
Abstract
To investigate differences in gut microbiota composition and short-chain fatty acids (SCFAs) metabolism between patients with Chronic Fatigue Syndrome (CFS) and Healthy Controls (HC), and to explore their associations with the CFS pathogenesis. This case–control study included 80 subjects, comprising 40 patients with CFS and 40 age- and sex-matched HC. Fecal microbial community structure was analyzed using 16S rRNA gene high-throughput sequencing. Fecal SCFAs concentrations were quantified using Gas Chromatography–Mass Spectrometry (GC–MS). Spearman correlation analysis with false discovery rate (FDR) adjustment was performed to elucidate associations among gut microbiota, SCFAs, and clinical scores. Compared to the HC group, the CFS group exhibited reduced gut microbiota α-diversity (e.g., ACE, Chao1, Shannon indices, all P < 0.01) and significantly altered β-diversity (ADONIS, P = 0.006). After FDR adjustment, fecal levels of acetate, butyrate, isobutyrate, and isovalerate remained significantly lower in the CFS group (all q < 0.05). Differential abundance analysis revealed a significant reduction in key taxa including the phylum Firmicutes (q = 0.010), class Verrucomicrobiae (q = 0.038), order Clostridiales (q = 0.043), and families Rikenellaceae (q = 0.011) and Ruminococcaceae (q = 0.049). Spearman correlation analysis solidified functional connections: key SCFA-producing taxa (e.g., Faecalibacterium, Subdoligranulum, Ruminococcaceae) were positively correlated with butyrate levels (r = 0.52–0.56, all q < 0.05). Furthermore, reduced abundances of Rikenellaceae and Alistipes were associated with lower SF-36 scores (r = 0.26, q = 0.032) and higher fatigue scores (FSS/FS-14, r = − 0.28 to − 0.30, q < 0.05). Isovalerate levels were negatively correlated with FS-14 scores (r = − 0.307, q = 0.014). Among CFS patients, those with higher dietary fiber intake had significantly higher levels of acetate and isovalerate than those with lower intake (both q < 0.05). Patients with CFS exhibit significant gut dysbiosis and abnormal SCFA metabolism. The reduction in key SCFA-producing taxa, their positive correlations with SCFAs levels, and the negative correlations of both with fatigue severity solidify a functional link between gut microbial depletion, reduced SCFAs, and clinical symptoms in CFS. Higher dietary fiber intake may partially ameliorate SCFAs metabolic disturbances in CFS patients.
Similar content being viewed by others
Introduction
Chronic Fatigue Syndrome (CFS) is a complex disorder characterized by persistent or recurrent severe fatigue as its core symptom, often accompanied by cognitive impairment, sleep disturbances, muscle and joint pain, and orthostatic intolerance, significantly impairing patients’ quality of life1. Epidemiological data indicate a global CFS prevalence of approximately 0.2–2.5%, with distinct gender and age distribution patterns: the incidence is significantly higher in women than in men (approximately 1.5- to twofold) and peaks among individuals aged 20 to 50 years 2. The long-term effects of the Coronavirus Disease 2019 (COVID-19) pandemic have further heightened the urgency of CFS research. The increased incidence of post-COVID-19 condition (“long COVID”) following the pandemic has refocused attention on CFS, as approximately 10–30% of COVID-19 survivors experience persistent fatigue, post-exertional malaise, and cognitive impairment meeting CFS diagnostic criteria3,4,5. Although the etiology of CFS remains incompletely understood, current research suggests potential associations with mechanisms such as immune dysregulation6,7, mitochondrial dysfunction8,9, and chronic viral infections10. Current clinical management primarily involves symptomatic and supportive approaches, including cognitive behavioral therapy11,12, graded exercise therapy13, and pharmacological interventions. However, treatment efficacy is limited and lacks specific targets, highlighting the critical need to explore objective biomarkers and novel therapeutic strategies.
In recent years, the role of the gut microbiota as the “second genome” in chronic diseases has garnered significant attention14. Research indicates that gut microbes can influence host physiological functions through various pathways, including microbial metabolites15, gut-brain axis signaling15,16, and immune modulation17. The high prevalence of gastrointestinal dysfunction in CFS patients suggests potential involvement of the microbiota in disease pathogenesis18. Short-chain fatty acids (SCFAs), the primary end products of dietary fiber fermentation by gut bacteria, not only serve as an energy source for colonic epithelial cells and maintain barrier integrity but also exert systemic anti-inflammatory and neuroprotective effects by inhibiting histone deacetylases and activating G protein-coupled receptors19. Although a few studies have reported alterations in gut microbiota composition in CFS patients, current evidence lacks systematic analysis linking microbial functional changes to community structure.
The Fatigue Severity Scale (FSS)20, the 36-Item Short Form Health Survey (SF-36)21, and the Fatigue Scale-14 (FS-14)22 are core assessment tools in CFS research. The FSS focuses on evaluating the subjective severity of fatigue and its interference with daily functioning. The SF-36 comprehensively measures the impact of illness on multidimensional health-related quality of life. The FS-14 distinguishes between physical fatigue and mental fatigue dimensions. This study employs 16S rRNA gene sequencing and targeted SCFAs quantification to analyze differences in gut microbiota composition and SCFAs metabolism in fecal samples between CFS patients and healthy individuals. Subgroup analysis of CFS patients based on dietary fiber intake was performed, and correlations between differential microbial taxa, SCFAs levels, and scores from the three assessment scales were analyzed to identify diagnostically significant biomarkers. The study aims to provide a theoretical foundation for the development of non-invasive diagnostic markers and microbiota-targeted interventions for CFS (Fig. 1).

Study flow chart.
Materials and methods
Research subjects
A total of 80 subjects were included in this case–control study. Participants were recruited between April 2023 and April 2024 from the Endocrine Clinic and Chronic Disease Management Clinic of the Affiliated Hospital of Shandong University of Traditional Chinese Medicine. CFS patients were consecutively recruited from outpatients presenting at these clinics, while HC were recruited from community volunteers and hospital staff and were matched for age and sex. The study comprised 40 CFS patients who met the diagnostic criteria and 40 age- and sex-matched HC. The inclusion criteria for the case group were based on the “Diagnostic Criteria for Systemic Exertion Intolerance Disease (SEID)” developed by the National Academy of Medicine (IOM) in 201523 and the clinical criteria in the 2023 international consensus guideline “Diagnosis and Management of Chronic Fatigue Syndrome”1. For the 40 CFS patients, according to the “Reference Intake of Dietary Nutrients of the Chinese Nutrition Society” (2023 edition)24, they were further grouped based on their habitual daily dietary fiber intake: intake < 25 g was defined as the CFS low-fiber diet group (LFD, n = 24), and ≥ 25 g was defined as the CFS high-fiber diet group (HFD, n = 16). Exclusion criteria included: the presence of definite organic diseases (such as thyroid dysfunction, autoimmune diseases); acute infection period or abnormal inflammatory indicators (CRP > 5 mg/L); use of antibiotics, probiotics, or immunomodulatory drugs in the past 3 months; combined with severe mental disorders (e.g., major depressive disorder, schizophrenia). The healthy control group had to meet the following conditions: no history of chronic disease and fatigue-related complaints; body mass index (BMI) within the normal range (18.5–24.9 kg/m2); no gastrointestinal function-modifying drugs taken in the past 1 month. Given their generally healthy and regular dietary patterns, the HC group exhibited a narrow range of dietary fiber intake. Therefore, subgroup analysis based on fiber intake was not conducted for the HC group, as it would lack the statistical power to reveal meaningful associations. The analytical focus was placed on the CFS group, which demonstrated sufficient heterogeneity in dietary fiber intake for such comparisons.
Fecal sample collection and processing
Following a 12-h fast, participants provided 5–10 g fecal samples using sterile collection tubes (Sarstedt). Samples were immediately flash-frozen in liquid nitrogen and subsequently stored at − 80 °C. Samples designated for SCFAs analysis were protected from repeated freeze–thaw cycles. Samples for 16S sequencing were transported on dry ice throughout the process.
Clinical assessment
All participants underwent a comprehensive clinical assessment prior to sample collection. Fatigue severity and health-related quality of life were evaluated using three standardized self-report questionnaires.
The Fatigue Severity Scale (FSS) contains 9 items assessing the impact of fatigue on daily functioning, rated on a 7-point Likert scale (1 = strongly disagree, 7 = strongly agree). The total score is calculated as the mean of the 9 items, with higher scores (range 1–7) indicating greater fatigue severity.
The Fatigue Scale-14 (FS-14) is a 14-item dichotomous (yes/no) scale measuring two dimensions: physical fatigue (8 items) and mental fatigue (6 items). The total score ranges from 0 to 14, with higher scores indicating more severe fatigue.
The 36-Item Short Form Health Survey (SF-36) assesses eight health domains: physical functioning, role limitations due to physical health, bodily pain, general health perceptions, vitality, social functioning, role limitations due to emotional problems, and mental health. Scores for each domain range from 0 to 100, with higher scores indicating better health status. The Physical Component Summary (PCS) and Mental Component Summary (MCS) scores were calculated according to the standard algorithm.
Assessments were conducted in a quiet, private room at the clinical center. A trained research coordinator was present to provide standardized instructions, answer questions, and ensure the consistency and reliability of the data collection process.
Daily dietary fiber intake was assessed using a validated food frequency questionnaire (FFQ)25. Participants reported their average consumption frequency over the past month. Portion sizes were estimated using standard household measures (e.g., bowls, grams). Dietary fiber intake was calculated based on the China Food Composition Table26.
16S rRNA gene sequencing and analysis
DNA Extraction and Quality Control: Total genomic DNA was extracted from 200 mg of fecal material using the E.Z.N.A.® Soil DNA Kit. DNA purity was assessed using a NanoDrop 2000 spectrophotometer (A260/A280 > 1.8, A260/A230 > 1.5). DNA integrity was verified by 1% agarose gel electrophoresis.
PCR Amplification and Library Construction: The V3-V4 hypervariable regions of the bacterial 16S rRNA gene were amplified using primers 338F and 806R. PCR reactions (20μL total volume) contained FastPfu buffer, dNTPs (0.8 μM each), primers (0.8 μM each), and template DNA (10 ng). Amplification conditions were: 95 °C for 3 min; 27 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 45 s; followed by a final extension at 72 °C for 10 min. PCR amplicons were purified using 2% agarose gels, quantified with the Quantus™ Fluorometer, pooled in equimolar amounts, and libraries were constructed using the NEXTFLEX® Rapid DNA-Seq Kit.
Sequencing and Bioinformatics Analysis: Sequencing was performed on the Illumina NovaSeq 6000 platform (PE250 mode). Raw reads were quality-filtered using fastp and assembled using FLASH. Operational Taxonomic Units (OTUs) were clustered at 97% sequence similarity using USEARCH (v11), followed by chimera removal and filtering of host-derived sequences. Taxonomic classification was performed against the SILVA database (v138). Results were analyzed and reported across multiple taxonomic levels, including phylum, class, order, family, and genus, as specified in the Results section. Prior to downstream diversity (alpha and beta diversity) and differential abundance analyses (such as LEfSe), a prevalence filter was applied to exclude rare taxa; specifically, Operational Taxonomic Units (OTUs) with a relative abundance less than 0.1% across all samples were removed. Alpha diversity indices (Chao1, Shannon) and beta diversity (Bray–Curtis dissimilarity, visualized via Principal Coordinates Analysis (PCoA)) were calculated. Linear discriminant analysis Effect Size (LEfSe) was employed to identify differentially abundant bacterial genera (Linear Discriminant Analysis (LDA) score > 3, P < 0.05).
Targeted SCFAs quantification
Sample Preparation: Fecal samples (50 mg) were homogenized with a solution of 15% phosphoric acid containing 10 μM hexanoic acid-d3 as an internal standard. The homogenate was centrifuged at 12,000 rpm for 10 min at 4 °C. The supernatant was filtered through a 0.22 μm membrane and stored at − 80 °C until analysis.
LC–MS/MS Analysis: Analysis was performed using a Waters UPLC system coupled to an AB Sciex 5500 Qtrap mass spectrometer. Chromatographic separation was achieved on an HSST3 column (2.1 × 100 mm, 1.8 μm particle size). The mobile phase consisted of (A) 0.05% formic acid in water and (B) 0.05% formic acid in acetonitrile. The gradient program was: 0–8 min (30% B to 55% B). The mass spectrometer operated in negative electrospray ionization (ESI) mode. Source parameters were: temperature 450 °C and spray voltage-4500 V. Seven SCFAs (acetate, propionate, butyrate, isobutyrate, valerate, isovalerate, hexanoate) were quantified using Multiple Reaction Monitoring (MRM). Calibration curves exhibited R2 values > 0.99.
Statistical analysis
All statistical analyses were performed using SPSS Statistics software (version 29.0). The normality of continuous data was assessed using the Shapiro–Wilk test. Normally distributed variables were presented as mean ± standard deviation and compared using the independent samples t-test. Non-normally distributed variables were expressed as median (interquartile range) and compared using the Mann–Whitney U test. Categorical data were expressed as percentages (%) and compared by the chi-square test. Correlations between microbial taxa, SCFAs, and clinical scores were analyzed using Spearman’s rank correlation coefficient. To account for multiple comparisons, the false discovery rate (FDR) was controlled using the Benjamini–Hochberg procedure, and the significance level was set at an FDR-adjusted *q*-value < 0.05. For 16S rRNA data processing, QIIME 2 (version 2022.11) was primarily used for downstream analyses, including diversity metrics and taxonomic composition. For the identification of differentially abundant taxa across groups, Linear Discriminant Analysis Effect Size (LEfSe) was performed, with the significance threshold set at an LDA score > 3 and an FDR-adjusted p-value < 0.05.
Results
Population and clinical characteristics
A total of 80 subjects were included in this case–control study, comprising 40 patients with CFS and 40 age- and sex-matched healthy controls. Based on their daily dietary fiber intake, the CFS patients were further subdivided into a low-fiber diet group (LFD, < 25 g/d, n = 24) and a high-fiber diet group (HFD, ≥ 25 g/d, n = 16). Baseline characteristics were comparable between the CFS and HC groups, with no significant differences observed in age, gender, or metabolic and endocrine indexes (all P > 0.05; Table 1). Similarly, the CFS subgroups (LFD and HFD) were well-matched in all demographic and lifestyle indicators, including age, gender, BMI, smoking status, and alcohol consumption (all P > 0.05), with the exception of dietary fiber intake, which was significantly higher in the HFD group (P < 0.01; Table 2). This confirmed the suitability of the subgroups for comparative analysis.
Analysis of intestinal microbiota diversity in CFS patients and HC
Fecal microbial communities from CFS patients and HC were analyzed via 16S rRNA gene sequencing. Alpha diversity indices, assessing microbial community richness and diversity, revealed significantly lower values in the CFS group compared to the HC group for the ACE index, Observed OTUs, Shannon index, and Chao1 index (all P < 0.01; Fig. 2A–E). These results indicate a significant reduction in species richness, overall diversity, and evenness within the gut microbiota of CFS patients. Beta diversity analysis, based on Bray–Curtis dissimilarity, demonstrated significant separation in microbial community structure between the CFS and HC groups (ADONIS test: Bray–Curtis distance, R2 = 0.05, P = 0.006; weighted UniFrac distance, R2 = 0.045, P = 0.013). Samples from the CFS group exhibited significantly greater dispersion in ordination space compared to the tighter clustering characteristic of HC group samples (Fig. 2F), suggesting increased inter-individual variation in gut microbiota composition among CFS patients. Notably, the overall statistical significance of the group separation was robust to the presence of the more dispersed CFS samples, as confirmed by a sensitivity analysis.

Comparison of intestinal microbiota diversity between CFS group and HC group. (A) ACE Index, (B) Observed OTUs, (C) Simpson Index, (D) Chao1 Index, (E) Shannon Index, (F) Principal coordinate analysis (PCoA) based on Bray–Curtis distance.
The Observed OTUs index represents the absolute number of distinct species detected. The Chao1 and ACE indices are non-parametric estimators of total species richness, with Chao1 being more sensitive to singletons (species with only one read) and ACE more sensitive to rare species (e.g., singletons and doubletons). The Shannon index reflects community diversity by considering both species richness and evenness.
Differences in species composition analysis between CFS and HC
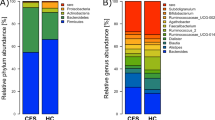
At the phylum level, Firmicutes and Bacteroidetes were the dominant phyla in both groups. Compared to the HC group, the CFS group exhibited a significant reduction in Firmicutes (q = 0.010) (Fig. 3A). At the class level, a significant reduction in Verrucomicrobiae (q = 0.038) was observed in the CFS group. At the order level, Clostridiales showed a marked reduction in the CFS group (q = 0.043). At the family level, taxa including Rikenellaceae (q = 0.011) and Ruminococcaceae (q = 0.049) were depleted in the CFS group.

LEfSe analysis of differential flora between CFS group and HC group. (A) Histogram of relative abundance of community composition, (B) Linear Discriminant Analysis (LDA) Effect Size (LEfSe) bar plot, (C) Evolutionary cladogram.
Linear discriminant analysis Effect Size (LEfSe) identified taxa that contributed most significantly to the differences between groups (LDA score > 3, P < 0.05; Fig. 3B, C). The HC group was characterized by an enrichment of commensal and SCFA-producing bacteria, primarily within the phylum Firmicutes. In contrast, the CFS group was significantly enriched in the phylum Verrucomicrobia.
Differential abundance testing corroborated these findings, revealing significant reductions in the CFS group at the family level (Rikenellaceae, q = 0.011; Ruminococcaceae, q = 0.049). A notable discrepancy was observed regarding Verrucomicrobia: while LEfSe analysis indicated its significant enrichment in the CFS group at the phylum level (LDA > 3), analysis at the class level revealed a significant reduction in Verrucomicrobiae (q = 0.038). This inconsistency may arise from divergent behaviors of other classes within the phylum (e.g., Lentisphaeria), or it could reflect the low overall abundance and high inter-individual variability of Verrucomicrobia, which can affect analytical consistency across taxonomic levels. The biological implications of these changes warrant further functional investigation.
Differential bacterial taxa comparison between CFS and HC
Analysis of 16S rRNA gene sequencing data, with FDR adjustment for multiple comparisons, revealed significant differences in the relative abundance of several bacterial taxa between CFS patients and HC across multiple taxonomic levels (Fig. 4). At the phylum level, the relative abundance of Firmicutes was significantly reduced in the CFS group (q = 0.010). At the class level, a significant decrease was observed for Verrucomicrobiae (q = 0.038). Furthermore, at the order level, the abundance of Clostridiales was notably lower in patients (q = 0.043). Significant reductions were also identified at the family level, including Rikenellaceae (q = 0.011) and Ruminococcaceae (q = 0.049).In contrast, at the genus level, Peptostreptococcus showed an increasing trend in the CFS group, though this difference was not significant after FDR correction.Other genera such as Alistipes, Faecalibacterium, and Subdoligranulum also exhibited reductions before multiple-testing correction, but these did not remain significant thereafter (all q > 0.05). These findings, with full statistical details for all tested taxa provided in Supplementary Table 3, collectively indicate a marked reduction in key commensal and SCFA-producing bacterial taxa within the gut microbiota of CFS patients.

Differences in key microbial taxa between CFS patients and HC at various taxonomic levels. (A–E) represent the following taxa: (A) Verrucomicrobiae (class), (B) Firmicutes (phylum), (C) Ruminococcaceae (family), (D) Clostridiales (order), (E) Alistipes (genus). Relative abundances of these taxa differed significantly between CFS and HC groups (q < 0.05).
Altered fecal short-chain fatty acid levels in CFS patients
Figure 5 demonstrates the differences in fecal SCFAs between CFS patients and healthy controls, as well as between HFD and LFD subgroups within the CFS cohort. Fecal SCFAs quantification revealed significantly lower levels in the CFS group compared to the HC group after FDR correction. The concentrations of butyrate (q = 0.044), acetate (q = 0.032), isobutyrate (q = 0.039), and isovalerate (q = 0.008) were all significantly reduced in the CFS group, with isovalerate showing the strongest association. No significant differences were observed for caproate (q = 0.190), propionate (q = 0.325), or valerate (q = 0.952) (Fig. 5A, B).

Comparison of fecal SCFAs concentrations between CFS group and HC group. (A) Concentrations of SCFAs that were significantly reduced in the CFS group, presented in order of ascending carbon number: acetate (C2), butyrate (C4), isobutyrate (C4), and isovalerate (C5). (B) Concentrations of SCFAs that showed no significant difference between groups, presented in order of ascending carbon number: propionate (C3) and valerate (C5). (C) Concentrations of acetate and isovalerate were significantly higher in the CFS high-fiber diet group (HFD) than in the low-fiber diet group (LFD). Data are presented as box plots. *P < 0.05; **P < 0.01; NS: No statistical difference.
Further subgroup analysis indicated that dietary fiber intake significantly modulated SCFAs profiles within the CFS cohort. The HFD subgroup exhibited significantly higher fecal levels of acetate and isovalerate compared to the LFD subgroup (q = 0.042; 0.035), suggesting that dietary fiber may partially restore SCFAs metabolic deficits in CFS patients (Fig. 5C).
Association analysis of microbiota and SCFAs with clinical symptoms
To elucidate the potential mechanistic links, Spearman correlation analysis with FDR adjustment revealed significant associations among gut microbiota, SCFAs, and clinical scores (Fig. 6A–F). Key SCFA-producing taxa were positively correlated with SCFAs levels: Faecalibacterium and Subdoligranulum with butyrate (r = 0.55, q = 0.032; r = 0.52, q = 0.038), Ruminococcaceae with butyrate (r = 0.56, q = 0.028), Alistipes with acetate and isovalerate (r = 0.54, q = 0.035; r = 0.51, q = 0.040), and Rikenellaceae with acetate and isovalerate (r = 0.53, q = 0.040; r = 0.55, q = 0.035). Clinically, Rikenellaceae and Alistipes were positively correlated with SF-36 scores (r = 0.26, q = 0.032) and negatively correlated with FSS and FS-14 scores (Rikenellaceae: r = − 0.28, q = 0.024; Alistipes: r = − 0.28, q = 0.045). Ruminococcaceae was negatively correlated with FSS scores (r = − 0.30, q = 0.016), and isovalerate was negatively correlated with FS-14 scores (r = − 0.307, q = 0.014). These results solidify the functional connection between depleted microbial taxa, reduced SCFAs, and fatigue severity in CFS patients.

Correlation heatmap among gut microbiota, SCFAs, and clinical symptom scale scores. The color scale represents the actual Spearman’s correlation coefficient (r-value), with red indicating positive correlations and blue indicating negative correlations. The color intensity corresponds to the strength of the correlation. Asterisks indicate statistical significance after FDR adjustment for multiple comparisons: *FDR-adjusted P < 0.05; **FDR-adjusted P < 0.01.
Discussion
Comparison of key findings with previous studies
This study, utilizing 16S rRNA gene sequencing and gas chromatography–mass spectrometry, confirmed the presence of structural dysbiosis in the gut microbiota and metabolic disturbances in short-chain fatty acids in patients with chronic fatigue syndrome. Consistent with most reports18,27,28, CFS patients exhibited significantly reduced gut microbial alpha diversity, while beta diversity analysis indicated clear separation of community structures between groups. In terms of taxonomic composition, the CFS group showed decreased abundances of beneficial bacterial groups such as Firmicutes and Clostridia, alongside an increased abundance of potential pathobionts like Proteobacteria, which aligns with the findings of Giloteaux et al.29. However, the interpretation of Actinobacteria alterations in CFS appears context-dependent. While an increase in this phylum has been reported in some fatigue-associated conditions30, our study did not find a significant enrichment after FDR correction. This discrepancy may be partly explained by the dual nature of Actinobacteria, which encompasses renowned beneficial genera like Bifidobacterium but also includes potential pathobionts. Therefore, the net effect of Actinobacteria abundance changes likely depends on the specific shifts occurring at lower taxonomic levels (e.g., genus or species), which could be influenced by cohort-specific factors such as geography, diet, or disease subtype, leading to inconsistent observations across studies. Notably, we identified several differential taxa, including Verrucomicrobiae, Rikenellaceae, and Ruminococcaceae, which have been relatively under-investigated in CFS research. These alterations in the microbiota are not merely a passive response to the disease milieu but may engage in bidirectional interactions with the inherent immune dysregulation state in CFS patients. Research suggests that host immune status, particularly the Th1/Th2 balance, is a key driver shaping gut microbiota structure31. Consequently, the observed dysbiosis in CFS should be interpreted as a manifestation of a disrupted dynamic equilibrium within the complex “host-microorganism” interaction network under disease conditions.
Novel findings and the gut microbiota–metabolite–symptom axis
The novelty of this study lies in systematically constructing an association network linking the gut microbiota, SCFAs metabolism, and clinical fatigue symptoms. Correlation analyses revealed that Faecalibacterium and Subdoligranulum were positively correlated with butyrate; Alistipes was positively correlated with butyrate, acetate, and isovalerate; and Rikenellaceae was positively correlated with acetate and isovalerate. These associations provide functional evidence that the capacity of the gut microbiota to ferment dietary fiber and produce beneficial metabolites is significantly compromised in CFS patients.
More clinically relevant is the significant correlation between these beneficial bacterial taxa and their associated SCFAs with core clinical symptoms.The abundance of Rikenellaceae showed a positive correlation with SF-36 scores and negative correlations with FSS and FS-14 scores. Similarly, Ruminococcaceae and Alistipes abundances were negatively correlated with FSS scores, and isovalerate level was negatively correlated with FS-14 scores. These results suggest a potential link between gut microbial metabolites and clinical manifestations in CFS (For detailed results, see Supplementary Table 1).
Discussion on pathogenesis
Our findings suggest that gut microbiota disruption contributes to the onset and progression of CFS through SCFAs metabolic dysfunction. Acetate, a crucial energy source for colonocytes32, its deficiency may lead to impaired intestinal barrier function, lipopolysaccharide translocation, and systemic inflammation activation33,34,35. Concurrently, acetate, acting as a histone deacetylase inhibitor, influences regulatory T-cell differentiation and anti-inflammatory signaling36,37,38,39,40. Its decreased levels might also affect HPA axis function via the vagus nerve-hypothalamus pathway, exacerbating stress responses and neuroendocrine disturbances41. In this study, the relative abundances of taxa with key SCFA-producing functions, such as Ruminococcaceae, Rikenellaceae, and Alistipes, were significantly reduced and positively correlated with various SCFAs, further supporting the notion of impaired SCFAs production capacity in CFS at the community functional level. Particularly noteworthy is the positive correlation of Alistipes and Rikenellaceae with multiple SCFAs and their association with symptomatic improvement, consistent with their recognition as potentially beneficial bacteria. Conversely, the negative correlation between Lachnospiraceae and isovalerate hints at functional differentiation in SCFAs metabolic contributions among different bacterial groups within the specific context of CFS. This inference aligns with the observed overall decline in multiple SCFAs concentrations, collectively pointing towards a continuum of pathological changes from structural dysbiosis to metabolic impairment in CFS. The negative correlation between isovalerate and fatigue severity is a finding warranting further investigation. Although often regarded as a marker of protein fermentation42, its physiological significance might be more complex in the setting of low-grade inflammation and energy metabolism dysfunction in CFS.Our observation that CFS patients with higher dietary fiber intake had relatively higher isovalerate levels suggests it might indirectly reflect functional remodeling of the microbiota. Furthermore, holistic lifestyle interventions, such as exercise, have been shown to promote SCFAs production via microbiota reshaping and improve cognition43, providing indirect support for the clinical relevance of our findings.
Clinical implications
The findings provide a rationale for microbiota-targeted intervention strategies in CFS. Several studies indicate that probiotics can alleviate CFS symptoms by restoring microbial balance and reducing pro-inflammatory cytokine levels44,45, mechanisms thought to involve promoting SCFAs production and repairing intestinal barrier function46,47,48.In our study, CFS patients with high dietary fiber intake exhibited significantly higher fecal acetate and isovalerate levels than the low-fiber group, consistent with the mechanism whereby prebiotics selectively increase SCFA-producing bacteria to elevate SCFAs concentrations48,49. Beyond directly supplying SCFAs precursors,dietary interventions can also indirectly improve energy metabolism and mitigate neuroinflammation by modulating the Firmicutes/Bacteroidetes ratio50,51 and reducing intestinal permeability52. Therefore, microbiota-targeted strategies for CFS could be based on dietary modifications, potentially combined with specific probiotics or prebiotics, to synergistically correct the core pathological aspect of SCFAs deficiency.
Limitations
Several limitations of this study must be acknowledged. Although the identified correlations were statistically significant, their effect sizes were generally small; for instance, the inter-group difference in beta diversity explained only a limited proportion of variance. Key limitations include: the cross-sectional design precludes causal inference; the total sample size and subgroup sizes were limited, potentially constraining the power to detect associations with small-to-moderate effect sizes; fecal SCFAs concentrations may not fully reflect host absorption and tissue utilization efficiency; and the specific downstream signaling pathways of SCFAs remain unelucidated. Although major confounders were controlled for, residual confounding from dietary recording, medication use, and potential environmental variables requires more granular handling. Furthermore, given the greater dietary heterogeneity among CFS patients and their potential heightened sensitivity to dietary factors, the subgroup analysis on dietary fiber intake focused solely within the CFS group, rather than including healthy controls who exhibited higher homogeneity in fiber intake. While this design allows for a more targeted examination of regulatory effects under pathological conditions, it limits direct comparison of the relationships between fiber, microbiota, and SCFAs in health versus disease states. Additionally, the study did not differentiate the potential distinct effects of various dietary fiber types on the SCFAs profile. Therefore, the current results should be interpreted cautiously as evidence of association. Future prospective studies in larger cohorts, encompassing both healthy individuals and patients with a wider range of fiber intake, are needed to clarify the temporal sequence of microbiota-SCFAs dynamics in relation to CFS development and to provide higher-level evidence for microbiota-targeted interventions.
Data availability
The raw sequence data generated in this study have been deposited in the National Genomics Data Center (GSA accession: CRA028172) and are publicly available at https://ngdc.cncb.ac.cn/gsa/s/7vrVNco0.
References
Grach, S. L., Seltzer, J., Chon, T. Y. & Ganesh, R. Diagnosis and management of myalgic encephalomyelitis/chronic fatigue syndrome. Mayo Clin. Proc. 98, 1544–1551 (2023).
Lim, E.-J. et al. Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). J. Transl. Med. 18, 100 (2020).
Sukocheva, O. A. et al. Analysis of post COVID-19 condition and its overlap with myalgic encephalomyelitis/chronic fatigue syndrome. J. Adv. Res. 40, 179–196 (2021).
Yong, S. J. Long COVID or post-COVID-19 syndrome: Putative pathophysiology, risk factors, and treatments. Infect. Dis. (Lond. Engl.) https://doi.org/10.1080/23744235.2021.1924397 (2021).
Kedor, C. et al. A prospective observational study of post-COVID-19 chronic fatigue syndrome following the first pandemic wave in Germany and biomarkers associated with symptom severity. Nat. Commun. 13, 5104 (2022).
Bjørklund, G. et al. Environmental, neuro-immune, and neuro-oxidative stress interactions in chronic fatigue syndrome. Mol. Neurobiol. 57, 4598–4607 (2020).
Bansal, A. S., Bradley, A. S., Bishop, K. N., Kiani-Alikhan, S. & Ford, B. Chronic fatigue syndrome, the immune system and viral infection. Brain Behav. Immun. 26, 24–31 (2012).
Wood, E., Hall, K. H. & Tate, W. Role of mitochondria, oxidative stress and the response to antioxidants in myalgic encephalomyelitis/chronic fatigue syndrome: A possible approach to SARS-CoV-2 ‘long-haulers’?. Chronic Dis. Transl. Med. 7, 14–26 (2021).
Syed, A. M., Karius, A. K., Ma, J., Wang, P.-Y. & Hwang, P. M. Mitochondrial dysfunction in myalgic encephalomyelitis/chronic fatigue syndrome. Physiology https://doi.org/10.1152/physiol.00056.2024 (2025).
Rasa, S. et al. Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J. Transl. Med. 16, 268 (2018).
Van Oudenhove, L., Debyser, S., Vergaelen, E., Claes, S. & Van Den Houte, M. Predictors of treatment response trajectories to cognitive behavioral therapy for chronic fatigue syndrome: A cohort study. J. Psychosom. Res. 187, 111954 (2024).
Kuut, T. A., Buffart, L. M., Braamse, A. M. J., Müller, F. & Knoop, H. Is the effect of cognitive behaviour therapy for chronic fatigue syndrome (ME/CFS) moderated by the presence of comorbid depressive symptoms? A meta-analysis of three treatment delivery formats. J. Psychosom. Res. 184, 111850 (2024).
Larun, L., Brurberg, K. G., Odgaard-Jensen, J. & Price, J. R. Exercise therapy for chronic fatigue syndrome. Cochrane Db. Syst. Rev. 12, CD003200 (2024).
Martel, J. et al. Gut barrier disruption and chronic disease. Trends Endocrinol. Metab. TEM 33, 247–265 (2022).
O’Riordan, K. J. et al. Short chain fatty acids: Microbial metabolites for gut-brain axis signalling. Mol. Cell. Endocrinol. 546, 111572 (2022).
König, R. S. et al. The gut microbiome in myalgic encephalomyelitis (ME)/chronic fatigue syndrome (CFS). Front. Immunol. 12, 628741 (2021).
Kim, C. H. Complex regulatory effects of gut microbial short-chain fatty acids on immune tolerance and autoimmunity. Cell. Mol. Immunol. 20, 341–350 (2023).
Xiong, R. et al. Multi-’omics of gut microbiome-host interactions in short- and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients. Cell Host Microbe 31, 273-287.e5 (2023).
Rekha, K. et al. Short-chain fatty acid: An updated review on signaling, metabolism, and therapeutic effects. Crit. Rev. Food Sci. Nutr. 64, 2461–2489 (2024).
Krupp, L. B., LaRocca, N. G., Muir-Nash, J. & Steinberg, A. D. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch. Neurol. 46, 1121–1123 (1989).
Stewart, A. L. et al. Functional status and well-being of patients with chronic conditions. Results from the medical outcomes study. JAMA 262, 907–913 (1989).
Chalder, T. et al. Development of a fatigue scale. J. Psychosom. Res. 37, 147–153 (1993).
Clayton, E. W. Beyond myalgic encephalomyelitis/chronic fatigue syndrome: An IOM report on redefining an illness. JAMA 313, 1101–1102 (2015).
Chinese Nutrition Society (CORP). Chinese Dietary Reference Intakes (2023 Edition) (Hardcover) (People’s Medical Publishing House, Beijing, 2023).
Pérez Rodrigo, C., Aranceta, J., Salvador, G. & Varela-Moreiras, G. Food frequency questionnaires. Nutr. Hosp. 31(Suppl 3), 49–56 (2015).
Yang, Y. China Food Composition Table (Peking University Medical Press, 2002).
Nagy-Szakal, D. et al. Fecal metagenomic profiles in subgroups of patients with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 5, 44 (2017).
Guo, C. et al. Deficient butyrate-producing capacity in the gut microbiome is associated with bacterial network disturbances and fatigue symptoms in ME/CFS. Cell Host Microbe 31, 288–304 (2023).
Giloteaux, L. et al. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 4, 30 (2016).
Borren, N. Z. et al. Alterations in fecal microbiomes and serum metabolomes of fatigued patients with quiescent inflammatory bowel diseases. Clin. Gastroenterol. Hepatol. 19, 519-527.e5 (2021).
Van Averbeke, V. et al. Host immunity influences the composition of murine gut microbiota. Front. Immunol. 13, 828016 (2022).
Zambell, K. L., Fitch, M. D. & Fleming, S. E. Acetate and butyrate are the major substrates for de novo lipogenesis in rat colonic epithelial cells. J. Nutr. 133, 3509–3515 (2003).
Wang, P.-Y. et al. WASF3 disrupts mitochondrial respiration and may mediate exercise intolerance in myalgic encephalomyelitis/chronic fatigue syndrome. PNAS 120, e2302738120 (2023).
Jonsjö, M. A. et al. Patients with ME/CFS (myalgic encephalomyelitis/chronic fatigue syndrome) and chronic pain report similar level of sickness behavior as individuals injected with bacterial endotoxin at peak inflammation. Brain Behav. Immun. Health 2, 100028 (2020).
Holzer, P. et al. Visceral inflammation and immune activation stress the brain. Front. Immunol. 8, 1613 (2017).
Soliman, M. L. & Rosenberger, T. A. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol. Cell. Biochem. 352, 173–180 (2011).
Penney, J. & Tsai, L.-H. Histone deacetylases in memory and cognition. Sci. Signal. 7, re12 (2014).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science 341, 569–573 (2013).
Park, J. et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 8, 80–93 (2015).
Corrêa-Oliveira, R., Fachi, J. L., Vieira, A., Sato, F. T. & Vinolo, M. A. R. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 5, e73 (2016).
Wu, M. et al. Associations between disordered gut microbiota and changes of neurotransmitters and short-chain fatty acids in depressed mice. Transl. Psychiatry 10, 350 (2020).
Xie, L., Alam, M. J., Marques, F. Z. & Mackay, C. R. A major mechanism for immunomodulation: Dietary fibres and acid metabolites. Semin. Immunol. 66, 101737 (2023).
Chen, Q. et al. Gut microbiota dysbiosis in patients with Alzheimer’s disease and correlation with multiple cognitive domains. Front. Aging Neurosci. 16, 1478557 (2024).
Groeger, D. et al. Bifidobacterium infantis 35624 modulates host inflammatory processes beyond the gut. Gut Microbes 4, 325–339 (2013).
Rao, A. V. et al. A randomized, double-blind, placebo-controlled pilot study of a probiotic in emotional symptoms of chronic fatigue syndrome. Gut Pathogens 1, 6 (2009).
Chelakkot, C. et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp. Mol. Med. 50, e450 (2018).
Lim, S.-M., Jeong, J.-J., Woo, K. H., Han, M. J. & Kim, D.-H. Lactobacillus sakei OK67 ameliorates high-fat diet-induced blood glucose intolerance and obesity in mice by inhibiting gut microbiota lipopolysaccharide production and inducing colon tight junction protein expression. Nutr. Res. 36, 337–348 (2016).
Boudry, G. et al. Bovine milk oligosaccharides decrease gut permeability and improve inflammation and microbial dysbiosis in diet-induced obese mice. J. Dairy Sci. 100, 2471–2481 (2017).
Nettleton, J. E. et al. Low-dose stevia (rebaudioside A) consumption perturbs gut microbiota and the mesolimbic dopamine reward system. Nutrients 11, 1248 (2019).
Do, M. H., Lee, E., Oh, M.-J., Kim, Y. & Park, H.-Y. High-glucose or -fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients 10, 761 (2018).
Snelson, M. et al. Long term high protein diet feeding alters the microbiome and increases intestinal permeability, systemic inflammation and kidney injury in mice. Mol. Nutr. Food Res. 65, e2000851 (2021).
Nilholm, C., Roth, B. & Ohlsson, B. A dietary intervention with reduction of starch and sucrose leads to reduced gastrointestinal and extra-intestinal symptoms in IBS patients. Nutrients 11, 1662 (2019).
Funding
This work was supported by the Shandong Province Key R&D Program (Major Science and Technology Innovation Project 2021CXGC010510).
Author information
Authors and Affiliations
Contributions
Xinxin Cheng proposed the primary research objectives, conceived and designed the study, and drafted the manuscript; Wenkuan Wang, Tingting Xu, and Yanjie Wang conducted data collection and collation, as well as the creation and presentation of figures and tables; Xina Zhen, Wenxuan Man, and Shuo Gao participated in manuscript revision; Yonghui Yin was responsible for quality control and review of the article, assumed overall responsibility for the work, and oversaw the project management.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and informed consent
This research protocol was approved by the Medical Ethics Committee of the Affiliated Hospital of Shandong University of Traditional Chinese Medicine (approval number: (2023) Lunshen No. (053)-KY), and strictly followed the ethical guidelines of the Declaration of Helsinki. All participants signed a written informed consent form.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, X., Wang, W., Xu, T. et al. Alterations in gut microbiota and associated metabolites in patients with chronic fatigue syndrome. Sci Rep 15, 43681 (2025). https://doi.org/10.1038/s41598-025-27564-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-27564-y
