
Abstract
Neuromyelitis optica spectrum disorder (NMOSD) is a rare neurological autoimmune disease caused by autoantibodies targeting the astrocytic water channel aquaporin-4 (AQP4). Binding to AQP4 initiates the activation of innate immune components, especially the complement system. Both in-vivo and in-vitro models have been developed to study the molecular pathophysiology of NMOSD. Our study aimed to characterize the molecular response of four human cell lines (AQP4-ECFP expressing U-87MG glioblastoma cells, U-87MG expressing only ECFP, HEK293 cells expressing AQP4-EmGFP, and human primary astrocytes) to a treatment with AQP4 antibody E5415A and human complement. Complement-dependent cytotoxicity was induced by this treatment in AQP4-expressing cells by the terminal complement pathway. Transcriptomic profiles of the in-vitro U-87MG-AQP4-ECFP model and an in-vivo rat model shared a proinflammatory shift towards NF-κB and interleukin-6 pathways. These findings were confirmed at both the mRNA and protein levels, and treatment with serum samples from AQP4 antibody seropositive NMOSD patients resulted in a similar response. Additionally, NF-κB upregulation was shown by immunohistochemistry in medulla oblongata lesions of NMOSD patients. In conclusion, interleukin-6 and NF-κB pathways play a key role in inflammation caused by the activation of the terminal complement pathway in a human cellular model of NMOSD using U-87MG-AQP4-ECFP cells.
Similar content being viewed by others


Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a rare neuroinflammatory disease that primarily affects the spinal cord and optic nerves1. The majority of patients are seropositive for autoantibodies targeting the water channel aquaporin-4 (AQP4) on astrocyte endfeet2. AQP4 has two major isoforms produced by alternative splicing, M1 and M23. At the astrocytic foot processes, the isoform AQP4M23 is more abundant than AQP4M1, and is more prone to form supramolecular aggregates called orthogonal arrays of particles, which are targeted by anti-AQP4 immunoglobulin G (AQP4-IgG) autoantibodies3. Upon binding, immune responses are predominantly mediated by complement-dependent cytotoxicity (CDC) through the classical pathway, resulting in astrocyte damage and blood-brain barrier (BBB) disruption4. Several studies have shown that AQP4-IgG titers in patients correlate with the intensity of complement activation, but not with disease severity5,6,7. Antibody-dependent cellular cytotoxicity (ADCC) has also been implicated, as it activates various leukocytes or natural killer cells via Fcγ receptors, damaging astrocytes and adjacent non-AQP4 expressing cells8.
The most important diagnostic tool to identify AQP4-IgG serostatus of NMOSD patients is a cell-based assay using human embryonic kidney 293 (HEK293) cells with a transiently transfected AQP4 construct and fluorescent label9,10. Beyond diagnostics, these cells are also used to study NMOSD pathophysiological mechanisms at the cellular level. To date, pathophysiological mechanisms mediated by AQP4-IgG have been studied in-vitro primarily on AQP4-overexpressing HEK293A cells5,6,11,12,13,14, Chinese hamster ovary (CHO) cells15,16,17,18,19,20,21, or primary astrocytes with human14,22,23,24,25,26 or rodent origin4,8,15,27,28,29,30,31,32.
Recently, we have established a cellular model for CDC activation after AQP4-IgG exposure in HEK293 expressing AQP4 and found a strong AQP4-IgG titer-dependent activation of CDC6. Here, we further investigate our findings by molecular profiling of cellular models of CDC activation in stably transduced U-87 MG glioblastoma cells with AQP4M23-ECFP overexpression (U-87MG-AQP4-ECFP), a non-AQP4 overexpressing counterpart (U-87MG-ECFP), in addition to AQP4M23-EmGFP-transfected HEK293 cells (HEK293-AQP4-EmGFP) and human primary astrocytes (HA). We exposed these cell lines to human complement and the monoclonal mouse anti-AQP4 antibody E5415A, analyzing cytotoxicity and transcriptomic changes post-treatment33,34. Furthermore, we compared our transcriptomic findings to data from spatial transcriptomics from an NMOSD in-vivo model to identify and validate overlapping upregulated proinflammatory pathways.
Methods
Cells
Human primary astrocytes (HA), isolated from the human cerebral cortex of a female donor, gestational age 22 weeks, were purchased by ScienCell (Lot. No. 33619, ScienCell Research Laboratories, San Diego, CA). For experiments, exclusively passages 3 to 5 were used. HA were cultured in 2 µg/cm2 poly-L-lysine-coated flasks (ScienCell Research Laboratories, San Diego, CA) in astrocyte medium AM (Innoprot, Derio, Spain), containing 2% fetal calf serum, 1% P/S, and 1% astrocyte growth supplement. Medium was exchanged every other day, and cells were passaged when 90% dense.
The glioblastoma cell line U-87 MG (ATCC; LGC Standards GmbH, Wesel, Germany) was genetically modified for a stable protein overexpression. Thereby, an overexpression of enhanced cyan fluorescent protein (ECFP) with (U-87MG-AQP4-ECFP) or without (U-87MG-ECFP) AQP4M23 in U-87 MG cells was obtained via viral infection as described elsewhere35,36. The vectors were pLIB-MCS-ECFP-iresNeo and pLIB-MCS-AQP4m23-ECFP-iresNeo. The plasmid maps are shown in Fig. S1. Cells were selected using 10 µg/mL neomycin for 72 h. HEK293A and U-87 MG cell lines were cultured in DMEM high glucose (all culture medium components: Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 10% fetal calf serum, and 1% non-essential amino acids and passaged twice per week.
Three days before the complement and antibody treatment, HEK293A (ATCC; LGC Standards GmbH, Wesel, Germany) cells were seeded. After 24 h, they were transiently transfected with a pcDNA 6.2 AQP4M23-EmGFP (HEK293-AQP4-EmGFP) plasmid with polyethylenimine (Sigma, St. Louis, Missouri) in a ratio of 1:3.6 plasmid: transfection reagent. Then, cells were incubated for two days until treatment.
Patient serum samples and ethical approval
As a proof of concept, serum samples from three NMOSD patients with (titers 1:20,480, 1:1,280, and 1:5,120) and from three NMOSD patients without AQP4-IgG positivity were provided by Romana Höftberger from the Division of Neuropathology and Neurochemistry, Department of Neurology, Medical University of Vienna, Austria. The use of these serum samples from a biobank was approved by the ethical committee of the Medical University of Vienna (EK 1636/2019 and 1123/2015). All patients or their legal representatives gave written informed consent to participate in the study, and all methods were performed in accordance with the relevant guidelines and regulations.
All samples were tested for complement activation and cytotoxicity on U-87MG-AQP4-ECFP cells. The test was performed blinded. The complement treatment and following procedures were performed the same way as with E5415A, but instead of 10 µg/mL of the monoclonal antibody, 10% of the end volume of serum was applied. Additionally, the sera were heat-inactivated for 30 min (min) at 56 °C to inactivate complement before usage.
AQP4 antibody and complement treatment and assessment of cytotoxicity
Treatment with the mouse monoclonal AQP4 antibody E5415A (isolated from the hybridoma cell line AQP4 E5415A-1H6-68, Resource no. RCB4883, provided by the Riken BRC through the National BioResource Project of the MEXT/AMED, Japan)30 and human complement was done as recently described6. Briefly, U-87MG-ECFP, U-87MG-AQP4-ECFP, HEK293-AQP4-EmGFP cells, or HA were grown in a 12- or 96-well plate until confluent. Then, they were treated with 40% active or heat-inactivated (45 min at 56 °C) pooled human complement serum (Cedarlane, Burlington, Ontario, Canada) in X-VIVO 15 medium (Lonza, Basel, Switzerland) and with or without E5415A (10 µg/mL; in-house production from E5415A-1H6-68 hybridoma cells, #RCB4883, Riken, Tsukubashi, Ibaraki, Japan), followed by incubation for 90 min at 37 °C33. Afterwards, the cell supernatant was taken to assess cytotoxicity, and cells were harvested for RNA isolation, or immunofluorescence staining was performed.
The CDC was assessed with a lactate dehydrogenase (LDH) assay (CytoTox 96 Nonradioactive Cytotoxicity Assay; Promega, Madison, WI). Thereby, the amount of cytosolic LDH, which is released after cell damage, can be measured indirectly via an enzymatic assay, resulting in a reduction of tetrazolium salt into a red formazan. Cells were treated as stated above. As a positive control, additional wells with cells were treated with the provided lysis buffer (1×, diluted from a 10× concentration in X-VIVO 15 medium) for the last 45 min of the incubation. As a negative control served untreated cells in X-VIVO 15 medium. Cell supernatant was taken and incubated with the same volume of substrate for 30 min at room temperature, according to the manufacturer. The reaction was stopped with the provided stop solution, and the absorption was measured with a plate reader at 492 nm (DTX880; Beckman Coulter, Brea, CA). The results were first normalized to the respective IC-treated samples to correct for the LDH background reactivity of human complement serum and then to the respective lysis buffer-treated samples to assess the percentage of cell lysis. Per treatment, three technical replicates were done for each of the three biological replicates.
mRNA transcriptomic analysis
Cells were treated with complement and E5415A as described above. After removing the supernatant to perform cytotoxicity assays, cells were washed with PBS and harvested with RLT buffer. Further steps were performed according to the protocol of the RNeasy Mini Kit (QIAGEN, Hilden, Germany). After quantity and quality determination by a NanoDrop System (Thermo Fisher Scientific, Waltham, MA, USA), total RNA samples were sent for sequencing by the Illumina PE150 technology to Novogene (Martinsried, Germany). A description of all samples sent for sequencing is shown in Table S1.
Messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, the first strand cDNA was synthesized using random hexamer primers, followed by the second strand cDNA synthesis using either dUTP for a directional library or dTTP for a non-directional library37. The non-directional library was prepared by end repair, A-tailing, adapter ligation, size selection, amplification, and purification. For the directional library, it was ready after end repair, A-tailing, adapter ligation, size selection, USER enzyme digestion, amplification, and purification. The library was checked using Qubit and real-time PCR for quantification, and a Bioanalyzer for size distribution detection. Quantified libraries were pooled and sequenced on Illumina platforms according to effective library concentration and data amount.
The raw FASTQ format data were initially processed using fastp software by Novogene. In this step, clean data (clean reads) were obtained by removing reads containing adapters, reads containing ploy-N and low-quality reads from the raw data. At the same time, Q20, Q30, and GC content of the clean data were calculated. All the downstream analyses were based on clean data of high quality. A summary of the sequencing data quality of all samples is shown in Table S2 and Fig. S2 (sample-to-sample correlation heatmap).
Reference genome and gene model annotation files were downloaded from the genome website directly. An index of the reference genome was built using Hisat2 v2.0.5, and paired-end clean 1 reads were aligned to the reference genome using Hisat2 v2.0.5. Hisat2 was used by Novogene as the mapping tool for that Hisat2 can generate a database of splice junctions based on the gene model annotation file, and thus, a better mapping result than other non-splice mapping tools38. A comparison of samples and reference genomes and the sequencing reads in the genomic region are shown in Tables S3 and S4.
The program featureCounts v1.5.0-p3 was used to count the reads numbers mapped to each gene39. Then, fragments per kilobase million (FPKM) of each gene were calculated based on the length of the gene and the reads count mapped to this gene. FPKM, expected number of Fragments Per Kilobase of transcript sequence per Million base pairs sequenced, considers the effect of sequencing depth and gene length for the reads count at the same time, and is currently the most commonly used method for estimating gene expression levels.
The acquired data were deposited in the Gene Expression Omnibus database under dataset accession number GSE291954.
Reverse transcription-quantitative polymerase chain reaction
Selected differentially regulated genes identified by transcriptomic analysis were validated by reverse transcription-quantitative polymerase chain reaction (RT-qPCR). First, 1 µg of RNA was retro-transcribed into cDNA with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosciences, Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer. The product was diluted with 480 µL of DEPC water and stored at -20 °C until needed. Then, TaqMan Gene Expression Assay probes (Applied Biosciences) and iTaq Universal Probes Supermix (Bio-Rad, Hercules, CA, USA) were mixed with 5 µL of cDNA each and run with a CFX96 RT-PCR machine (Bio-Rad) with CFX maestro software for 40 cycles (initial denaturation at 95 °C for 30 s; second denaturation at 95 °C for 5 s, annealing/extension at 60 °C for 30 s). GAPDH served as a housekeeping gene, and delta-Ct values were calculated for each gene. All TaqMan Gene Expression Assay probes are listed in Table S5.
Immunocytochemistry
For immunocytochemistry (ICC), cells were grown on a 0.1% gelatin solution (Sigma-Aldrich, St. Louis, MO, USA) coated ibidi µ-Slide 18 Well Glass Bottom (ibidi, ibidi GmbH, Gräfelfing, Germany). When reaching confluence, cells were exposed to human complement with or without E5415A or human serum, as described above. Live cell staining was performed on cells for terminal complement complex (TCC), as well as C3/C3b/iC3b deposition on the cell surface. Therefore, 10% heat-inactivated fetal calf serum in PBS served as a washing and antibody buffer. Mouse anti-C5b-C9 neo (aE11) AlexaFluor594 (Novus Biologicals, Centennial, CO, USA) or mouse IgG1 anti-C3/C3b/iC3b (Cedarlane, Burlington, Ontario, Canada) was applied at 5 µg/mL for 60 min at 4 °C. As the secondary antibody for C3/C3b/iC3b served goat-anti-mouse IgG1 AlexaFluor594 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) which was applied for 30 min at room temperature in the dark. Thereafter, cells were fixed with 4% paraformaldehyde for 10 min. After washing, nucleus staining with 4′,6-diamidino-2-phenylindole (DAPI) and mounting was performed in one step by applying Immunoselect Antifading Mounting Medium DAPI (Dianova, BIOZOL, Hamburg, Germany) to the cells. For intracellular ICC, cells were fixed with 4% paraformaldehyde and permeabilized with 1% Triton X-100 for 10 min each at room temperature before blocking in 1% bovine serum albumin and 5% normal goat serum in PBS. Then, 2 µg/mL rabbit monoclonal anti-p65 (D14E12; Cell Signaling Technology, Danvers, MA, USA) was added to the antibody buffer (1% bovine serum albumin and 1% normal goat serum in PBS). Cells were incubated at 4 °C overnight. Goat-anti rabbit AlexaFluor647 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) in antibody buffer was incubated on the cells for 60 min at room temperature in the dark. Cells were mounted the same way as for the extracellular ICC. Microscopy was performed with the Zeiss LSM700 microscope (Zeiss Plan-Apochromat 63×/1.40 oil M27 objective). Fiji ImageJ (version 1.64) was used for background subtraction and adjustments for contrast and brightness for the ICC figures displayed. For the analysis of TCC deposition on U-87MG-AQP4-ECFP cells, raw images were opened as standard ImageJ files with split channels using the BioFormats import tool. A threshold was set in the ECFP channel to recognize all AQP4-ECFP-expressing cells. The same threshold was used for all figures of one approach. Then, a selection in the ECFP channel was created based on the threshold. The selection was then applied to the TCC-AlexaFluor594 channel, and the mean grey value was measured. Mean grey values of untreated cells were subtracted to eliminate background signals. Photos of three biological replicates per condition were analyzed. For the analysis of p65 translocation from the cytosol into the nucleus, raw images were opened as a hyperstack image in ImageJ using the BioFormats application. Then, a pre-defined macro was run (code available in the supplementary file) that automatically recognized all nuclei and calculated the p65-fluorescence signal ratio between cytosol and nucleus. Ratios > 1 were defined as nuclear translocation. At least three images/biological replicates per condition were analyzed.
C3a enzyme-linked immunosorbent assay
C3a levels in different cell lines were evaluated by enzyme-linked immunosorbent assay (ELISA) with the Human C3a ELISA Kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Supernatants of AQP4 antibody and complement treatment experiments were collected and stored at − 20 °C. Before usage, the cell supernatants were thawed and centrifuged briefly (10 min at 2000 g). The ELISA was performed according to the manufacturer’s instructions with a sample dilution of 1:2000 and a 25 min incubation time of the substrate solution before adding the stop solution. The plate was measured in single values at 450 nm and 620 nm with a plate reader (DTX880; Beckman Coulter, Brea, CA).
Interleukin-6 enzyme-linked immunosorbent assay
To assess interleukin-6 (IL6) levels in cell supernatants after complement and E5415A exposition, an ELISA was used. The Human IL6 DuoSet ELISA with DuoSet Ancillary Reagent Kit 2 (R&D Systems, Minneapolis, MN, USA) was performed according to the manufacturer. Dilutions of 1:4 in reagent diluent were used for supernatants of U-87MG-AQP4-ECFP and HEK293-AQP4-EmGFP cells, and 1:2 dilutions for supernatants of HA and U-87MG-ECFP cells. The optical density of the samples was measured in technical duplicates at 450 nm.
Terminal complement pathway inhibition with eculizumab
Inhibition of the terminal complement activation in E5415A + AC-treated U-87MG-AQP4-ECFP cells was achieved by the C5 inhibitor eculizumab (MedChemExpress, Monmouth Junction, NJ, USA). Eculizumab (1–100 µg/mL) was added to the prepared AC or IC in XVIVO for 15 min at 37 °C. Thereafter, E5415A was added, and the mixtures were transferred onto the cells and incubated for 90 min at 37 °C, the same as without inhibitor. For experiments with eculizumab, complement controls without inhibitor were pre-incubated at 37 °C for 15 min as well.
NMOSD animal model and spatial transcriptomics
Spatial transcriptomics data deposited at the Sequence Read Archive (SRA) database (NCBI/SRA accession numbers PRJNA1258753, PRJNA1262739, PRJNA1262139, PRJNA1262895, PRJNA1262368, and PRJNA1263155) were re-analyzed from the inflamed medulla oblongata (MO) of a female 7-week-old Lewis rat that received daily intraperitoneal injections of 1 mg E5415A in PBS for two consecutive days, inducing experimental NMOSD. The animal was euthanized by CO2 inhalation 24 h after the final antibody injection, and the MO was dissected for Visium spatial transcriptomics according to protocols from 10× Genomics (10× Genomics B.V, Leiden, The Netherlands). Data were processed using Space Ranger (10× Genomics) to generate gene expression profiles per gene and spot, overlaid on HE-stained tissue images. Loupe Browser 6.5.0 (10× Genomics) was used to define a perivascular lesion area and identify differentially upregulated genes compared to the remaining MO of the same tissue section through the “categories,” “globally distinguishing,” and “significant feature comparison” functions. Genes with low average counts were not excluded. Log2-fold changes and p-values were calculated in Space Ranger, with p-values adjusted for multiple comparisons using the Benjamini–Hochberg method. The experiments were approved by the Ethics Commission of the Medical University of Vienna and performed with the license of the Austrian Ministry for Science and Research (GZ: BMBWF-66.009/0107-V/3b/2018). The study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
NF-κB activation in human brain tissue
Neuropathological analysis was performed on two human autopsy cases of patients with AQP4-IgG seropositive NMOSD. The use of these tissue samples from a biobank was approved by the ethical committee of the Medical University of Vienna (EK 1636/2019 and 1123/2015). All patients or their legal representatives gave written informed consent to participate in the study.
Formalin-fixed and paraffin-embedded tissue sections were stained for hematoxylin and eosin (H&E) and Luxol fast blue-Periodic acid Schiff (LFB/PAS). Immunohistochemistry was performed on the automated platform Autostainer Link 48 using EnVision FLEX + secondary system (Dako, Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer´s protocol using the following primary antibodies: rabbit polyclonal anti-GFAP (1:3000; Dako/Agilent) or mouse monoclonal anti-GFAP (1:800; Millipore, Merck KGaA, Darmstadt, Germany), rabbit monoclonal anti-p65 (1:800; D14E12; Cell Signaling), and mouse monoclonal anti-SMI31 (phosphorylated neurofilament; 1:25,000; Sternberger Monoclonals Inc. Lutherville, MD, USA). Manual immunohistochemistry was performed in a humidified chamber for complement C9 neoantigen (C9neo; complement-mediated tissue injury; rabbit polyclonal; 1:2000, from Professor Paul Morgan, Cardiff, UK). Double labeling of GFAP and NF-κB (p65) was performed with the chromogenic reactions with Fast Blue (blue) and 3-amino-9-ethylcarbazole (AEC; BioSB (red)). Image acquisition was performed using a NanoZoomer 2.0-HT digital slide scanner C9600 (Hamamatsu Photonics, Hamamatsu, Japan).
Bioinformatic and statistical analysis
Statistical analyses (2-way ANOVA with Šídák’s and Kruskal-Wallis multiple comparisons test, principal component analysis) and drawing of figures (bar and scatter graphs, volcano plots, principal component plots, and heat maps) were prepared using GraphPad Prism 10.4 (GraphPad Software Inc., La Jolla, California, United States). All tests were done using a significance threshold of p < 0.05 with correction for multiple comparisons if applicable. Data are expressed as mean ± standard deviation. For all experiments, at least three biological replicates were analyzed.
Differential expression analysis of two conditions/groups (three biological replicates per condition) was performed using the DESeq2 R package using the WebMev platform40,41,42. WebMev and DESeq2 provide statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting p-values were adjusted using Benjamini and Hochberg’s approach to control the false discovery rate. Genes with an adjusted p-value ≤ 0.001 and log2-fold change ≥ 1 or ≤ − 1 found by DESeq2 were assigned as differentially expressed. The overlap of differentially expressed genes between different treatments was analyzed using Venn diagrams and the WebMev platform. All application codes are available at https://github.com/web-mev/.
To functionally annotate the most significant genes, gene ontology (GO) analysis was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) using the official gene symbols of Homo Sapiens of genes with a statistical significance of p < 0.05 (adjusted using the Benjamini–Hochberg correction for multiple tests)43,44. The DAVID annotation chart for the Gene Ontology Term “biological processes” (GOTERM-BP) was used. Enrichment of genes in annotation terms was evaluated using the EASE Score, a one-tailed Fisher’s exact test p-value, with p-values ≤ 0.05 indicating strong enrichment. Moreover, significantly altered genes were analyzed and visualized by STRING network analysis45.
Biological annotations were confirmed by rank-based enrichment analysis. Therefore, raw read count tables were processed in R (Bioconductor package) using the DESeq2 package42. Genes with zero counts across all samples and all genes with a mean expression < 10 were removed. A design matrix was specified to compare Group 1 (E5415A + AC replicates) versus Group 2 (all other treatments: E5415A + IC, AC, IC, and untreated) for each cell line. Differential expression analysis was performed with DESeq2’s Wald test, producing log2-fold changes and associated adjusted p-values (Benjamini–Hochberg correction). For pathway analysis, genes were ranked by the DESeq2 Wald test statistic (or log2-fold change if unavailable). Gene set enrichment analysis (GSEA) was performed using the fgsea package with MSigDB Hallmark and Reactome collections as the reference gene sets46. Pathways of interest (NF-κB signaling, IL1 and IL6 signaling, chemokine signaling, complement cascade, apoptosis and stress response) were selected by keyword filtering. Enrichment results were reported as normalized enrichment scores (NES) with associated FDR-adjusted p-values. Visualization of results included (i) volcano plots of differential expression, and (ii) enrichment plots of significant pathways using plotGseaTable for combined summaries. Only pathways with FDR < 0.05 were considered significantly enriched. All codes used for this analysis are provided in the supplementary file.
Results
Transcriptomic changes in-vitro after anti-AQP4 antibody and human complement treatment indicate an inflammatory stress response in human U-87MG-AQP4-ECFP cells
We analyzed the transcriptomic changes after treatment of four established NMOSD cellular models (U-87MG-AQP4-ECFP, U-87MG-ECFP, HEK293-AQP4-EmGFP, and HA) with active or heat-inactivated human complement and with or without the monoclonal AQP4 antibody E5415A, as previously described6. Cytotoxicity assessed by LDH release quantification was significantly increased in AQP4-expressing cells, whereas AQP4-negative U-87MG-ECFP cells showed less cell death (Fig. 1A). The highest level of cytotoxicity (mean 37.2%) was observed in U-87MG-AQP4-ECFP after E5415A + AC treatment. This effect was significantly different to all other treatments, and to E5415A + AC-treated U-87MG-ECFP cells (all p < 0.001). In contrast, U-87MG-ECFP cells, which lack AQP4 expression, exhibited a lower level of cytotoxicity (9.6% E5415A + AC, 11.2% AC), with a statistically significant difference between E5415A + AC to IC ± E5415A, and untreated cells (p < 0.05) (Fig. 1A).

Cytotoxicity assay after human complement and E5415A treatment. The cell lines U-87MG-AQP4-ECFP, U-87MG-ECFP, HEK293-AQP4-EmGFP, and HA were treated with or without the monoclonal AQP4 antibody E5415A (10 µg/mL) in combination with active or heat-inactivated human complement. (A) Cytotoxicity was assessed with the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega). Values were normalized to cells treated with lysis buffer, which served as a positive control (= 100% cell lysis), and human complement. (B) Levels of C3a were determined by ELISA (Invitrogen, Thermo Fisher Scientific). Values were normalized to the C3a levels present in the human complement serum. Bar charts (means with standard deviation and individual values) were created with GraphPad Prism 10.4. Groups (for (A) n = 9, for (B) n = 3) were compared with a 2-way ANOVA with Šídák’s multiple comparisons test, all treatments per cell line against E5415A + AC. ns , not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
To validate that terminal complement pathway activation caused the cytotoxicity, ICC for the terminal complement complex (TCC, C5b-C9) was performed. TCC deposition was exclusively observed in U-87MG-AQP4-ECFP cells treated with E5415A + AC, while no staining was detected in cells treated with E5415A + IC, complement-only treatment, or untreated controls (Fig. 2A). The background level of CDC observed in controls (E5145A + IC, AC, and IC) was already seen in a previous study in HEK-AQP4-EmGFP cells and was associated with opsonization with C3b and associated effector functions6. Bound C3/C3b on AC-only-treated cells was observed for U-87MG-AQP4-ECFP cells as well, although to a smaller extent (Fig. 2B). In contrast, C3/C3b opsonization on U-87MG-ECFP was seen in comparable quantities in E5415A + AC or AC-only-treated cells (Fig. 2C). As no TCC deposition on the cellular surface of U-87MG-ECFP cells could be observed for any of the treatments, this demonstrates the alternative pathway contribution in the U-87MG cell line. Moreover, C3a levels in both U-87MG-AQP4-ECFP and U-87MG-ECFP cells were not significantly different between E5415A + AC and AC treatments, indicating an additional AQP4 antibody-independent activation of the alternative complement pathway (Fig. 1B).

Complement component deposition on the surface of different cell lines after application of the monoclonal AQP4 antibody E5415A (10 µg/mL) and active or heat-inactivated human complement. (A) Terminal complement complex (TCC) formation on U-87MG-AQP4-ECFP cells. After the treatments, cells were washed with PBS with 10% heat-inactivated FCS, and mouse anti-human C5b-C9 neo (aE11) AlexaFluor594 (Novus Biologicals) was applied with 5 µg/mL to visualize TCC deposition on the cell surface. (B–D) Complement component C3/C3b/iC3b deposition. After the treatments, cells were washed with PBS with 10% heat-inactivated FCS, and mouse anti-human/mouse C3/C3b/iC3b purified (6C9) (Cedarlane) was applied with 5 µg/mL, followed by 2 µg/mL goat-anti mouse IgG1 Cross-Adsorbed Secondary Antibody, AlexaFluor594 (Invitrogen, Thermo Fisher Scientific) to visualize opsonization by C3/C3b/iC3b on the cell surface of (B) U-87MG-AQP4-ECFP cells, (C) U-87MG-ECFP cells and (D) primary human astrocytes. Scale bar = 20 μm. AF , Alexa Fluor; AQP4, aquaporin-4; DAPI, 4′,6-diamidino-2-phenylindole; ECFP, enhanced cyan fluorescent protein.
Transcriptomic analysis was performed by pairwise comparisons of E5415A + AC-treated U-87MG-AQP4-ECFP cells, U-87MG-AQP4-ECFP, and U-87MG-ECFP cells with the other treatments using Dseq2 analysis. The volcano plot shown in Fig. 3A and the pie chart in Fig. 3B revealed that amongst all detectable transcripts, 13,224 (99.1%) genes remained unchanged. In comparison, 96 genes were upregulated (0.7%), and 20 genes were downregulated (0.2%) when comparing U-87MG-AQP4-ECFP cells treated with E5415A + AC and E5415A + IC. From Fig. 3C,D it is evident that 59 genes were significantly changed in U-87MG-AQP4-ECFP cells after E5415A + AC exposure compared to all other treatments. Functional annotation using DAVID revealed a strong enrichment of genes associated with inflammation, cell death, and stress responses, and NF-κB signaling pathways (Fig. 3E), which is comparable to previous findings32,47.

RNA-seq analysis revealed that treatment with the monoclonal AQP4 antibody E5145A and human complement induces the expression of inflammatory and reactive genes in U-87MG cells expressing AQP4. (A) Volcano plot of differentially expressed genes between U-87MG-AQP4-ECFP cells treated with E5145+AC compared to E5145+IC; green indicates upregulated genes and red indicates downregulated genes. Groups (n=3) were compared using DSeq2 analysis, and changes with an adjusted p-value <0.001 and a two-fold change were considered statistically significant. (B) Pie chart of the results shown in (A) summarizing the upregulated, downregulated, and unchanged genes. (C) Pairwise comparisons using DSeq2 analysis of E5145+AC-treated U-87MG-AQP4-ECFP cells with U-87MG-AQP4-ECFP and U-87MG-ECFP cells after other treatments indicated 59 specifically changed genes. (D) A heat map (created with GraphPad Prism 10.4.) showing the mean standardized expression (z-scores, according to decreasing principal component 1 scores) of these 59 differential genes. Each row shows the relative expression level for a single sample, and each column shows the expression level of single genes. (E) A bubble plot (created with GraphPad Prism 10.4) showing the functional annotation by DAVID of the 59 specifically changed genes, showing the most significant associations indicated three functional clusters. P-values are visualized by a heatmap and different sizes. AC, active complement; AQP4, aquaporin-4; ECFP, enhanced cyan fluorescent protein; EmGFP, emerald green fluorescent protein; IC, inactive complement.
Next, a similar analysis of HEK293-AQP4-EmGFP cells was performed, comparing E5415A + AC-treated cells with the other treatments, respectively. HEK293-AQP4-EmGFP cells also showed significant cytotoxicity after E5415A + AC (mean 32.7%) compared to all other treatments (all p < 0.001, Fig. 1A). Background levels of complement-derived cell lysis were, as in previous findings, likely caused by a partially activated alternative pathway. C3a levels detected in differently treated HEK293-AQP4-EmGFP cells showed an AC-specific effect (mean of 32.39 µg/mL), but this effect was not higher when adding E5415A (mean of 38.82 µg/mL) (Fig. 1B). Only 6 genes were significantly changed in E5415A + AC-treated HEK293-AQP4-EmGFP cells compared with the other treatments (Fig. 4A), thereof 4 genes overlapped with the genes changed in U-87MG-AQP4-ECFP cells after E5415A + AC treatment (Fig. 4B). The expression of all 59 differentially regulated genes from U-87MG-AQP4-ECFP cells after E5415A + AC treatment in HEK293-AQP4-EmGFP cells is shown in Fig. 4C.

RNA-seq analysis revealed that treatment with the monoclonal AQP4 antibody E5145A and human complement does not induce the expression of a specific response in HEK293 cells expressing AQP4 and human astrocytes. (A) Pairwise comparisons using DSeq2 analysis of E5145+AC-treated HEK293A-AQP4-EmGFP cells with other treatments indicated only 6 specifically changed genes. (B) A heat map (created with GraphPad Prism 10.4.) showing the mean standardized expression (z-scores, according to decreasing principal component 1 scores) of the 59 differential genes from U-87MG-AQP4-ECFP cells in HEK293A-AQP4-EmGFP cells. (C) From the 59 differential genes from U-87MG-AQP4-ECFP cells, only 4 genes were also differentially expressed in E5145+AC-treated HEK293A-AQP4-EmGFP cells. (D) Pairwise comparisons using DSeq2 analysis of E5145+AC-treated HA with other treatments indicated no specifically changed genes. AC, active complement; AQP4, aquaporin-4; ECFP, enhanced cyan fluorescent protein; EmGFP, emerald green fluorescent protein; HA, human astrocytes; IC, inactive complement.
Finally, a comparable analysis was performed of primary HA. Similarly, HA cells displayed moderate cytotoxicity after treatment with E5415A + AC (mean 11.9%) and AC (12.6%), but with a large variation between replicates, and a significant difference to untreated cells (p < 0.001) (Fig. 1A). Furthermore, HA treated with AC alone or in combination with E5415A showed similar mean C3a levels (22.44 µg/mL and 24.87 µg/mL) (Fig. 1B). The lack of an E5415A + AC-specific effect could be explained by the lower level of AQP4 gene expression (mean gene counts 970.9, standard deviation 37.4) as compared to U-87MG-AQP4-ECFP (50,645.9 ± 11,295.7) or HEK293-AQP4-EmGFP (324,388.8 ± 108,263.6). Moreover, HA showed a higher level of background cytotoxicity, which is related to C3 cleavage and opsonization (Figs. 1B and 2D), and the differential expression of complement associated genes (Fig. S3). While HA showed a higher proportion of differentially expressed genes (n = 808) compared to untreated cells, no specific effect of E5415A + AC treatments was detectable (Fig. 4D,E). To summarize, despite high expression of glial fibrillary acidic protein (GFAP) and other astrocyte-specific genes, the HA used in this study were not useful as an in-vitro NMOSD model for our research question. The AQP4 levels were found to be insufficient. Nevertheless, similar AQP4-unspecific effects could be observed, which were probably due to the activation of the alternative complement pathway and C3 and C5 receptors or other antibodies in the complement serum that bind to HA.
Rank-based enrichment analysis was then used to confirm coordinated shifts in NF-κB, IL1, or IL6 signaling pathways, chemokines, complement, apoptosis, and stress responses in all cell lines treated with E5415A + AC versus the other treatments (Fig. 5 and Figs. S4 and S5). For this analysis, all ranked individual genes were included, and again the strongest response was seen for NF-κB signaling in U-87MG-AQP4-ECFP cells (NES = 3.57, p = 4.5 × 10− 49). Additionally, we observed strong enrichment of the IL1 and IL6 signaling pathways, as well as the complement, apoptosis, and stress response pathways. U-87MG-ECFP and HEK293-AQP4-EmGFP cells were mainly characterized by an enrichment of apoptosis and stress response genes, whereas HA also showed an, although less pronounced, enrichment of NF-κB and IL6 signaling pathways.

Rank-based enrichment analysis to confirm coordinated shifts in NF-κB, IL1, or IL6 signaling pathways, chemokines, complement, apoptosis, and stress responses in all cell lines treated with E5415A + AC versus the other treatments (E5415A + IC, AC, IC, and untreated). Treatments were compared using DESeq2 analysis, and ranked genes were analyzed by gene set enrichment analysis using GSEA for pathways of interest (NF-κB signaling, IL1 and IL6 signaling, chemokine signaling, complement cascade, apoptosis, and stress response). Pathways with a positive normalized enrichment score (NES > 0) are enriched among genes upregulated after E5415A + AC treatment, whereas pathways with a negative NES (NES < 0) are enriched among genes downregulated after E5415A + AC treatment (i.e., preferentially upregulated in the other treatments). AC = , active complement,; AQP4 = aquaporin-4,; ECFP, = enhanced cyan fluorescent protein,; EmGFP; = emerald green fluorescent protein;, IC, = inactive complement.
Transcriptomic changes in U-87MG-AQP4-ECFP cells with anti-AQP4 antibody and human complement treatment compared to an NMOSD in-vivo model
To validate these findings with a NMOSD in-vivo model, we compared the transcriptomic changes of the treated U-87MG-AQP4-ECFP cells with the results of a spatial transcriptomic analysis of MO tissue from Lewis rats treated with AQP4 antibody E5415A (Fig. S6A). 322 genes were significantly upregulated in the inflammatory experimental NMOSD lesion (Fig. 6A). As can be seen in a Venn diagram, 15 of these genes were shared with the 59 differentially expressed genes from U-87MG-AQP4-ECFP E5415A + AC-treated cells (Fig. 6B; ATF3, CEBPD, CXCL1, CXCL2, GEM, IL11, IL6, IRAK2, IRF1, JUNB, NFKB2, NFKBIA, NFKBIZ, NR4A2, and PTX3). These genes were associated with inflammation (CXCL1, CXCL2, IL11, IL6, IRAK2, NFKB2, NFKBIZ, PTX3), cell death and stress responses (ATF3, CEBPD, CXCL1, CXCL2, IL6, IRAK2, IRF1, JUNB, NFKB2, NFKBIA, NFKBIZ, NR4A2, PTX3), and NF-κB signaling pathways (IRAK2, NFKB2, NFKBIA, NFKBIZ) identified by functional annotation with DAVID (Fig. 3E) and ranked enrichment analysis (Fig. 5). The summative expression of these genes in tissue from Lewis rats treated with E5415A for two days, one day and an untreated control is shown in Fig. S6A–C, and the expression of the individual genes is shown in Fig. S6D. STRING pathway analysis (Fig. S7) revealed potential connections among these genes, except GEM (a guanidine triphosphate-binding protein that might be participating in receptor-mediated signal transduction at the plasma membrane), which showed no associations with other genes and no association with the GO terms mentioned above and was therefore excluded from further analysis48.

RNA-seq validation by qRT-PCR in cell lines treated with AQP4 antibody E5415A and complement. (A) Volcano plot of differentially regulated genes identified by spatial transcriptomics in a MO blood perivascular lesion from rats treated with E5415A (rat E5415A MO). The area was compared to a control area using DSeq analysis, and changes with an adjusted p-value < 0.05 and a two-fold change were considered statistically significant. (B) Pie chart showing the overlap of significantly altered genes between U-87MG-AQP4-ECFP and the rat E5415A MO. (C) A heat map showing the standardized expression analyzed by RNA-Seq (log2-fold change to E5415A + AC) of the 14 differentially expressed genes overlapping between U-87MG-AQP4-ECFP and the rat E5415A MO lesion. Additionally, AQP4 was selected as an internal control. Each row shows the mean expression level for a single gene, and each column shows the mean expression level of 3 replicates. (D) A heat map showing the standardized expression (log2-fold change to E5415A + AC) analyzed by RT-qPCR as validation of the 15 genes from RNA-seq. Gene expression was first normalized to the housekeeping gene GAPDH (ΔCt), followed by normalization to E5415A + AC treatment (ΔΔCt). The data represent six biological replicates from two independent experiments. AC , active complement; AQP4, aquaporin-4; ECFP, enhanced cyan fluorescent protein; EmGFP, emerald green fluorescent protein; HA, human astrocytes; IC, inactive complement; MO, medulla oblongata.
The transcriptomic analysis comparing the expression changes of these genes in treated cells compared with the rat E5415A MO lesion is visualized in the heat map in Fig. 6C. As an internal control, we added AQP4 to our gene list as the target protein of AQP4-IgG. The difference in expression is expressed with a log2-fold change of E5415A + AC-treated cells compared to the other treatments. E5415A + AC-treated U-87MG-AQP4-ECFP cells showed an upregulation in selected genes compared with all other treatments, which was comparable to the changes seen in the rat E5415A MO lesion. In HA, these gene expression changes were further accentuated. However, they were only upregulated after E5415A + AC compared to untreated cells. This pattern was not observed in HEK293-AQP4-EmGFP cells treated with E5415A + AC compared to the other treatments.
To validate these findings, we performed RT-qPCR on cDNA synthesized from RNA used for transcriptomic analysis of all four cell lines. The results were visualized in a heat map showing the standardized expression (log2-fold change of E5415A + AC-treated cells compared to the other treatments, Fig. 6D). Overall, gene expression changes were comparable to the transcriptomic analysis. For better visualization of total gene expression levels, results are additionally shown as ΔCt values (Fig. S8). In U-87MG-AQP4-ECFP cells, highly significant gene expression differences after E5415A + AC treatment compared to other treatments were observed. Expression levels of AQP4 were significantly downregulated, NFKB2 was only upregulated compared to IC or untreated cells, and all other tested genes were significantly upregulated compared to all other treatments. In contrast, HA and U-87MG-ECFP cells showed less significant differences in expression between E5415A + AC and the different treatments, only compared to untreated cells, there was a significant upregulation of most genes. In HEK293-AQP4-EmGFP cells, NR4A2 and CXCL1 were upregulated significantly after E5415A + AC treatment compared to all other treatments, whereas some of the other genes were only upregulated compared to untreated cells. No significant changes in any treatment of HEK293-AQP4-EmGFP cells were observed in AQP4, CEBPB, IL6, IL11, IRAK2, and NFKB2.
Validation of transcriptomic changes of cells with anti-AQP4 antibody and human complement treatment compared to an NMOSD in-vivo model on the protein level
In general, most of the differentially expressed genes were involved in NF-κB signaling, stress and cell death, and inflammation. For the analysis on protein level, two key proteins were selected: (i) of the differentially expressed genes in the gene set are directly or indirectly related to NF-κB signaling. To efficiently test NF-κB activation, we decided to investigate RelA (p65) translocation upon E5415A and complement treatment in U-87MG-AQP4-ECFP cells since they showed the most pronounced difference on the mRNA level regarding their NF-κB activation. For the ICC of RelA, U-87MG-AQP4-ECFP were treated with E5415A in combination with active or heat-inactivated human complement. Untreated cells served as a negative control, and tumor necrosis factor-alpha (TNF-α)-treated cells as a positive control. Thereafter, an intracellular ICC was performed. Representative confocal microscopy images are shown in Fig. 7A, and results from statistical analysis are shown in Fig. S9A,B. As expected, no translocation into the nucleus was observed in untreated cells (0%), whereas the majority of nuclei in TNF-α-treated cells showed translocated p65 (89.7%). In E5415A + AC-treated cells, 65.9% showed NF-κB activation with RelA nuclear translocation, which was significantly elevated compared to the remaining treatments (all p < 0.001) except for the TNF α-treated positive control. Treatments with E5415A + IC or complement only showed RelA translocation in some cells, but in a smaller proportion of 2.7–4.2% (Fig. S7B).

Validation of RNA-seq compared to in-vivo transcriptomics on protein level. (A) U-87MG-AQP4-ECFP were treated with E5415A (10 µg/mL) in combination with active or heat-inactivated human complement. Thereafter, immunocytochemistry was performed for NF-κB component RelA (p65). As a positive control served cells that had been treated with TNF-α (10 ng/mL). Cells were fixed, permeabilized, and blocked. Then, 2 µg/mL rabbit anti-p65 (D14E12) (Cell Signaling) served as primary antibody, followed by goat-anti rabbit AlexaFluor647 (Invitrogen, Thermo Fisher Scientific). The white arrows in the composite panel indicate translocation of p65 into the nucleus. Scale bar = 20 μm. (B) U-87MG-AQP4-ECFP, HA, and HEK293-AQP4-EmGFP cells were treated as in (A), and the supernatant was assessed for IL6 levels by human IL6 ELISA (R&D Systems). The bar graph (means, standard deviations, and individual values) was created with GraphPad Prism 10.4. Groups (n = 3) were compared with 2-way ANOVA with Dunnett’s multiple comparisons test. *** p < 0.001. AF, Alexa Fluor; DAPI, 4′,6-diamidino-2-phenylindole; ECFP, enhanced cyan fluorescent protein; EmGFP, emerald green fluorescent protein; HA, human astrocytes; NF-κB, nuclear factor kappa B; IC, inactive complement.
(ii) IL6 is one of the key players in NMOSD and was significantly upregulated on the mRNA level in U-87MG-AQP4-ECFP cells treated with E5415A + AC. For the assessment of IL6 production after treatment, a human IL6 ELISA was used. U-87MG-AQP4-ECFP, HA, and HEK293-AQP4-EmGFP cells were treated as before; the cell supernatant was collected after the end of incubation and analyzed for IL6 levels. Results are shown in Fig. 7B. Similar to the gene expression results, treatment with E5415A + AC resulted in the significantly increased secretion of IL6 only in U-87MG-AQP4-ECFP cells (mean of 284.6 pg/mL) compared to the other treatments (IC ± E5415A approximately 120 pg/mL, AC 68.8 pg/mL, untreated 21.1 pg/mL; all p < 0.001). Importantly, IL6 was released into the cell supernatant in higher amounts by U-87MG-AQP4-ECFP cells as well as in complement-only treated cells compared to the other cell lines. Whereas HA showed no IL6 production in untreated cells, they had low IL6 levels in treated cells (all between 18.85 and 33.11 pg/mL), which did not show significant changes between E5415A + AC-treated cells and the other conditions. No IL6 secretion was found in HEK293-AQP4-EmGFP cells.
Effects of terminal complement pathway inhibition on a human cell NMOSD model
To investigate if observed effects in U-87MG-AQP4-ECFP cells were exclusively mediated by the activation of the terminal complement pathway, eculizumab was applied additionally with the other treatments. Eculizumab is an inhibitor of complement component C5, thereby inhibiting the formation of the terminal complement complex, and is approved as a relapse prevention agent for AQP4-IgG seropositive NMOSD patients49. Regarding cytotoxicity, the extent of cell lysis reduction in cells was highly significant (both p < 0.001) when comparing E5415A + AC only-treated U-87MG-AQP4-ECFP cells (mean 37.3%) to cells that were additionally treated with eculizumab (1.5% mean lysis), visible in Fig. 8A. There were no significant differences between samples treated with or without inhibitor and IC + E5415A. IL6 levels of AC/IC + E5415A only-treated cells were, in general, slightly higher with the additional 15 min preincubation at 37 °C (Fig. 8B and 295.5 and 176.1 pg/mL mean IL6 for AC/IC + E5415A, respectively). Furthermore, the addition of eculizumab reduced the mean IL6 levels released from IC + E5415A treated cells 74.4 pg/mL (p = 0.001). The effect on AC + E5415A treated cells was even higher, with a reduction of 288.2 pg/mL mean difference for eculizumab (p < 0.001). Figure 8C summarizes log2-fold transcriptomic changes of E5415A + AC/IC with or without eculizumab-treated U-87MG-AQP4-ECFP cells compared to untreated controls. Transcriptomic changes on selected genes caused by E5415A + AC on U-87MG-AQP4-ECFP cells were significantly decreased when adding the complement inhibitor. The expression levels of AQP4 remained unchanged, while the upregulation of all other genes was blocked. In contrast, there were no significant changes between IC + E5415A treated cells with or without eculizumab. When analyzing the extent of TCC deposition on the surface of U-87MG-AQP4-ECFP cells (Fig. 8D), eculizumab already showed significant effects at 3 µg/mL (p < 0.05). From a concentration of 30 µg/mL and higher, there was only a minimal amount of TCC detected (p < 0.001). This effect was not observed when analyzing C3/C3b on the cellular surface (Fig. 8E). There were no significant differences at any concentration of eculizumab compared to E5415A + AC only-treated cells.

Effects of eculizumab on U-87MG-AQP4-ECFP cells after complement and E5415A treatment. Cells were treated with active or heat-inactivated human complement (AC/IC) in combination with E5415A (10 µg/mL). In addition to E5415A + AC, complement inhibitors against C5 (eculizumab, 30 µg/mL in (A–C), 1–100 µg/mL in D + E) were added to the complement 15 min before the application on cells. (A) LDH assay results with cell lysis normalized to lysis buffer-treated cells and additional AC/IC-only background absorbance subtraction. (B) IL6 ELISA results of IL6 levels in cell supernatants of treated samples. IL6 production of untreated cells was subtracted. (C) qPCR results of genes differently expressed in U-87MG-AQP4-ECFP cells after complement and E5415A treatment. Log2-fold changes of E5415A + AC/IC with or without eculizumab-treated cells were compared to untreated controls. GAPDH served as a housekeeping gene, and all ΔCt values of all genes were calculated before the normalization to untreated cells. After treatments, cells were stained for (D) terminal complement complex deposition on their surface or (E) complement component C3/C3b. The mean fluorescence of complement component-bound fluorescent antibodies was measured. Fluorescence of untreated samples was subtracted. Additionally, the fluorescence levels of all samples were normalized to E5415A + AC-only treated cells. All experiments (A-E) were performed in biological triplicates and technical duplicates. (A + B) 2-way ANOVA with Šídák’s multiple comparisons test between treatments with and without eculizumab. (D + E) An ordinary one-way ANOVA with Dunnett’s multiple comparisons test was performed, comparing all complement inhibitor treatments to E5415A + AC-only treated samples * p < 0.05 **p < 0.01, ***p < 0.001.
Confirmatory translational analysis of NMOSD patient samples
Since U-87MG-AQP4-ECFP proved to be the most applicable cell line for AQP4-IgG seropositive NMOSD modeling on the cellular level, we next investigated the effects of human AQP4-IgG from NMOSD patients in this model. Therefore, we applied six NMOSD patient sera to the cells; three of them were AQP4-IgG seropositive (NMOSD#1, #4, and #5), and three were seronegative (NMOSD#2, #3, and #6). The binding of AQP4-IgG and AQP4-IgM was confirmed retrospectively by cell-based assays (Figs. S10 and S11). Cells were treated with 10% human serum sample and AC or IC. Cell supernatants were analyzed for LDH to assess the degree of cytotoxicity and for IL6 production. Furthermore, cells were either lysed for RNA isolation and analysis or stained for TCC deposition or p65 translocation.
Results of gene expression levels are shown as a heat map with log2-fold change to complement only controls (Fig. 9A). Two-fold changes in gene expression were only found in the AQP4-IgG seropositive samples NMOSD#1 (AQP4-IgG titer 1:20,480), #4 (1:1,280), and #5 (1:5,120) in combination with AC, but not IC. In general, the most pronounced changes were found for NMOSD#1 + AC treatment. Next, a 2-way ANOVA with Šídák’s multiple comparisons test was performed comparing each serum treatment combined with AC vs. IC. The results are shown in Fig. S13. For AQP4, only NMOSD#1 showed significant differences (adj. p-value < 0.01). All gene expressions were significantly upregulated between NMOSD#1 with AC vs. IC treatment, with a p < 0.001, except for JUNB and NFKB2 (both not significant). NMOSD#2 serum-treated cells showed no significance in any of the tested genes between AC or IC addition. Similarly, NMOSD#3 + AC -treated cells showed no significant changes, except for a downregulation of IL11 (p = 0.03) and JUNB (p = 0.01). NMOSD#4 + AC treated cells showed gene upregulation in CXCL1, CXCL2, IL6, IRAK2, NFKBIA, NFKBIZ, NR4A2, and PTX3 (all p < 0.001), as well as in IRF1 (p = 0.007). Likewise, CXCL1, CXCL2, IL6, NFKBIZ, and NR4A2 were highly upregulated in NMOSD#5 + AC treated cells (p < 0.001). IL11 (p = 0.001), NFKBIA, and PTX3 (both p = 0.003) were upregulated as well. NMOSD#6 serum with AC treatment led to the upregulation of three genes, CXCL2 (p = 0.002), IL6 (p = 0.008), and NR4A2 (p < 0.001).

Effects of treatment with AQP4-IgG positive and negative NMOSD serum samples on U-87MG-AQP4-ECFP. U-87MG-AQP4-ECFP cells were treated with human NMOSD patient sera #1–6 in combination and active or heat-inactivated human complement. After the treatment, cells were harvested for RNA isolation (A), and the cell supernatant was used for cytotoxicity (B) and IL6 (C) assessment. (A) Log2-fold changes of 2−ΔΔCt values of human sera plus complement (AC/IC). ΔCt values were calculated with the Ct values of the respective genes minus the Ct values of the housekeeping gene GAPDH. To assess serum-specific effects (ΔΔCt), ΔCt values of complement-only samples were subtracted from those with serum plus complement. (B) Cytotoxicity was assessed with the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega). Afterwards, each mean value of active or inactive complement only was subtracted from the respective values with serum. These values were normalized to those of lysis buffer-treated cells (set to 100%), to obtain serum-specific impact on cell lysis. (C) IL6 levels of the cell supernatants after treatment were measured by human IL6 ELISA (R&D Systems). Afterwards, each median value of active or inactive complement-only was subtracted from the respective values with serum. Negative values were set to zero. (A) The heat map was created with GraphPad Prism 10.4. (B,C) Bar charts (means, standard deviation, and individual values) were created with GraphPad Prism 10.4. Groups (n = 3) were compared by 2-way ANOVA with Šídák’s multiple comparisons test * p < 0.05, **p < 0.01, ***p < 0.001. AC, active complement; AQP4, aquaporin-4; IC,inactive complement.
The results from the cytotoxicity assay are shown in Fig. 9B. U-87MG-AQP4-ECFP treated with the AQP4-IgG positive serum samples NMOSD#1, #4, and #5 plus AC showed the highest cytotoxicity (p < 0.001), but also the AQP4-IgG seronegative sample NMOSD#6 + AC was cytotoxic (p < 0.01). Again, the greatest effect was seen for NMOSD#1. Additionally, ICC for TCC of treated cells was performed to further assess CDC (Fig. 10A). Thereby, only cells treated with AQP4-IgG seropositive NMOSD patient sera showed TCC deposition on the cellular surface, and only when combined with AC. NMOSD#6 serum-treated cells here did not show any TCC deposition. Furthermore, the intensity of TCC was higher in NMOSD#1-treated cells, in line with the LDH assay results.

Immunocytochemistry of U-87MG-AQP4-ECFP cells after treatment with NMOSD patient sera and human complement. U-87MG-AQP4-ECFP cells were treated with 10% NMOSD patient sera (with or without AQP4-IgG) in combination with active or heat-inactivated human complement. Then, the cells were washed with PBS with 10% heat-inactivated fetal calf serum, and immunocytochemistry was performed. (A) Terminal complement complex (TCC) staining. The cellular surface was stained for TCC deposition by applying mouse anti-C5b-C9 neo (aE11) AlexaFluor594 (Novus Biologicals) with 5 µg/mL. (B) NF-κB component p65 staining. Translocation of p65 from the cytosol into the nucleus was visualized by intracellular immunocytochemistry. Cells were fixed, permeabilized, and blocked. Then, 2 µg/mL rabbit anti-human p65 (D14E12) (Cell Signaling) served as the primary antibody, followed by goat anti-rabbit AlexaFluor647 (Invitrogen, Thermo Fisher Scientific). White arrows indicate p65 translocation into the nucleus. Scale bar = 20 μm. AF, Alexa Fluor; DAPI, 4′,6-diamidino-2-phenylindole; TCC, terminal complement complex.
IL6 levels were significantly increased after treatment of serum samples NMOSD#1, #4, and #5 in combination with AC, but not after treatment with AQP4-IgG seronegative samples (Fig. 9C). Again, NMOSD#1 and AC-treated cells produced the highest levels of IL6 (252.7 pg/mL).
To assess NF-κB activation in NMOSD patient sera and human complement-treated U-87MG-AQP4-ECFP cells, ICC for RelA (p65) was performed (Fig. 10B). Although NF-κB-related genes were highly upregulated in AQP4-IgG seropositive NMOSD patient sera on the mRNA level, the nuclear translocation of p65 was not so pronounced. NMOSD#1 + AC treated cells showed the highest proportion of p65 in the nuclei at approximately 46.2%, which was highly significant compared to NMOSD#1 + IC treated controls (p < 0.001; statistical analysis see Fig. S9C + D), whereas NMOSD#4 and NMOSD#5 showed smaller proportions of p65 translocation in approximately 15.0% (p < 0.001) and 9.7% (p < 0.01) of nuclei. There was a weak signal of p65 observed in a few nuclei of cells exposed to AQP4-IgG seronegative NMOSD patient sera plus AC (NMOSD#2, #3, and #6) and with all sera plus IC. However, the signals did not exceed the nucleus: cytosol ratio of > 1 and were therefore accounted as negative in these samples (Fig. S9C + D).
Finally, we examined whether the NF-κB pathway, previously implicated experimentally, is likewise activated in human NMOSD neuropathology. Therefore, we investigated the MO from two NMOSD patients who presented different lesion types: one patient showed acute astrocyte loss (Fig. S14A,B), extensive complement deposition C9neo (Fig. S14C) and acute axonal injury in the lesion (Fig. S14D); the other patient revealed pronounced tissue vacuolization along with selective loss of AQP4 immunoreactivity and some axonal spheroids, but without complement deposition. Both lesions were located in the raphe and affected axons crossing the midline, derived from the inferior olivary nucleus. Some neurons in the inferior olivary nucleus showed strong nuclear translocation of p65 (Fig. S14E, arrows), while astrocytes in the olivary nucleus (Fig. S14E, arrowheads) and lesion rim (Fig. S14F, arrowheads) were p65 negative.
Discussion
It has been shown both in-vitro and in-vivo that the combination of antibodies against AQP4 and human complement induces CDC in astrocytes4,6,8,11,12,13,14,15,16,17,19,20,21,22,23,24,25,26,27,28,29,30,31,−32,34,50,51,52,53,54,55. We treated four human cellular models with E5415A, a monoclonal antibody against AQP4, in combination with human complement. Then, we performed mRNA-sequencing and compared transcriptomic changes to those of an NMOSD in-vivo rat model.
To ensure that mRNA changes after E5415A and AC treatment were associated with CDC, we tested all four cell lines for cytotoxicity. To confirm that cell lysis resulted from the terminal complement pathway activation, we checked for TCC and C3/C3b deposition. First, we used a cellular model previously established in our lab by Lerch et al. using transfected HEK293 cells6. Moreover, we used a stable AQP4-ECFP expressing U-87MG cell line, with an ECFP-only overexpressing equivalent to compare AQP4-specific effects. CDC was observed in HEK293-AQP4-EmGFP cells and in U-87MG-AQP4-ECFP cells, confirming previous findings3,15,55. Importantly, U-87MG-AQP4-ECFP showed the highest toxicity after incubation, followed by HEK293-AQP4-EmGFP cells, which might be explained by AQP4 expression (stable in U-87MG cells and transient in HEK293 cells). However, partial activation of the alternative complement pathway was observed as well in AC-only-treated cells. Levels of C3a were elevated in AC-treated cells in the cell supernatant, and C3/C3b deposition was found on U-87MG-AQP4-ECFP cells in our study, and in the previous study of Lerch et al.6. Typically, the alternative pathway is additionally eventually activated, although the classical pathway is the primary cause of CDC, leading to complement amplification accompanied by inflammation56. The alternative complement pathway was confirmed not to be the major pathway in mediating cell lysis in U-87MG-AQP4-ECFP cells. The inhibition of terminal complement activation in E5415A + AC-treated cells with 30 µg/mL eculizumab, a C5 convertase inhibitor, prevented both cell lysis and TCC deposition. This confirms that cell lysis is dependent on the terminal complement pathway (C5 cleavage). In contrast, the extent of C3/C3b deposition on the surface of U-87MG-AQP4-ECFP cells remained unchanged, regardless of eculizumab addition or the occurrence of cell lysis.
As we wanted to compare the cellular effects of these immortalized cell lines with those of physiologically relevant cells, we additionally used HA. However, the HA chosen for our study turned out to be inappropriate for our research question. First, cell culture with primary cells is limited to a few passages, as they rapidly alter their morphology, growth behavior, and gene expression, including AQP4 levels. Second, cytotoxicity was induced in these cells with no significant alterations between the different treatments, and they showed high variation between replicates. Although the gene expression levels of our target genes were altered compared to untreated cells, differences between treatments could only be observed in some genes. Furthermore, no TCC deposition was detected on HA after complement treatment, but C3/C3b on cells treated with AC ± E5415A. Additionally, increased C3a levels in HA indicate the activation of the alternative complement pathway. Since gene counts of general astrocyte markers, such as GFAP and S100B, were high in HA samples, and AQP4 was moderately expressed across treatments as well, we wondered which developmental state the cells had at the point of isolation. Upon request at ScienCell, we got the information that the Lot we had purchased (#33619) was derived from the cerebral cortex of a female donor with a gestational age of 22 weeks. At this stage of development, AQP4 expression is only rarely found in the neocortex, and astrocyte differentiation and blood-brain barrier encirclement occur postnatally57,58,59. On the one hand, the overexpression of AQP4 in immortalized cell lines led to a 100-fold higher expression, which might explain the majority of why changes were more pronounced in the immortalized cell lines. On the other hand, a small proportion might have been caused by the immaturity of our HA as well. This limitation should be addressed in future studies by differentiating primary astrocytes to promote maturation. However, the question remains if a sufficient AQP4 expression at the astrocyte endfeet could be achieved without co-culturing the astrocytes with brain microvascular endothelial cells.
The treatment with E5415A + AC led to CDC in AQP4-expressing cells and profound transcriptomic changes on the mRNA level. As expected, AQP4 expression levels were downregulated after treatment with E5415A and AC in U-87MG-AQP4-ECFP and HEK293-AQP4-EmGFP cells, indicating a specific cell loss and confirming previous findings31,60. Moreover, most changes of E5415A + AC treatment compared to all other treatments led to most genes being changed in U-87MG-AQP4-ECFP cells (compared to all other treatments in AQP4-positive and -negative cells; n = 59). In contrast, only 6 genes changed compared to differently treated cells in HEK293-AQP4-EmGFP cells, and none in HA. Interestingly, most genes changed in HA compared to the untreated control (n = 808), but the antibody addition did not alter any gene expression compared to AC-only treated cells. This was surprising, since when compared to IC with or without E5415A-treated cells, the number of changed genes deviated greatly. This could be due to the low AQP expression level and the high variability in HA replicates.
Importantly, although HEK293-AQP4-EmGFP cells underwent CDC, IL6, and NF-κB pathways were not activated in this cell line, as confirmed by rank-based enrichment analysis. Their transcriptomic profile was characterized by apoptosis and stress response enrichment. This highlights the need for a cellular model with glial origin to study cytokine pathways relevant to lesion propagation. HEK293-AQP4-EmGFP cells showed an upregulation of NR4A1-3 and downregulation of TXNIP when treated with E5415A and AC, which were overlapping genes that had been altered after the same treatment in U-87MG-AQP4-ECFP as well. NR4A1-3 encode for nuclear receptor 4 A family proteins and are immediate early genes that are upregulated during inflammation and cellular stress61,62. In astrocytes, NR4A2 is induced via pro-inflammatory cytokines and binds NF-κB component p65 on the target inflammatory gene promoter and resulting in transcriptional repression63. Upregulation of NR4A2 suggests a cellular response to the NF-κB activation overshoot that the cells experience after treatment. TXNIP, an antioxidant and key protein in regular stress response that usually promotes apoptosis, was downregulated in both AQP4 overexpressing cell lines, indicating an anti-apoptotic gene regulation64. CGA upregulation and ARRDC3 downregulation were detected in HEK293-AQP4-EmGFP cells exclusively.
Differently expressed genes were then compared to the spatial transcriptomic results of an in-vivo rat model (MO inflammatory NMOSD lesion) after peripheral injection of E5415A antibody. Most of the overlapping differently expressed genes were directly or indirectly associated with stress responses, inflammation, and NF-κB signaling (interactions are represented schematically in Fig. 11). Moreover, activation of NF-κB signaling was also observed in human NMOSD pathological tissue. However, differentially expressed genes of the in-vivo model did not only involve astrocytes. The brain area used for comparison comprised a perivascular lesion. Although astrocytes (including their endfeet with a high AQP4 expression) are highly enriched around vasculature in the brain, other cell types are present at this site, including endothelium, neurons, and oligodendrocytes. Therefore, it can be concluded that there is a shift towards a proinflammatory pattern rather than towards precisely upregulated genes. However, the shared differentially expressed individual genes from the in-vitro and in-vivo overlap are all directly or indirectly involved in NF-κB and IL6 signaling pathways, which was clearly evident in the rank-based enrichment analysis. Therefore, a closer look into single upregulated genes might nevertheless provide valuable insights into potential novel treatment possibilities.

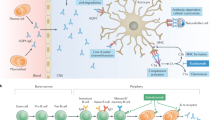
Schematic representation of differently expressed genes after AQP4-IgG and active human complement treatment. Differently expressed genes observed in this study are highlighted in red. Grey elements were not investigated in the study. Binding of AQP4-IgG to AQP4 on U-87MG-AQP4-ECFP, primary human astrocytes, or on rat astrocytes enables complement C1q initiation of the classical complement cascade, resulting in the terminal complement complex (TCC). Moreover, the alternative complement pathway and cellular stress are induced. Differently expressed genes are primarily involved in interleukin (IL)-6 and nuclear factor kappa B (NF-κB) activation. AQP4, aquaporin-4; ATF-3, activating transcription factor-3; CEBPB, CCAAT/enhancer-binding protein beta; ERK, extracellular-signal regulated kinase; IRAK2, interleukin-1 receptor-associated kinase 2; IRF1, interferon regulatory factor-1; Jak, Janus kinase; JNK, c-jun N-terminal kinases; JUNB, transcription factor jun-B; MAPK, mitogen-activated protein kinase; NFKB2, NF-κB subunit 2; NFKBIA, NF-κB inhibitor alpha; NFKBIZ, NF-κB inhibitor zeta; NR4A2, nuclear receptor 4A2; P, phosphorylation; PI3K, phosphoinositide 3-kinase; STAT3, signal transducers and activators of transcription-3; Th, T helper; TLR, toll like receptor; TNFR, tumor necrosis factor receptor; PTX-3, pentraxin-3; Ub, ubiquitin. Created in BioRender. Brandl, S. (2025) https://BioRender.com/1x2unnn.
Although NF-κB is also activated due to the glioblastoma origin of U-87 MG cells and activating transcription factor-3 (ATF3) and CCAAT/enhancer-binding protein beta (CEBPB) are transcription factors that are associated with malignant glioblastoma, they are likewise activated under general cellular stress and inflammation65,66,67. These genes were elevated in treated HA compared to untreated cells as well. ATF3 and CEBPB work downstream of NF-κB to amplify or modulate immune responses68,69,70. Our findings are in line with a previous study of Walker-Caulfield and colleagues, which investigated primary astrocyte-enriched mouse cultures after stimulation with AQP4 antibody-positive serum samples from NMOSD patients for their transcriptomic changes47. This study found an upregulation of mouse equivalents of genes detected in our study (CXCL1, CXCL2, IL6, Cebpb, Nfkbia, Nfkbiz). The authors highlighted a “NMOSD granulocytic footprint”, which they indicated is activated not only downstream via CDC but also as an early event in the onset of NMOSD pathology, creating a pro-granulocytic inflammatory environment. Furthermore, this study demonstrated the efficiency of bortezomib, a small-molecule proteasome inhibitor, which is currently approved for myeloma treatment, to successfully inhibit NF-κB signaling in astrocytes47. Similar results of a predominantly NF-κB and IL6-driven response to AQP4 antibodies and complement were seen by two other studies in primary rat astrocytes: Du et al. found an IL6 upregulation after NMOSD patient serum application, which was assigned to Janus kinase/signal transducer and activator of transcription 3-dependent inflammatory response, as they were able to decrease IL6 levels with a Janus kinase1/2 specific inhibitor AZD148061. Additionally, Wang et al. were able to prevent IL6 increase after NMOSD patient AQP4 antibody exposure by blocking NF-κB with the inhibitor S3633, indicating the contribution of NF-κB to elevated IL6 in NMOSD29. IL6 has often been highlighted as a leading inflammatory cytokine in NMOSD, given its abundance in the blood and cerebrospinal fluid of affected patients72. Its major impact on the disease, as well as its efficient effects on relapse prevention when blocked, mark IL6 as one of the major cytokines that should be inducible in any NMOSD model71. Importantly, we observed an upregulation of IL6 at both mRNA and protein levels in U-87MG-AQP4-ECFP cells after treatment with E5415A or AQP4 antibody-positive NMOSD patient serum samples, but not in HEK293-AQP4-EmGFP cells.
Regarding the other changed genes in our set, NF-κB pathways are highlighted by direct or indirect involvement even more: C-X-C motif chemokine ligand 1 and 2 (CXCL1/2) are pro-inflammatory chemokines, which recruit neutrophils to sites of inflammation and are directly regulated by NF-κB. IL6 and IL11 are cytokines involved in acute and chronic inflammatory processes, with IL6 being a well-established downstream target of NF-κB73. Interleukin-1 receptor-associated kinase-like-2 (IRAK2) plays a role in Toll-like receptor (TLR) and IL1 receptor signaling, which activates NF-κB73,74. Interferon regulatory factor-1 (IRF1) cooperates with NF-κB in regulating genes involved in immune defense74. NF-κB subunit-2/p100 (NFKB2) and NF-κB inhibitor alpha (IκBα; NFKBIA) encode components of the NF-κB pathway itself. NFKB2 contributes to the non-canonical NF-κB pathway75. NFKBIA is an inhibitor that regulates NF-κB activity through feedback in the canonical pathway75. Upon activation by pro-inflammatory stimuli such as TNF-α, IL1β, or pathogen-associated molecular patterns, IκBα is phosphorylated and degraded. This frees the NF-κB dimer, comprising p50/ NF-κB1 and p65/RelA, allowing it to translocate into the nucleus75. NFKBIA is downregulated in peripheral blood mononuclear cells (PBMCs) from NMOSD patients during NMOSD relapses, perhaps reflecting the underlying inflammatory pathway in NMOSD during a disease flare76. NF-κB inhibitor zeta (IκBζ; NFKBIZ) functions as a co-activator for specific NF-κB target genes, particularly during inflammatory responses75. RelA, when unshed from IκBα, plays a pivotal role by binding to κB sites in the promoters of target genes and driving the transcription of pro-inflammatory mediators, such as CXCL1, IL6, and pentraxin-3 (PTX3). When NF-κB signaling was tested via RelA translocation in this study, it was more elevated in our U-87MG-AQP4-ECFP cells treated with E5415A + AC combination treatment. Nevertheless, it needs to be mentioned that this effect was only as pronounced when adding the monoclonal antibody or anti-AQP4 IgG seropositive human serum with a high titer (1:20,480). PTX3, an acute-phase protein, is involved in innate immunity and complement component interaction (C1q, factor H, ficolins, mannan-binding lectin)77. PTX3 has also been shown to be associated with inflammatory responses in NMOSD patients78. NR4A2, as mentioned before, plays an anti-inflammatory role by suppressing RelA binding to the target inflammatory gene promoter63. Therefore, NR4A2 would be a possible candidate for NF-κB blockade by upregulating its expression. Indeed, we have seen E5415A + AC-specific upregulation of NR4A2 in AQP4-overexpressing cells. Its NF-κB downregulating effect, however, was not observed in this study.
Transcription factor jun-B (JUNB), part of the AP-pathogenicity and alongside NF-κB in cytokine expression, is essential for IL23-dependent Th17 pathogenicity and is itself induced by IL680,81. Nishiyama et al. observed the release of pro-inflammatory Th17 cytokines in AQP4-IgG seropositive NMOSD patient PBMCs following incubation with AQP4-immunocomplexes and complement. Notably, upregulation of IL17A and IL23 was detected only in treatment-naïve PBMCs that had not undergone B cell depletion, whereas Rituximab-treated PBMCs exhibited enhanced IL6 production. These findings underscore the necessity of IL6-activated B and T cells for downstream Th17 cytokine production81. This was highlighted before by Agasing et al. in transcriptomic analysis of NMOSD patient PBMCs as well, as they showed the co-upregulation of IL6 and interferon type I, thereby interferon type I being essential for B cell activation, and IL6 for further Th17 differentiation82.
Our study has some limitations. First, cell lysis was also observed to a lower degree in samples with complement only or E5415A with IC as well. TCC staining showed that this cell lysis was not due to TCC formation. A possible explanation for this could be unspecific binding or alternative pathway activation by C3 opsonization after the addition of human complement. Chen and colleagues saw C3 upregulation in mouse astrocytes after NMO-IgG exposure that led to microglial interactions via C3a receptors83. We detected high gene counts of C3 in U-87MG cell lines independent of treatment. In contrast, gene counts were lower in E5415A + AC-treated HEK293-AQP4-EmGFP cells compared to the other treatments, and higher in E5415A + AC-treated HA compared to the remaining samples. C1R and C1S components were elevated in these cells as well, but gene counts were moderate to low in all four cell lines and did not correlate notably with different treatments (Fig. S3). Our finding that the activation of the terminal complement (shown by C5 cleavage inhibition) is much stronger in E5415A + AC treated cells compared to AC treatment alone indicates a major contribution of the classical complement pathway. However, additional experiments with specific depletion/reconstitution of C1q or perturbation of C4/C1r/s would be necessary to clarify the role of classical complement pathway activation and will be subject of future studies. Second, besides complement components being directly involved in the classical complement pathway, there are additional complement regulatory proteins such as CD46 (membrane cofactor protein), CD55 (decay accelerating factor), and CD59 (protectin), which either inhibit the C3 convertase, or the TCC formation84. A study by Saadoun and Papadopoulus revealed that complement regulatory proteins are expressed in human astrocytes, but not in NMOSD lesions, and after co-culture of astrocytes with endothelial cells26. These complement inhibitors, CFH (complement factor H) and CLU (clusterin), were expressed at different levels in the cell lines used in our study (Fig. S3). The presence of these complement inhibitors might explain why relatively high amounts of human complement were necessary to induce CDC. Third, findings were rather unspecific in HA, which is explained by their low AQP4 expression. These primary HA were obtained from human cerebral cortex (purchased via ScienCell) and cultured for up to 5 passages. To obtain more physiologically relevant results, it would be preferable to test mature induced pluripotent stem cell-derived HA or mature HA with higher AQP4 expression in future studies. Fourth, the stringent cutoffs used for differential expression analysis in this study are very stringent (adjusted p-value ≤ 0.001 and log2-fold change ≥ 1 or ≤ − 1). While this approach minimizes false positives in the context of multiple treatment comparisons, it also reduces sensitivity and may underestimate coordinated pathway-level changes. Consequently, subtle but biologically relevant shifts in proinflammatory signaling (e.g. NF-κB, IL6, IL1, chemokines, and complement) may not be fully captured at the single-gene level. However, we approached this limitation using rank-based enrichment analysis to improve the changes in enriched pathways seen after E5415A + AC treatment for all four cell lines. Finally, the results from human serum samples were AQP4 antibody titer dependent. Most genes were significantly changed after treatment with human AQP4 antibodies and AC. Cytotoxicity also correlated with AQP4 antibody levels. Surprisingly, serum from the AQP4-antibody negative NMOSD patient #6 induced some CDC, probably due to the presence of other factors in the serum of this patient. Finally, in the human neuropathological sections investigated in our study, strong nuclear translocation of NF-κB was only seen in some neurons, while astrocytes were negative. This could be explained by the selective loss of astrocytes in the lesions and the activation of NF-κB signaling pathways by the alternative complement pathway in neurons, similar as seen in other cells in this study.
Conclusions
Our study demonstrates that AQP4-expressing U-87MG and HEK293 cells exhibit significant CDC upon exposure to the monoclonal AQP4 antibody E5415A in combination with active human complement. However, transcriptomic analysis revealed an upregulation of genes directly or indirectly linked to IL6 and NF-κB after this treatment only in U-87MG-AQP4-ECFP cells. A similar transcriptomic profile was seen in the perivascular lesion tissue of an experimental NMOSD rat model. This suggests that U-87MG-AQP4-ECFP cells effectively mimic CDC-related astrocytic responses in AQP4-IgG seropositive NMOSD and enable to testing of new treatment strategies, especially regarding the NF-κB pathway, on a cellular level.
Data availability
The datasets used and/or analyzed during the current study and all R scripts are available at the Gene Expression Omnibus database (GEO) under dataset accession number GSE29195 or are shown in the manuscript and supplementary file.
Abbreviations
- AC:
-
Active complement
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- AQP4:
-
Aquaporin-4
- ATF3:
-
Activating transcription factor-3
- BBB:
-
Blood–brain barrier
- CDC:
-
Complement-dependent cytotoxicity
- CEBPB:
-
CCAAT/enhancer-binding protein beta
- CFH:
-
Complement factor H
- CHO:
-
Chinese hamster ovary
- CXCL:
-
C-X-C motif chemokine ligand
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DAVID:
-
Database for annotation, visualization, and integrated discovery
- DEG:
-
Differentially expressed gene
- ECFP:
-
Enhanced cyan fluorescent protein
- ELISA:
-
Enzyme-linked immunosorbent assay
- EmGFP:
-
Emerald green fluorescent protein
- FPKM:
-
Fragments per kilobase million
- GFAP:
-
Glial fibrillary acidic protein
- GO:
-
Gene ontology
- HEK293:
-
Human embryonic kidney 293
- IL:
-
Interleukin
- IC:
-
Inactive complement
- ICC:
-
Immunocytochemistry
- IgG:
-
Immunoglobulin-G
- IRAK2:
-
Interleukin-1 receptor-associated kinase-like-2
- IRF1:
-
Interferon regulatory factor-1
- JUNB:
-
Transcription factor jun-B
- LDH:
-
Lactate dehydrogenase
- MO:
-
Medulla oblongata
- min:
-
Minutes
- NES:
-
Normalized enriched score
- NF-κB:
-
Nuclear factor K-light-chain-enhancer of activated B cells
- NFKB2:
-
NF-κB subunit-2
- NFKBIA:
-
NF-κB inhibitor alpha
- NFKBIZ:
-
NF-κB inhibitor zeta
- NR4A:
-
Nuclear receptor subfamily 4 group A protein
- PBMCs:
-
Peripheral blood mononuclear cells
- PC:
-
Principal component
- PTX3:
-
Pentraxin-3
- RT-qPCR:
-
Reverse transcription-quantitative polymerase chain reaction
- TCC:
-
Terminal complement complex
- TNF-α:
-
Tumor necrosis factor-alpha
References
Jarius, S. et al. Neuromyelitis optica. Nat. Rev. Dis. Primers. 6, 85. https://doi.org/10.1038/s41572-020-0214-9 (2020).
Wingerchuk, D. M. et al. International consensus diagnostic criteria for neuromyelitis Optica spectrum disorders. Neurology 85, 177–189. https://doi.org/10.1212/wnl.0000000000001729 (2015).
Crane, J. M. et al. Binding affinity and specificity of neuromyelitis Optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J. Biol. Chem. 286, 16516–16524. https://doi.org/10.1074/jbc.M111.227298 (2011).
Hinson, S. R. et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc. Natl. Acad. Sci. 109, 1245–1250. https://doi.org/10.1073/pnas.1109980108 (2012).
Jitprapaikulsan, J. et al. Clinical utility of AQP4-IgG titers and measures of complement-mediated cell killing in NMOSD. Neurology(R) Neuroimmunol. Neuroinflamm. 7 https://doi.org/10.1212/nxi.0000000000000727 (2020).
Lerch, M. et al. More efficient complement activation by Anti-Aquaporin-4 compared with Anti-Myelin oligodendrocyte glycoprotein antibodies. Neurology(R) Neuroimmunol. Neuroinflamm. 10 https://doi.org/10.1212/nxi.0000000000200059 (2023).
Yandamuri, S. S. et al. MOGAD patient autoantibodies induce complement, phagocytosis, and cellular cytotoxicity. JCI Insight. 8 https://doi.org/10.1172/jci.insight.165373 (2023).
Duan, T., Smith, A. J. & Verkman, A. S. Complement-independent bystander injury in AQP4-IgG seropositive neuromyelitis Optica produced by antibody-dependent cellular cytotoxicity. Acta Neuropathol. Commun. 7, 112. https://doi.org/10.1186/s40478-019-0766-7 (2019).
Waters, P. J. et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 78, 665–671. https://doi.org/10.1212/WNL.0b013e318248dec1 (2012). discussion 669.
Waters, P. et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis Optica. J. Neurol. Neurosurg. Psychiatry. 87, 1005–1015. https://doi.org/10.1136/jnnp-2015-312601 (2016).
Ishikura, T. et al. Anti-AQP4 autoantibodies promote ATP release from astrocytes and induce mechanical pain in rats. J. Neuroinflamm. 18, 181. https://doi.org/10.1186/s12974-021-02232-w (2021).
Lennon, V. A., Kryzer, T. J., Pittock, S. J., Verkman, A. S. & Hinson, S. R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 202, 473–477. https://doi.org/10.1084/jem.20050304 (2005).
Kaneko, K. et al. Different complement activation patterns following C5 cleavage in MOGAD and AQP4-IgG + NMOSD. Neurol. Neuroimmunol. Neuroinflamm. 11, e200293. https://doi.org/10.1212/NXI.0000000000200293 (2024).
Yamashita, K. et al. Cerebrospinal fluid mitochondrial DNA in neuromyelitis Optica spectrum disorder. J. Neuroinflamm. 15, 125. https://doi.org/10.1186/s12974-018-1162-0 (2018).
Soltys, J. et al. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis Optica. J. Clin. Invest. 129, 2000–2013. https://doi.org/10.1172/jci122942 (2019).
Ratelade, J. et al. Neuromyelitis Optica IgG and natural killer cells produce NMO lesions in mice without Myelin loss. Acta Neuropathol. 123, 861–872. https://doi.org/10.1007/s00401-012-0986-4 (2012).
Zhu, W. et al. Human C5-specific single-chain variable fragment ameliorates brain injury in a model of NMOSD. Neurol. Neuroimmunol. Neuroinflamm. 6, e561. https://doi.org/10.1212/nxi.0000000000000561 (2019).
Tradtrantip, L., Yeaman, M. R. & Verkman, A. S. Cytoprotective IgG antibodies in Sera from a subset of patients with AQP4-IgG seropositive neuromyelitis Optica spectrum disorder. Sci. Rep. 11, 21962. https://doi.org/10.1038/s41598-021-01294-3 (2021).
Phuan, P. W., Ratelade, J., Rossi, A., Tradtrantip, L. & Verkman, A. S. Complement-dependent cytotoxicity in neuromyelitis Optica requires aquaporin-4 protein assembly in orthogonal arrays. J. Biol. Chem. 287, 13829–13839. https://doi.org/10.1074/jbc.M112.344325 (2012).
Tradtrantip, L., Ratelade, J., Zhang, H. & Verkman, A. S. Enzymatic deglycosylation converts pathogenic neuromyelitis Optica anti-aquaporin-4 Immunoglobulin G into therapeutic antibody. Ann. Neurol. 73, 77–85. https://doi.org/10.1002/ana.23741 (2013).
Mader, S., Brimberg, L., Soltys, J. N., Bennett, J. L. & Diamond, B. Mutations of Recombinant Aquaporin-4 antibody in the Fc domain can impair complement-dependent cellular cytotoxicity and transplacental transport. Front. Immunol. 9, 1599. https://doi.org/10.3389/fimmu.2018.01599 (2018).
Vincent, T. et al. Functional consequences of neuromyelitis Optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte Recruitment1. J. Immunol. 181, 5730–5737. https://doi.org/10.4049/jimmunol.181.8.5730 (2008).
Haruki, H. et al. NMO Sera down-regulate AQP4 in human astrocyte and induce cytotoxicity independent of complement. J. Neurol. Sci. 331, 136–144. https://doi.org/10.1016/j.jns.2013.05.035 (2013).
Nishiyama, S. et al. Complement-dependent and -independent Aquaporin 4-antibody-mediated cytotoxicity in human astrocytes: pathogenetic implications in neuromyelitis Optica. Biochem. Biophys. Rep. 7, 45–51. https://doi.org/10.1016/j.bbrep.2016.05.012 (2016).
Piatek, P. et al. C5a-Preactivated neutrophils are critical for autoimmune-induced astrocyte dysregulation in neuromyelitis Optica spectrum disorder. Front. Immunol. 9, 1694. https://doi.org/10.3389/fimmu.2018.01694 (2018).
Saadoun, S. & Papadopoulos, M. C. Role of membrane complement regulators in neuromyelitis Optica. Multiple Scler. J. 21, 1644–1654. https://doi.org/10.1177/1352458515571446 (2015).
Sabater, L. et al. Cytotoxic effect of neuromyelitis Optica antibody (NMO-IgG) to astrocytes: an in vitro study. J. Neuroimmunol. 215, 31–35. https://doi.org/10.1016/j.jneuroim.2009.07.014 (2009).
Alexopoulos, H. et al. Anti-aquaporin-4 autoantibodies in systemic lupus erythematosus persist for years and induce astrocytic cytotoxicity but not CNS disease. J. Neuroimmunol. 289, 8–11. https://doi.org/10.1016/j.jneuroim.2015.10.007 (2015).
Wang, Y. et al. NMO-IgG induce Interleukin-6 release via activation of the NF-κB signaling pathway in astrocytes. Neuroscience 496, 96–104. https://doi.org/10.1016/j.neuroscience.2022.05.038 (2022).
Huang, P. et al. The binding property of a monoclonal antibody against the extracellular domains of aquaporin-4 directs aquaporin-4 toward endocytosis. Biochem. Biophys. Rep. 7, 77–83. https://doi.org/10.1016/j.bbrep.2016.05.017 (2016).
Hinson, S. R. et al. Aquaporin-4-binding autoantibodies in patients with neuromyelitis Optica impair glutamate transport by down-regulating EAAT2. J. Exp. Med. 205, 2473–2481. https://doi.org/10.1084/jem.20081241 (2008).
Howe, C. L. et al. Neuromyelitis Optica IgG stimulates an immunological response in rat astrocyte cultures. Glia 62, 692–708. https://doi.org/10.1002/glia.22635 (2014).
Kurosawa, K. et al. Severely exacerbated neuromyelitis Optica rat model with extensive astrocytopathy by high affinity anti-aquaporin-4 monoclonal antibody. Acta Neuropathol. Commun. 3, 82. https://doi.org/10.1186/s40478-015-0259-2 (2015).
Hillebrand, S. et al. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis Optica spectrum disorder in the rat. Acta Neuropathol. 137, 467–485. https://doi.org/10.1007/s00401-018-1950-8 (2019).
Hagenbuchner, J. et al. The tubulin inhibitor MG-2477 induces autophagy-regulated cell death, ROS accumulation and activation of FOXO3 in neuroblastoma. Oncotarget. 8 (2017).
Nothdurfter, D. et al. 3D bioprinted, vascularized neuroblastoma tumor environment in fluidic chip devices for precision medicine drug testing. Biofabrication. 14, 035002. https://doi.org/10.1088/1758-5090/ac5fb7 (2022).
Parkhomchuk, D. et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37, e123. https://doi.org/10.1093/nar/gkp596 (2009).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 5, 621–628. https://doi.org/10.1038/nmeth.1226 (2008).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. https://doi.org/10.1093/bioinformatics/btt656 (2014).
Wang, Y. E. et al. A cloud platform for analyzing and visualizing cancer genomic data. Cancer Res. 77, e11–e14. https://doi.org/10.1158/0008-5472.Can-17-0802 (2017).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11 (R106). https://doi.org/10.1186/gb-2010-11-10-r106 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–w221. https://doi.org/10.1093/nar/gkac194 (2022).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. https://doi.org/10.1038/nprot.2008.211 (2009).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–d646. https://doi.org/10.1093/nar/gkac1000 (2023).
Korotkevich, G. et al. Fast gene set enrichment analysis. BioRxiv 060012 https://doi.org/10.1101/060012 (2021).
Walker-Caulfield, M. E. et al. NFκB signaling drives pro-granulocytic astroglial responses to neuromyelitis Optica patient IgG. J. Neuroinflamm. 12, 185. https://doi.org/10.1186/s12974-015-0403-8 (2015).
Maguire, J. et al. Gem: an induced, immediate early protein belonging to the Ras family. Science 265, 241–244. https://doi.org/10.1126/science.7912851 (1994).
Wingerchuk, D. M. et al. Long-term safety and efficacy of eculizumab in aquaporin-4 IgG-Positive NMOSD. Ann. Neurol. 89, 1088–1098. https://doi.org/10.1002/ana.26049 (2021).
Kinoshita, M. et al. Astrocytic necrosis is induced by anti-aquaporin-4 antibody-positive serum. Neuroreport 20, 508–512. https://doi.org/10.1097/wnr.0b013e32832776f4 (2009).
Stathopoulos, P. & Dalakas, M. C. The role of complement and complement therapeutics in neuromyelitis Optica spectrum disorders. Expert Rev. Clin. Immunol. 18, 933–945. https://doi.org/10.1080/1744666X.2022.2105205 (2022).
Bennett, J. L. et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis Optica. Ann. Neurol. 66, 617–629. https://doi.org/10.1002/ana.21802 (2009).
Bradl, M. et al. Neuromyelitis optica: pathogenicity of patient Immunoglobulin in vivo. Ann. Neurol. 66, 630–643. https://doi.org/10.1002/ana.21837 (2009).
Saadoun, S. et al. Intra-cerebral injection of neuromyelitis Optica Immunoglobulin G and human complement produces neuromyelitis Optica lesions in mice. Brain 133, 349–361. https://doi.org/10.1093/brain/awp309 (2010).
Rossi, A., Moritz, T. J., Ratelade, J. & Verkman, A. S. Super-resolution imaging of aquaporin-4 orthogonal arrays of particles in cell membranes. J. Cell. Sci. 125, 4405–4412. https://doi.org/10.1242/jcs.109603 (2012).
Ho, B. H. T., Spicer, B. A. & Dunstone, M. A. Action of the terminal complement pathway on cell membranes. J. Membr. Biol. https://doi.org/10.1007/s00232-025-00343-6 (2025).
Gömöri, E. et al. Fetal development of membrane water channel proteins aquaporin-1 and aquaporin-4 in the human brain. Int. J. Dev. Neurosci. 24, 295–305. https://doi.org/10.1016/j.ijdevneu.2006.05.003 (2006).
Holst, C. B., Brøchner, C. B., Vitting-Seerup, K. & Møllgård, K. Astrogliogenesis in human fetal brain: complex Spatiotemporal immunoreactivity patterns of GFAP, S100, AQP4 and YKL-40. J. Anat. 235, 590–615. https://doi.org/10.1111/joa.12948 (2019).
Goasdoué, K., Miller, S. M., Colditz, P. B. & Björkman, S. T. Review: The blood-brain barrier; protecting the developing fetal brain. Placenta 54, 111–116. https://doi.org/10.1016/j.placenta.2016.12.005 (2017).
Du, L. et al. Effect of NMO-IgG on the interleukin-6 cascade in astrocytes via activation of the JAK/STAT3 signaling pathway. Life Sci. 258, 118217. https://doi.org/10.1016/j.lfs.2020.118217 (2020).
Chen, L., Fan, F., Wu, L. & Zhao, Y. The nuclear receptor 4A family members: mediators in human disease and autophagy. Cell. Mol. Biol. Lett. 25 https://doi.org/10.1186/s11658-020-00241-w (2020).
Safe, S. et al. Nuclear receptor 4A (NR4A) family-orphans no more.
Saijo, K. et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death.
Choi, E. H. & Park, S. J. TXNIP: A key protein in the cellular stress response pathway and a potential therapeutic target. Exp. Mol. Med. 55, 1348–1356. https://doi.org/10.1038/s12276-023-01019-8 (2023).
Ji, J. et al. TRIM22 activates NF-κB signaling in glioblastoma by accelerating the degradation of IκBα. Cell. Death Differ. 28, 367–381. https://doi.org/10.1038/s41418-020-00606-w (2021).
Nagai, S. et al. Aberrant nuclear factor-kappaB activity and its participation in the growth of human malignant Astrocytoma. J. Neurosurg. 96, 909–917. https://doi.org/10.3171/jns.2002.96.5.0909 (2002).
Li, W. et al. Sphingosine kinase 1 promotes growth of glioblastoma by increasing inflammation mediated by the NF-κB /IL-6/STAT3 and JNK/PTX3 pathways. Acta Pharm. Sinica B. 12, 4390–4406. https://doi.org/10.1016/j.apsb.2022.09.012 (2022).
Ko, C. Y., Chang, W. C. & Wang, J. M. Biological roles of CCAAT/Enhancer-binding protein delta during inflammation. J. Biomed. Sci. 22, 6. https://doi.org/10.1186/s12929-014-0110-2 (2015).
Nyunt, T. et al. Mitochondrial oxidative stress-induced transcript variants of ATF3 mediate lipotoxic brain microvascular injury. Free Radic. Biol. Med. 143, 25–46. https://doi.org/10.1016/j.freeradbiomed.2019.07.024 (2019).
Holland, S. D. & Ramer, M. S. Microglial activating transcription factor 3 upregulation: an indirect target to attenuate inflammation in the nervous system. Front. Mol. Neurosci. 16 https://doi.org/10.3389/fnmol.2023.1150296 (2023).
Fujihara, K. et al. Interleukin-6 in neuromyelitis Optica spectrum disorder pathophysiology. Neurol. Neuroimmunol. Neuroinflamm. 7 https://doi.org/10.1212/nxi.0000000000000841 (2020).
Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 33, 127–148. https://doi.org/10.1093/intimm/dxaa078 (2021).
Pereira, M. & Gazzinelli, R. T. Regulation of innate immune signaling by IRAK proteins. Front. Immunol. 14 https://doi.org/10.3389/fimmu.2023.1133354 (2023).
Feng, H., Zhang, Y. B., Gui, J. F., Lemon, S. M. & Yamane, D. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PLoS Pathog. 17, e1009220. https://doi.org/10.1371/journal.ppat.1009220 (2021).
Guo, Q. et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal. Transduct. Target. Therapy. 9 https://doi.org/10.1038/s41392-024-01757-9 (2024).
Yamamura, R. et al. Transcriptome signature in the blood of neuromyelitis Optica spectrum disorder under steroid tapering. Front. Immunol. 16, 1508977. https://doi.org/10.3389/fimmu.2025.1508977 (2025).
Inforzato, A. et al. PTX3 as a paradigm for the interaction of pentraxins with the complement system. Semin. Immunol. 25, 79–85. https://doi.org/10.1016/j.smim.2013.05.002 (2013).
Wang, H. et al. Increased plasma levels of pentraxin 3 in patients with multiple sclerosis and neuromyelitis Optica. Mult Scler. 19, 926–931. https://doi.org/10.1177/1352458512457845 (2013).
Ren, F. J., Cai, X. Y., Yao, Y. & Fang, G. Y. JunB: a paradigm for Jun family in immune response and cancer. Front. Cell. Infect. Microbiol. 13, 1222265. https://doi.org/10.3389/fcimb.2023.1222265 (2023).
Hasan, Z. et al. JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat. Commun. 8, 15628. https://doi.org/10.1038/ncomms15628 (2017).
Nishiyama, S. et al. Anti-aquaporin-4 immune complex stimulates complement-dependent Th17 cytokine release in neuromyelitis optica spectrum disorders. Sci. Rep. 14, 3146. https://doi.org/10.1038/s41598-024-53661-5 (2024).
Agasing, A. M. et al. Transcriptomics and proteomics reveal a Cooperation between interferon and T-helper 17 cells in neuromyelitis optica. Nat. Commun. 11, 2856. https://doi.org/10.1038/s41467-020-16625-7 (2020).
Chen, T. et al. Astrocyte-microglia interaction drives evolving neuromyelitis Optica lesion. J. Clin. Investig. 130, 4025–4038. https://doi.org/10.1172/JCI134816 (2020).
Zipfel, P. F. & Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 9, 729–740. https://doi.org/10.1038/nri2620 (2009).
Funding
This study was financially supported by the intramural funding program of the Medical University of Innsbruck Ph.D. Research Training Groups, Project 2022-1-2 “CONNECT” (to SB, JH and MR) and a restricted research grant from Roche Austria GmbH supporting this study (to MR). It was also supported by the Austrian Science Fund (FWF grant https://doi.org/10.55776/PAT6054424) to MB, the China Scholarship Council (CSC 202306170046) to QY, and the Austrian Research Promotion Agency (FFG, project number FO999920011) to RH and VE.
Author information
Authors and Affiliations
Contributions
S.B. analyzed and interpreted the data, wrote the manuscript, and performed all experiments. M.R. designed the study, supervised the work, analyzed and interpreted data, and participated in preparing the manuscript. J.H. produced the stable cell lines and contributed to the manuscript. Q.Y. and M.B. provided the spatial transcriptomics analysis of the experimental rat NMO model and helped with the analysis and interpretation of data. V.E. and R.H. provided serum samples of NMOSD patients and performed the immunohistochemistry of human NMOSD neuropathological sections. All authors reviewed the manuscript critically for important intellectual content and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Serum samples from six NMOSD patients and neuropathological tissue sections from two NMOSD patients were provided by the biobank of the Department of Neuropathology, Medical University of Vienna, Austria. The use of these samples from a biobank for research studies was approved by the ethical committee of the Medical University of Vienna (EK 1636/2019 and 1123/2015). All patients or their legal representatives gave written informed consent to participate in the study and all methods were performed in accordance with the relevant guidelines and regulation.
Competing interests
The authors declare that this study received funding from Roche Austria GmbH (to MR). The funder was not involved in the study design, analysis, or critical revision of the article for important intellectual content. RH reports speaker honoraria from UCB and BMS. The Medical Universities of Innsbruck (Austria; employer of MR) and Vienna (Austria; employer of RH) receive payments for antibody assays and for antibody validation experiments organized by Euroimmun (Lübeck, Germany). SB, QY, JH, VE and MB declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Brandl, S., Yu, Q., Hagenbuchner, J. et al. Inflammatory transcriptomic signatures in a human cellular NMOSD model reveal upregulation of NF-κB and IL6 pathways. Sci Rep 15, 43346 (2025). https://doi.org/10.1038/s41598-025-27335-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-27335-9
