incision model32. C57BL/6 J male mice injected intraperitoneally (i.p.) with 0.04–2.5 mg/kg of Fj1 and its effect on post-incision mechanical hypersensitivity was evaluated (Fig. 3A,B). Fj1 dose-dependently reduced post-incisional mechanical hypersensitivity, as measured by paw withdrawal thresholds in response to von Frey filament stimulation, with maximal effect observed at the dose of 2.5 mg/kg, and a peak effect observed at 1 h. The positive control used was morphine sulfate (5 mg/kg). While morphine exhibited higher peak efficacy than Fj1, the duration of action was shorter. Even at the 5 h time point, the mice treated with 2.5 mg/kg of Fj1 showed an increased paw withdrawal threshold of about 28% of maximum Fj1 response (P = 0.0284, as compared to vehicle). Both in the groups treated with 0.04 mg/kg and with 0.1 mg/kg of Fj1, there was a similar trend as for 2.5 mg/kg, though neither reached a statistically significant difference from the vehicle control at any time point (P = 0.2936 and P = 0.0565, respectively, at 2 h). When tested in female mice, we observed similar results with one noticeable difference (Fig. 3C,D). While the effect was statistically significant at 2.5 mg/kg of Fj1, it was not as robust as for the males with a lower effect size observed both at 0.5 h and 1 h. By administering 5 mg/kg to females, the effect more closely followed the effect of 2.5 mg/kg in males.

Consomatin Fj1 provided analgesia in two mouse models of pain. Post-surgical pain model (paw incision) in either male (A,B) or female (C,D) mice. Plantar incision surgery was performed 24 h post-surgery, and mice were administered morphine sulfate (5 mg/kg), indicated doses of Fj1, or vehicle (saline) by intraperitoneal (i.p.) injections, whereafter mechanical hypersensitivity was evaluated at indicated times (A,C). Values represent mean ± CI95 from six mice. A summary of the area under the curve is shown (B,D), where each data point represents one mouse, and the error bars represent CI95. (E,F) Neuropathic pain model (spared nerve injury, SNI). The peroneal and tibial branches of the sciatic nerve of the left hindleg were ligated, 16 days post-surgery, the mice were administered gabapentin (30 mg/kg), indicated doses of Fj1, or vehicle (phosphate buffered saline) by i.p. injections, whereafter mechanical hypersensitivity was evaluated at indicated times (E). Values represent the mean ± CI95 from five (gabapentin) or six (all other conditions) mice. A summary of the area under the curve is shown (F), where each data point represents one mouse, and the error bars represent CI95. Statistical analysis was performed as described in the methods section. Asterisks indicate P values < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****) as compared with the vehicle control.
Peripheral consomatin Fj1 administration provides analgesia in a neuropathic pain model
Activation of the SSTR4 has previously been shown to provide analgesia in rodent models of neuropathic pain14,33,34,35. Drugs that are effective in postoperative pain may not be efficacious in neuropathic pain states and vice a versa. In general, neuropathic pain is particularly difficult to treat; many current treatment options are only effective in a limited subset of patients and can elicit severe side effects36,37,38. To assess whether Fj1 could alleviate the mechanical hypersensitivity associated with neuropathic pain, the peptide was tested in the spared nerve injury (SNI) model of peripheral neuropathic pain using male C57BL/6 mice. The sciatic nerve was injured on one side of the mouse (ipsilateral side), whereas the other side (contralateral side) served as a control. On day 16 post-injury, either 0.5 mg/kg or 5 mg/kg of Fj1, 30 mg/kg of gabapentin, or vehicle was administered by i.p. injection (Fig. 3E,F). Both doses of Fj1 reduced mechanical hypersensitivity 30 min after injection, and this effect was more pronounced after 1 h. For the 0.5 mg/kg dose, the effect persisted at 2.5 h, while for the 5 mg/kg dose, the tendency was the same, albeit not reaching statistical significance (P = 0.0548 compared to vehicle). These results demonstrate that Fj1 can provide potent and efficacious analgesia in this model, although with a slower onset and shorter duration of action than 30 mg/kg of gabapentin.
Molecular dynamics simulation reveals the binding mode of Fj1 at the SSTR4
To investigate the binding mode of Fj1 at the SSTR4 and better understand the molecular basis for the peptide’s selectivity and potency, we performed molecular dynamics (MD) simulations in the µs range of Fj1 to the holo-SSTR4 embedded within a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer. The binding mode of consomatin Fj1 was stabilized and almost exclusively characterized through the side chain interactions of D-Trp4 and Lys5 with receptor residues (Fig. 4A). D-Trp4 buried deep within the hydrophobic core cavity formed by the side chains of Leu1233×29, Met1303×36, Phe1313×37, Ile1814×61, Leu20045×52, and Phe2756×51 (superscripts denote the GPCRdb numbering scheme outlined by Isberg et al.39). Interestingly, D-Trp4 occupied the same binding pocket as the corresponding L-Trp of SST-14 observed in the recent cryogenic electron microscopy (cryoEM) structure of SSTR4 (PBD ID: 7XMS)24 (Fig. 4B). Lys5 forms a salient salt bridge with the side chain of Asp1263×32, also mirroring the conformation and interactions of the corresponding Lys9 in SST-14. Additionally, Lys5 in Fj1 also demonstrated transient yet notable hydrogen bonding to the side chain of Ser3007×41 during simulation. On the opposite face of the side chain, Lys5 maintains a side-by-side stacking arrangement against the indole ring of D-Trp4, which is also present in the monomeric structure of Fj1, and is characteristic of SST receptor binding and activation6. Both D-Trp4 and Lys5 demonstrated limited sampling of other conformations throughout the simulation.

Molecular Dynamics (MD) simulations reveal contacts between Fj1 and SSTR4. (A) Representative orientation of Fj1 (orange stick representation) in complex with SSTR4 (gray ribbon representation). The membrane present within the simulation was removed for clarity. The interactions made by Fj1, which facilitated binding, are labeled. The representative binding mode was obtained by clustering all replicate trajectories, with a model representing the largest cluster displayed. (B) Overlay of Fj1 (orange) and SST-14 (blue, PDB 7XMS) in complex with SSTR4 (dark gray). (C) The orientation of Fj1 throughout a representative 1 µs simulation is shown with 10 snapshots colored from blue to white to red. (D) Representative orientation of Fj1 (orange) from the simulation when in complex with SSTR4, overlaid with SST-14 in complex with the same receptor construct (gray, PDB 7XMS).
In contrast to the persistent orientation adopted by D-Trp4 and Lys5, residues bordering and outside the cyclic core, Pro1, Cys3, Phe6, Cys8, Hyp9, and Leu10 (i.e., with the exception of Val2) displayed considerable mobility, demonstrating limited persistent contacts describing the ligand-bound orientation (Fig. 4C,D). Likewise, residues outside of the conserved pharmacophore (Phe7-Trp8-Lys9-Thr10) in SST-14 do not form significant side chain interactions with SSTR4, as has been observed with SSTR224, instead forming more backbone and intramolecular interactions (most notably between Phe6-Phe11). The short cyclic core (four residues in Fj1, compared to ten residues in SST-14) does not protrude significantly beyond the binding pocket and aligns well with the smaller four-residue pseudo-core formed between Phe6 and Phe11 of the SSTR-bound structure of SST-14. Val2 presents a somewhat smaller hydrophobic interface within the helical core, packing between residues Val2125×40 of helix 5 and Leu2836×59 of helix 6, occupying a position similar that to of Phe7 in SST-14. These simulations showed that Fj1 binds SSTR4 in a manner similar to SST-14, particularly in regard to the Trp-Lys motif, and that residues outside of this could be amenable to modification while retaining SSTR4 binding.
Analog design provides improved agonists and scaffolds for future drug development
To assess the molecular determinants of SSTR4 potency and selectivity in the G protein dissociation assay, we first performed alanine scanning of the four amino acids in the loop that were buried in the binding pocket of the receptor (Fig. 5). As expected, mutating key residues known to be important for the binding of somatostatin6, Trp4 (Fj1A1) and Lys5 (Fj1A2), to Ala markedly decreased potency by 5000-fold and 730-fold, respectively. The mutation of Phe6 (corresponding to Thr10 in SST-14, see Fig. 4D) to Ala (Fj1A3) also decreased the potency by 630-fold. However, mutating either Gly7 to an Ala (Fj1A4) or deleting it, thereby shortening the loop (Fj1A5), resulted in a slight increase in potency at SSTR4 (6.2-fold and 65-fold, respectively) with no discernable increase in potency at other SSTRs. Next, we assessed the role of the predicted post-translational modifications. Changing D-Trp and L-Hyp, to their unmodified equivalents, L-Trp and L-Pro (Fj1A6 and Fj1A7, respectively), had virtually no effect on the SSTR4 potency, although both showed an increase in SSTR1 potency. Another epimerization, the substitution of L-Phe6 to D-Phe (Fj1A8), on the other hand decreased the potency at SSTR4 by 42-fold. Since the MD simulations suggested that residues outside the cyclic core did not form significant and salient interactions with the receptor, we tested the “minimal core” peptide Cys3-D-Trp4-Lys5-Phe6-Gly7-Cys8 (Fj1A9). This decreased the potency by 47-fold, while deleting only the N-terminus (Fj1A10) resulted in a 13-fold decrease in potency. However, deletion of only the C-terminus (Fj1A11) resulted in an analog that was virtually equipotent at the SSTR4.

Analog design and testing identified improved ligands for SSTR4-selective drug development. (A) Identifiers and sequences of SST-14, Fj1, and the analogs of Fj1. Sequences (left) show the Cys residues forming the intramolecular disulfide in Fj1 and its analogs highlighted in yellow. For all sequences, the Trp residue is shown in blue, the Lys residue in green, and all modifications in the analogs, as compared to Fj1, are highlighted in red. Sequences use standard amino acid one-letter abbreviations extended with O = hydroxyproline and lower-case letters denoting D-amino acids. (B) Heatmap showing the potency of Fj1 and its analogs at the five human SSTRs in the G protein dissociation assay using GαoA. The text in each cell represents the EC50 value (first line, nanomolar, two significant digits) and the pEC50 ± CI95 (two significant decimals) with the number of independent repeats for each in parentheses (second line). Approximate values (~) or “higher than” (>) and “lower than” (<) are used when we did not obtain full curves within the concentrations tested (see Fig. S3 for representative curves). The known SSTR1 and SSTR4 peptide agonist, TT-232 was also tested. However, we note that we could not dissolve it at sufficiently high concentrations to fully test this peptide in the assay used).
Recently, we identified consomatin pG1 as a potent and selective agonist of the SSTR216,29. To investigate whether determinants from pG1 could be used to improve the potency of Fj1, we designed a hybrid analog combining the cyclic core residues of Fj1 with the amino acids flanking the pG1 core (Fj1A12). This hybrid analog showed a 92-fold increase in potency at the SSTR4, although with a corresponding increase at the SSTR1 and SSTR5. Interestingly, this did not lead to increased potency at the SSTR2. Introducing only the N-terminus of pG1 (Fj1A13) increased potency by 11-fold at the SSTR4, with SSTR1 activity similar to that of Fj1A12. With the suggestion from Fj1A11, that the Fj1 C-terminus was not required, we next tested the effect of adding the C-terminus from pG1, namely the single Trp residue, to Fj1. This analog (Fj1A14) showed an 85-fold increase in potency at the SSTR4, with no enhanced activity observed at the other receptor subtypes, including the SSTR1. Given the apparent redundancy of Gly7, we evaluated whether replacing Gly7 with hydrophilic amino acids to decrease the hydrophobicity of the peptide could be tolerated. Substituting Gly9 with Ser (Fj1A15) or Asn (Fj1A17) slightly increased the potency at SSTR4 (14-fold and 6.8-fold, respectively), whereas a Thr at this position (Fj1A16) retained the SSTR4 potency of Fj1, while showing a slight increase in SSTR1 activity. Based on these observations, we next assessed whether the increased potency of Fj1A14 could be combined with the polar substitutions of Gly7. While a Ser in place of Gly7 (Fj1A18) was well tolerated at the SSTR4, both the Thr and Asn substitutions (Fj1A19 and Fj1A20, respectively) showed a slight decrease in SSTR4 potency compared to Fj1A14. However, all three double substitutions increased SSTR1 activity, thereby decreasing selectivity.
These results show that while Fj1 was the most potent and selective SSTR4 peptide agonist identified from venom-encoding genes, there are clear opportunities for further optimization of the peptide’s potency, selectivity, and physiochemical properties such as hydrophilicity.
Discussion
Cone snail venoms are a rich source of bioactive peptides, several of which have become valuable tools in biomedical research and pain drug development (reviewed in40). One notable example is ω-MVIIA (ziconotide), a peptide from Conus magus that blocks the Cav2.2 calcium channel to inhibit pain signaling in the central nervous system (CNS)41. Ziconotide not only highlighted the role of Cav2.2 in pain but also became an FDA-approved drug for chronic, intractable pain42. Another analgesic peptide from Conus geographus, contulakin-G (CGX-1160), shares structural similarity with the human neuropeptide neurotensin43. Although its exact mechanism is unclear, evidence suggests contulakin-G modulates the neurotensin 2 receptor (NTSR2), leading to inhibition of the R-type calcium channel (Cav2.3) in the spinal cord44. Despite its promising pain-relieving effects, the development of contulakin-G was halted due to company closure42.
More recently, we identified consomatin Ro1, an analgesic peptide from C. rolani that activates the SSTR1 and SSTR416. Unlike ziconotide and contulakin-G, Ro1 acts peripherally, offering potential advantages such as non-invasive administration and reduced CNS side effects like addiction or respiratory depression. Building on this, we discover consomatin Fj1, a novel and potent SSTR4 agonist derived from C. fijisulcatus. Like Ro1, Fj1 belongs to a group of peptides called consomatins, which mimic the structure and function of somatostatin-like hormones. Fj1 demonstrates superior potency and selectivity for the SSTR4 receptor and shows therapeutic promise in preclinical pain models.
Conus rolani and C. fijisulcatus are members of the Asprella clade, deep-sea, fish-hunting cone snails45,46. Unlike most cone snails that induce rapid prey paralysis, Asprella snails use a delayed “ambush-and-assess” strategy, in which prey becomes incapacitated over 15 min to several hours16. This suggests the presence of venom components that suppress escape responses rather than fast-acting ion channel blockers, making these snails especially promising for the discovery of novel analgesics. In this study, we focused on consomatin sequences that activate SSTR4, however, Asprella venoms likely harbor additional analgesic compounds yet to be identified.
Consomatin Fj1 features a unique structural motif (Cys-D-Trp-Lys) that supports its high potency and SSTR4 selectivity. MD simulations revealed a binding mode similar to that of human SST-14, the natural ligand for somatostatin receptors (see Fig. 4B,D). Notably, Fj1’s D-Trp occupies the same binding pocket as SST-14’s L-Trp24,47, while the D-amino acid helps to stabilize the peptide and resist enzymatic degradation.
To further optimize Fj1, we performed structure–activity studies and generated analogs to explore the role of individual residues. A “core” linked by the disulfide loop and consisting of six amino acids (Fj1A9) retained activity at SSTR4, although it was less potent. Removing residues at the N-terminus reduced potency, while trimming the C-terminus had minimal impact. One analog, Fj1A12, showed improved potency when a Trp residue from another consomatin (pG1)29 was added to the C-terminus. Further analogs revealed that the Gly residue within the disulfide loop could be modified to enhance potency or solubility, though some modifications affected selectivity. Overall, our results indicate that Fj1 can be optimized further while maintaining or improving selectivity for the SSTR4.
The small molecule SSTR4 agonist, J-2156, which has been reported to be approximately 360-fold selective for SSTR4 over SSTR1 and 390-fold over SSTR5 in binding assays48, has been widely used in the literature14,15,33,35,48,49. However, peptide agonists that can distinguish between SSTR1 and SSTR4, two closely related subtypes, have not been previously reported. The most widely used peptide agonist at the SSTR4 is the heptapeptide, TT-232, which suffers from low potency and a 6.5-fold reported selectivity over SSTR150. Using the venom peptide Fj1 as an inspiration, this study provides several analogs with significantly improved potency and selectivity over previously described peptide agonists and suggests additional opportunities for further optimization.
Moreover, having a potent and selective peptide agonist of the SSTR4 expands the repertoire of existing drug leads for this target from small molecules to peptides. Peptides, especially those that have evolved by nature, often display better selectivity and fewer off-target effects or complications from hepatic metabolism51. They are also often easier to peripherally restrict, which is a strategy employed for instance in developing peripherally restricted opioid receptor agonists52, and was observed for Fj1 (Supplemental Tables S1–S2). One challenge with peptide drugs is their short in vivo half-life. Fj1, for example, has a half-life of 8.7 min after intraperitoneal injection (Supplemental Tables S3–S4). However, various peptide conjugation strategies can extend duration of action—many modern peptide drugs now have once-weekly dosing regimens53. Given the rising success of peptide-based therapeutics54,55, Fj1’s profile is promising for future development.
We tested Fj1 in two rodent models of pain: the incision model (for postoperative pain) and the spared nerve injury (SNI) model (for neuropathic pain). Both models showed significant reductions in mechanical hypersensitivity following a single dose of Fj1. In the incision model, injury activates both C- and Aδ-fibers and triggers a local inflammatory response56. Fj1’s effect was more sustained than morphine’s, possibly due to its action on inflammation or a slower receptor dissociation rate. SSTR4 expression is upregulated during inflammation57, and activation of this receptor has been shown to reduce inflammatory pain in preclinical studies10,58,59. The prolonged analgesic effect of Fj1 may therefore result from SSTR4-mediated inhibition of inflammation following injury, though differences in receptor binding kinetics or pharmacokinetics could also contribute. In the SNI model, both 0.5 and 5.0 mg/kg doses of Fj1 were effective at the 1-h mark, with no further benefit at the higher dose, indicating that 0.5 mg/kg may be saturating. At 2.5 h, the higher dose showed more pronounced effects, likely due to pharmacokinetic factors. These findings suggest that Fj1 effectively alleviates pain behaviors in two distinct types of pain and holds potential for treating both inflammatory and neuropathic conditions.
Our study has several limitations. Although MD simulations support Fj1’s binding mode, an experimental structure is needed for full validation. Additionally, despite promising potency and selectivity, Fj1’s pharmacokinetic properties will need optimization before clinical development and the SSTR4-mediated mechanism of action proposed here should be confirmed using SSTR4 knockout mice in the future. Additionally, although we did not observe any obvious behavioral changes in animals injected with the peptide, dedicated assays will be needed to evaluate potential motor or other side effects of Fj1, such as rotarod or open-field tests. Most importantly, clinical validation of SSTR4 as a pain target is incomplete. While a small-molecule SSTR4 agonist showed efficacy in a phase II trial for diabetic neuropathy, trials for osteoarthritis and chronic low back pain did not meet endpoints60. No trials have yet investigated SSTR4-targeting drugs for postoperative pain, the setting in which Fj1 was most effective in our models. Further studies are needed to understand which preclinical models best predict human outcomes for SSTR4-targeted therapies. Additionally, in-depth exploration of sex differences in SSTR4 signaling may be important, as variations in treatment responses based on sex are common in pain research.
Conclusion
Consomatin Fj1 represents a new class of venom-derived, peripherally acting peptide agonists targeting SSTR4. Its potent analgesic effects in rodent models, coupled with its selectivity and modifiability, make it a promising drug lead for non-opioid pain treatment. As the search for effective, non-addictive pain therapies continues, venom-derived peptides like Fj1 may offer a novel path forward.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information file.
References
Murphy, M. Z., Jackson, T. P. & Mishra, P. Hospitalized Chronic Pain Patient: A Multidisciplinary Treatment Guide (Springer, 2022).
Cohen, S. P., Vase, L. & Hooten, W. M. Chronic pain: An update on burden, best practices, and new advances. Lancet 397, 2082–2097. https://doi.org/10.1016/s0140-6736(21)00393-7 (2021).
Queremel Milani, D. A. & Davis, D. D. Pain Management Medications (StatPearls, 2023).
CDC. Centers for Disease Control and Prevention: Understanding the Opioid Overdose Epidemic (2023). https://www.cdc.gov/opioids/basics/epidemic.html.
Tostivint, H. et al. Molecular evolution of GPCRs: Somatostatin/urotensin II receptors. J. Mol. Endocrinol. 52, T61-86. https://doi.org/10.1530/jme-13-0274 (2014).
Møller, L. N., Stidsen, C. E., Hartmann, B. & Holst, J. J. Somatostatin receptors. Biochem. Biophys. Acta 1616, 1–84. https://doi.org/10.1016/s0005-2736(03)00235-9 (2003).
Chalabi, M. et al. Somatostatin analogs: Does pharmacology impact antitumor efficacy?. Trends Endocrinol. Metab. 25, 115–127. https://doi.org/10.1016/j.tem.2013.11.003 (2014).
Hofland, L. J. & Lamberts, S. W. The pathophysiological consequences of somatostatin receptor internalization and resistance. Endocr. Rev. 24, 28–47. https://doi.org/10.1210/er.2000-0001 (2003).
Pless, J. From somatostatin to Sandostatin®: History and chemistry. Metabolism 41, 5–6. https://doi.org/10.1016/0026-0495(92)90023-4 (1992).
Elekes, K. et al. Inhibitory effects of synthetic somatostatin receptor subtype 4 agonists on acute and chronic airway inflammation and hyperreactivity in the mouse. Eur. J. Pharmacol. 578, 313–322. https://doi.org/10.1016/j.ejphar.2007.09.033 (2008).
Pinter, E., Helyes, Z. & Szolcsanyi, J. Inhibitory effect of somatostatin on inflammation and nociception. Pharmacol. Ther. 112, 440–456. https://doi.org/10.1016/j.pharmthera.2006.04.010 (2006).
Szolcsanyi, J. et al. Analgesic effect of TT-232, a heptapeptide somatostatin analogue, in acute pain models of the rat and the mouse and in streptozotocin-induced diabetic mechanical allodynia. Eur. J. Pharmacol. 498, 103–109. https://doi.org/10.1016/j.ejphar.2004.07.085 (2004).
Engström, M., Savola, J. M. & Wurster, S. Differential efficacies of somatostatin receptor agonists for G-protein activation and desensitization of somatostatin receptor subtype 4-mediated responses. J. Pharmacol. Exp. Ther. 316, 1262–1268. https://doi.org/10.1124/jpet.105.094128 (2006).
Sándor, K. et al. Analgesic effects of the somatostatin sst4 receptor selective agonist J-2156 in acute and chronic pain models. Eur. J. Pharmacol. 539, 71–75. https://doi.org/10.1016/j.ejphar.2006.03.082 (2006).
Gorham, L., Just, S. & Doods, H. Somatostatin 4 receptor activation modulates TRPV1[correction of TPRV1] currents in dorsal root ganglion neurons. Neurosci. Lett. 573, 35–39. https://doi.org/10.1016/j.neulet.2014.04.042 (2014).
Ramiro, I. B. L. et al. Somatostatin venom analogs evolved by fish-hunting cone snails: From prey capture behavior to identifying drug leads. Sci. Adv. 8, eabk1410. https://doi.org/10.1126/sciadv.abk1410 (2022).
Koch, T. L. et al. Reconstructing the origins of the somatostatin and allatostatin-C signaling systems using the accelerated evolution of biodiverse cone snail venoms. Mol. Biol. Evol. 39, msac075. https://doi.org/10.1093/molbev/msac075 (2022).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. https://doi.org/10.1038/nprot.2013.084 (2013).
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V. & Thirion, B. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Kroeze, W. K. et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369. https://doi.org/10.1038/nsmb.3014 (2015).
Masuho, I., Martemyanov, K. A. & Lambert, N. A. Monitoring G protein activation in cells with BRET. Methods Mol. Biol. 1335, 107–113. https://doi.org/10.1007/978-1-4939-2914-6_8 (2015).
Brennan, T. J., Zahn, P. K. & Pogatzki-Zahn, E. M. Mechanisms of incisional pain. Anesthesiol. Clin. N. Am. 23, 1–20. https://doi.org/10.1016/j.atc.2004.11.009 (2005).
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M. & Yaksh, T. L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. https://doi.org/10.1016/0165-0270(94)90144-9 (1994).
Zhao, W. et al. Structural insights into ligand recognition and selectivity of somatostatin receptors. Cell Res. 32, 761–772. https://doi.org/10.1038/s41422-022-00679-x (2022).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273. https://doi.org/10.1021/ct300400x (2012).
Guvench, O. et al. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 7, 3162–3180. https://doi.org/10.1021/ct200328p (2011).
Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122. https://doi.org/10.1021/ct700200b (2008).
Yeung, H. Y. et al. Fish-hunting cone snail disrupts prey’s glucose homeostasis with weaponized mimetics of somatostatin and insulin. Nat. Commun. 15, 6408. https://doi.org/10.1038/s41467-024-50470-2 (2024).
Buczek, O., Bulaj, G. & Olivera, B. M. Conotoxins and the posttranslational modification of secreted gene products. Cell. Mol. Life Sci. CMLS 62, 3067–3079. https://doi.org/10.1007/s00018-005-5283-0 (2005).
Ray, P. et al. Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq–based resource for pain and sensory neuroscience research. Pain 159, 1325–1345. https://doi.org/10.1097/j.pain.0000000000001217 (2018).
Cowie, A. M. & Stucky, C. L. A mouse model of postoperative pain. Bio Protoc. 9, e3140. https://doi.org/10.21769/BioProtoc.3140 (2019).
Shenoy, P. A. et al. The somatostatin receptor-4 agonist J-2156 alleviates mechanical hypersensitivity in a rat model of breast cancer induced bone pain. Front. Pharmacol. 9, 495. https://doi.org/10.3389/fphar.2018.00495 (2018).
Kántás, B. et al. Novel drug-like somatostatin receptor 4 agonists are potential analgesics for neuropathic pain. Int. J. Mol. Sci. 20, 6245. https://doi.org/10.3390/ijms20246245 (2019).
Szőke, É. et al. Small molecule somatostatin receptor subtype 4 (sst4) agonists are novel anti-inflammatory and analgesic drug candidates. Neuropharmacology 178, 108198. https://doi.org/10.1016/j.neuropharm.2020.108198 (2020).
Sindrup, S. H., Otto, M., Finnerup, N. B. & Jensen, T. S. Antidepressants in the treatment of neuropathic pain. Basic Clin. Pharmacol. Toxicol. 96, 399–409. https://doi.org/10.1111/j.1742-7843.2005.pto_96696601.x (2005).
Quintero, G. C. Review about gabapentin misuse, interactions, contraindications and side effects. J. Exp. Pharmacol. 9, 13–21. https://doi.org/10.2147/JEP.S124391 (2017).
van Velzen, M., Dahan, A. & Niesters, M. Neuropathic pain: Challenges and opportunities. Front. Pain Res. 1, 1. https://doi.org/10.3389/fpain.2020.00001 (2020).
Isberg, V. et al. Generic GPCR residue numbers—Aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 36, 22–31. https://doi.org/10.1016/j.tips.2014.11.001 (2015).
Olivera, B. M. et al. Peptide neurotoxins from fish-hunting cone snails. Science 230, 1338–1343. https://doi.org/10.1126/science.4071055 (1985).
Miljanich, G. P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 11, 3029–3040. https://doi.org/10.2174/0929867043363884 (2004).
Safavi-Hemami, H., Brogan, S. E. & Olivera, B. M. Pain therapeutics from cone snail venoms: From Ziconotide to novel non-opioid pathways. J. Proteom. 190, 12–20. https://doi.org/10.1016/j.jprot.2018.05.009 (2019).
Craig, A. G. et al. An O-glycosylated neuroexcitatory conus peptide. Biochemistry 37, 16019–16025. https://doi.org/10.1021/bi981690a (1998).
Martin, L. et al. Conotoxin contulakin-G engages a neurotensin receptor 2/R-type calcium channel (Cav2.3) pathway to mediate spinal antinociception. Pain 163, 1751–1762. https://doi.org/10.1097/j.pain.0000000000002561 (2022).
Puillandre, N. et al. Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol. Phylogenet. Evol. 78, 290–303. https://doi.org/10.1016/j.ympev.2014.05.023 (2014).
Olivera, B. M., Watkins, M., Puillandre, N. & Tenorio, M. J. Hidden diversity in the Asprella clade: description of Conus (Asprella) neocostatus sp. Nov. (Gastropoda, Conidae). Xenophora Taxon. 33, 22–29 (2021).
Robertson, M. J., Meyerowitz, J. G., Panova, O., Borrelli, K. & Skiniotis, G. Plasticity in ligand recognition at somatostatin receptors. Nat. Struct. Mol. Biol. 29, 210–217. https://doi.org/10.1038/s41594-022-00727-5 (2022).
Engström, M. et al. Superagonism at the human somatostatin receptor subtype 4. J. Pharmacol. Exp. Ther. 312, 332–338. https://doi.org/10.1124/jpet.104.075531 (2005).
Schuelert, N. et al. The somatostatin receptor 4 agonist J-2156 reduces mechanosensitivity of peripheral nerve afferents and spinal neurons in an inflammatory pain model. Eur. J. Pharmacol. 746, 274–281. https://doi.org/10.1016/j.ejphar.2014.11.003 (2015).
Helyes, Z., Pinter, E. & Szolcsanyi, J. TT-232. Drugs Future 30, 0558 (2005).
Lamers, C. Overcoming the shortcomings of peptide-based therapeutics. Future Drug Discov. 4, FDD75. https://doi.org/10.4155/fdd-2022-0005 (2022).
Albert-Vartanian, A. et al. Will peripherally restricted kappa-opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential?. J. Clin. Pharm. Ther. 41, 371–382. https://doi.org/10.1111/jcpt.12404 (2016).
Kurtzhals, P., Østergaard, S., Nishimura, E. & Kjeldsen, T. Derivatization with fatty acids in peptide and protein drug discovery. Nat. Rev. Drug Discov. 22, 59–80 (2023).
Muttenthaler, M., King, G. F., Adams, D. J. & Alewood, P. F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 20, 309–325. https://doi.org/10.1038/s41573-020-00135-8 (2021).
Wang, L. et al. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 7, 48. https://doi.org/10.1038/s41392-022-00904-4 (2022).
Kang, S. & Brennan, T. J. Mechanisms of postoperative pain. Anesth. Pain Med. 11, 236–248. https://doi.org/10.17085/apm.2016.11.3.236 (2016).
Varecza, Z. et al. Expression of the somatostatin receptor subtype 4 in intact and inflamed pulmonary tissues. J. Histochem. Cytochem. 57, 1127–1137. https://doi.org/10.1369/jhc.2009.953919 (2009).
Helyes, Z. et al. Effects of the somatostatin receptor subtype 4 selective agonist J-2156 on sensory neuropeptide release and inflammatory reactions in rodents. Br. J. Pharmacol. 149, 405–415. https://doi.org/10.1038/sj.bjp.0706876 (2006).
Silwal, A. et al. Novel somatostatin receptor-4 agonist SM-I-26 mitigates lipopolysaccharide-induced inflammatory gene expression in microglia. Neurochem. Res. 47, 768–780. https://doi.org/10.1007/s11064-021-03482-z (2022).
ClinicalTrials.gov. NCT04707157 (2023). https://clinicaltrials.gov/study/NCT04707157?term=NCT04707157&rank=1.
Acknowledgements
We thank Prof. Hans Bräuner-Osborne for help with establishing GPCR receptor assays, Dr. Joanna Gajewiak for illustrations of the chemical structure of consomatin Fj1, and Dr. Paula Flórez Salcedo for illustration of the cone snail shell. Funding of this work was provided by the Villum Foundation Young Investigator grant (19063 to HS-H), the Lundbeck Foundation Experiment grant (R400-2022-509 to WEB), the Independent Research Fund Denmark grant (3102-00006 to TLK), the Lundbeck Foundation Ascending Investigator grant (R344-2020-1063 to KLM), the Lundbeck Postdoc grant (R322-2019-1816 to KLJ), and the National Institutes of Health grants (R01NS116694 and Health K08NS104272 to AP). Part of this work was undertaken with the assistance of resources from the National Computational Infrastructure (NCI), which is supported by the Australian Government and provided through Intersect Australia Ltd.
Author information
Authors and Affiliations
Contributions
H.S.H. conceptualized the study. H.S.H., B.J.S., K.L.M., K.J.K, and A.P. provided supervision and funding. W.E.B.Y., I.B.L.R, T.L.K., E.E., H.Y.Y., K.K.S, C.M.G, K.L.J., N.A.S., and L.F.M. conducted experiments and analyzed data, W.E.B.Y. and H.S.H. wrote the initial manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
WEB, IBLR, TLK, and HSH are inventors of a patent application for Fj1 and its analogs (patent application # WO2023180125A1) and WEB and HSH are co-founders of a company focused on the development of novel SSTR4-targeting pain therapeutics.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bjørn-Yoshimoto, W.E., Ramiro, I.B.L., Koch, T.L. et al. Cone snail venom-inspired somatostatin receptor 4 (SSTR4) agonists as new drug leads for peripheral pain. Sci Rep 15, 42638 (2025). https://doi.org/10.1038/s41598-025-26820-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-26820-5
