
Abstract
Alport syndrome (AS) is a genetic disorder characterized by progressive nephritis, hearing loss, and visual impairment. Quantitative analyses of hearing in AS are rare. We conducted a retrospective chart review of clinical features and longitudinal follow-up of patients with AS, including multiple evaluations of pure-tone audiograms in each individual and examination of the relationships between hearing loss and genetic variants. Twelve AS cases from eight Japanese families were studied: one family had an autosomal dominant COL4A4 variant and seven carried X-linked variants in COL4A5; all cases presented with moderate hearing loss. Progression of hearing loss was 0.25, 0.29, 0.57, and 2.60 dB HL/year at 500, 1000, 4000, and 8000 Hz, respectively. During follow-up (1–10 years), all families exhibited mid-frequency hearing loss. Hearing loss onset occurred later than renal symptoms in all patients with COL4A5 variants, and the COL4A4 variant did not cause renal symptoms. Hearing loss severity and age of onset appeared to vary depending on the characteristics of the genetic variants, indicating genotype–phenotype correlations; however, hearing decline was predominantly at high frequencies, regardless of genetic variation. These findings provide valuable insights into genotype–phenotype correlations in AS, particularly regarding hearing loss progression and characteristics.
Similar content being viewed by others
Introduction
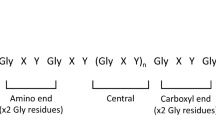
Alport syndrome (AS) is a nephropathy associated with hearing loss and eye disease caused by variants of genes encoding type IV collagen1,2. Type IV collagen molecules each comprise three helical α-chains, an N-terminal collagen domain of approximately 1400 residues with a Gly-X-Y repeat, and a C-terminal non-collagenous domain of approximately 230 residues3,4. Type IV collagen has subtypes based on its α3 (encoded by COL4A3), α4 (encoded by COL4A4), and α5 (encoded by COL4A5) chains. According to inheritance modes, AS is classified into the following types: X-linked, comprising 80% of cases and caused by COL4A5 variants; autosomal recessive, at 15% frequency, caused by COL4A3 or COL4A4 variants; and autosomal dominant, which makes up 5% of cases and is caused by COL4A3 or COL4A4 variants5,6,7. Type IV collagen monomers form reticular networks and are integral to the basilar membrane, sensory epithelium, blood vessels, Schwann cells, spiral ligament, spiral limbus, and Reissner’s membrane in the cochlea8,9.
Hearing impairment associated with AS typically becomes apparent in late childhood or early adolescence. During childhood, the degree of hearing loss tends to be mild to moderate, but severe hearing loss of ≥ 70 dB HL has been documented later, when renal failure becomes advanced10. Several studies have examined audiograms of individuals with AS. Some reports have indicated that hearing impairment in AS primarily affects the 2–4 kHz range11,12,13. In addition, various types of audiogram findings, including horizontal, mid-frequency, down-sloping, and low-frequency hearing loss were reported9.
The prevalence of hearing loss in AS varies depending on the mode of inheritance and specific genetic variants. In autosomal recessive AS caused by COL4A3 or COL4A4 variants, reported hearing loss prevalence ranges from 58.3% to 100%14,15,16, whereas in autosomal dominant AS associated with COL4A4 variants, hearing loss was reported in only 13.3% of cases13. In X-linked AS, associated with COL4A5 variants, hearing loss prevalence ranges from 16% to 82.5%12,17,18,19,20,21. Hearing loss progression in patients with AS is thought to result from the gradual degeneration of type IV collagen α3, α4, or α5 chains. The severity and age of onset of hearing loss are influenced by the type of genetic variant, with truncating variants leading to earlier onset and greater severity, while missense variants are associated with later onset and a milder severity of hearing loss22.
By 40 years old, approximately 80% of males and 10% of females with AS experience hearing loss20. In male patients with missense variants, around 60% develop hearing loss by age 30 years, whereas this percentage rises to 90% in those with truncating variants12. Similarly, a study by Zhang et al. on 87 pediatric male patients with X-linked AS found that indel, splice site, and nonsense variants, which are classified as having severe effects on proteins, were associated with earlier onset and more severe hearing loss, typically beginning between 5 and 15 years old23. Jais reported that 82.5% of 401 male patients with X-linked AS had hearing loss, and there was a clear correlation between variant type and age of onset12. Bekheirnia examined 681 male patients from 175 families with X-linked AS and found that those with splice or truncating variants were twice as likely to have progressive ocular symptoms and hearing loss as those with missense variants17. Additionally, variants located near the 5’ end of COL4A5 were more frequently associated with hearing loss and ocular involvement. Further, large deletions, small deletions, splice site variants, and truncating variants in COL4A5 were 4–20 times more likely to cause progressive hearing loss than missense variants.
Overall, the reported frequency and severity of hearing loss in AS vary widely, influenced by genetic variant type and the age at which hearing assessments were conducted. Renal symptoms also exhibit variability, and cases have been reported in which hearing loss occurs in the absence of renal impairment within affected families24. Although some studies have examined the association between causative genes and the long-term course of renal symptom progression in AS25,26,27,28,29,30,31, there has been no report describing long-term changes in hearing levels. In this study, we examined the associations between genotype and the long-term course of hearing loss in eight Japanese families diagnosed with AS.
Results
Genetic test results
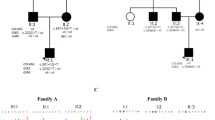
The pedigrees and genetic and clinical backgrounds of participants are presented in Fig. 1; Table 1, respectively. Mean age of the eight probands at the time of genetic testing was 30.3 (7–56) years. The genetic causes of AS identified in the probands of the eight families included in this study are detailed in Table 232,33,34,35,36,37,38. A pathological variant was found in COL4A4 in Family 1, while seven different variants in COL4A5 were detected in the remaining seven families (Families 2–8). Structure of the two genes and the positions of detected variants are shown in Supplementary Figure S1. Six of the identified variants were novel. Variant types detected included two nonsense changes in COL4A5 (families 5 and 7), one large exonic deletion in COL4A5 (Family 4), one frameshift variant in COL4A5 (Family 2), three missense changes affecting glycine (which has an important role in maintaining collagen structure) in COL4A5 (families 3, 6, and 8), and one in-frame deletion in COL4A4 (Family 1). The two nonsense variants (families 5 and 7), the frameshift variant (Family 2), and the large exonic deletion (Family 4) are predicted to produce a premature stop codon and undergo nonsense-mediated decay, resulting in the absence of protein synthesis, and considered to be truncating variants with high impacts on the protein (Table 232,33,34,35,36,37,38. Three types of glycine missense variants (families 3, 6, and 8) within the Gly-X-Y-repeat sequences in the collagenous domain and the in-frame deletion of six amino acids within the same domain in COL4A4 (Family 1) are expected to generate proteins with structural alterations, and considered as non-truncating variants with low impacts on the protein. The variants sites in COL4A4 and COL4A5 for each case are shown in Supplementary Figure S1.

Alport syndrome pedigrees. Black arrows indicate probands. Horizontal bars above circles and squares denote subjects who underwent genetic testing. The plus symbol (+) indicates subjects carrying a causative genetic variant. Black shading on the left side of circles and squares indicates hearing loss, while black shading on the right side indicates renal symptoms.
Clinical course
The probands of each of the eight families included in this study were diagnosed with AS between the ages of 7 and 56 years. Hearing loss was diagnosed between the ages of 6 and 50 years; none of the patients had hearing loss at birth (Table 1, Supplementary Figure S2). Patient follow-up duration ranged from 1 to 10 years (mean, 4.6 years). Pure-tone audiometry (PTA) results showed that all eight probands exhibited mid-frequency-dominant hearing loss in either one or both ears at some point during disease progression, regardless of the variants detected in either COL4A4 or COL4A5 (Fig. 2). All six male patients with COL4A5 variants exhibited renal symptoms before hearing loss onset (Table 1). The proband of Family 1 with the COL4A4 variant had no renal symptoms at last follow-up age 57 years. No cases of AS with ocular manifestations were observed in any of the families studied. These clinical courses are presented in Supplementary Figure S2.

Audiograms of families with Alport syndrome. The oldest and most recent audiograms of subjects with Alport syndrome are shown. The age at the time of audiometry is indicated below each audiogram. M, male; F, female; y, years.
Individual cases
Family 1 (COL4A4 c.1323_1340del): The proband of Family 1 was a 56-year-old male with an autosomal dominant COL4A4 variant. He became aware of hearing loss at around 50 years old and started wearing hearing aids aged 51 years. His renal function was normal at the time of the initial examination. Age-related hearing loss was suspected in both parents.
Family 2 (COL4A5 c.2246_2255del): The proband of Family 2 was a 40-year-old male who presented with hematuria at age 5 years and was subsequently diagnosed with AS. He began receiving renal dialysis at 17 years old. Hearing loss was first observed at 11 years old. The proband’s mother had nephritis and died of renal failure, but did not have hearing loss. His maternal grandmother also died of nephritis and similarly did not have hearing loss. The same variant was identified in his younger brother, younger brother’s daughter, and a female cousin. The younger brother had mid-frequency-dominant moderate hearing loss and nephritis, and started renal dialysis aged 30 years. The younger brother’s daughter had hematuria and proteinuria at 6 months of age, with normal distortion product otoacoustic emissions (DPOAE) at 4 years old. Hematuria was noted in the female cousin at 12 years old, but hearing was normal. The proband’s maternal aunt developed hearing loss and nephritis, started dialysis aged 41 years, and died at 60 years old.
Family 3 (COL4A5 c.866G > T): The proband of Family 3 was a 25-year-old male who had hematuria since the age of 5 years and was regularly followed up with urinalysis. He noted hearing loss at 20 years old, and the hearing loss progressed. The first PTA at age 25 years revealed moderate bilateral hearing loss. Increased proteinuria was detected at 36 years old, and he was diagnosed with renal dysfunction, with blood urea nitrogen (BUN) 43.3 mg/dL (normal range: 8.0–22.0 mg/dL), creatinine 3.94 mg/dL (normal range [males]: 0.61–1.04 mg/dL), and estimated glomerular filtration rate 15.5 mL/min/1.73 m2 (normal range: 63.1–134.7 ml/min/1.73 m2). A paternal male cousin had hearing loss but no renal dysfunction.
Family 4 (COL4A5 exon2_19del): The proband of Family 4 was a 15-year-old boy who was first diagnosed with proteinuria and hematuria aged 3 years. Renal biopsy revealed AS. He developed hearing loss at around 10 years old, but the hearing loss fluctuated. None of the other family members presented with hearing loss or renal symptoms.
Family 5 (COL4A5 c.4291 C > T): The proband of Family 5 was a 7-year-old boy who was found to have hematuria during a preschool physical examination aged 5 years. Hearing loss was noted during a school health examination at 6 years old. Renal function was within the normal range at 7 years old, with BUN 14 mg/dL and creatinine 0.26 mg/dL. His mother had hearing loss and chronic nephritis, while his sister had hearing loss alone.
Family 6 (COL4A5 c.1634G > A): The proband of Family 6 was an 11-year-old male with hematuria at 3 years old, plus proteinuria and large amounts of occult hematuria at 5 years old. At the time of genetic testing, age 11 years, his renal function was normal. At 8 years old, hearing loss was noted during a school health examination, and he started using hearing aids aged 11 years. His mother, who also harbored the same COL4A5 variant, had hematuria, and underwent a renal biopsy at 12 years old, which revealed a thin basement membrane, although her renal function was normal. Her hearing level was also within the normal range. The proband’s maternal grandmother had been on dialysis since her 60s.
Family 7 (COL4A5 c.3841 A > T): The proband of Family 7 was a 41-year-old female who had had hematuria since 6 years old. Aged 13 years, she underwent renal biopsy and was diagnosed with AS. Hearing loss was first noticed at 28 years old and progressed thereafter. Aged 41 years, she had moderate hearing loss along with hematuria and proteinuria. Renal function was mildly impaired. Her younger brother underwent kidney biopsy aged 6 years and was diagnosed with AS. He had been on dialysis since 17 years old and had experienced hearing loss since age 20 years. According to the proband, her brother’s hearing level was better than her own and he only used a hearing aid when necessary.
Family 8 (COL4A5 c.647G > C): The proband of Family 8 was a 47-year-old man who had hematuria aged 3 years, had been receiving dialysis since 35 years old, and had undergone kidney transplantation aged 45 years. Progressive hearing loss was noted at ages 45 and 47 years. His mother, sister, and the other 4 family members had renal symptoms, but no hearing loss. A male cousin had experienced renal failure and was preparing for a kidney transplant.
Longitudinal follow-up of hearing levels
To illustrate the long-term changes in hearing levels of the patients with AS, hearing levels at each frequency were plotted for individuals according to their age in years (Fig. 3). While hearing in the low and mid frequencies (500 and 1000 Hz) was relatively preserved, hearing at higher frequencies (4000 and 8000 Hz) tended to deteriorate gradually. The mean rates of changes in hearing loss were 0.25, 0.29, 0.57, and 2.60 dB HL/year at 500, 1000, 4000, and 8000 Hz, respectively.

Time-dependent changes in hearing levels of eight cases with Alport syndrome. Hearing in the low and mid frequencies (500 and 1000 Hz) was relatively preserved, whereas that in high frequencies (4000 and 8000 Hz) gradually deteriorated over time. Mean rate of hearing loss (dB HL/year) is shown above each plot. y/o, years old.
Genotype–phenotype correlations
To evaluate the correlation between genotypes and hearing phenotypes, we investigated the hearing levels of probands and the ages of hearing loss onset. The genotype of each patient was classified based on the variant evaluation described in the subsection “Genetic test results”: truncating variants were categorized as having a presumed high impact on protein, whereas non-truncating variants were categorized as having a presumed low impact. Then the relationship between these genotype classifications and both the hearing levels and the ages at onset of hearing loss were examined (Fig. 4).

Associations between genotypes and hearing phenotypes in eight cases with Alport syndrome. a Correlation between the presumed impacts of genetic variants on the protein and hearing levels measured at the time of genetic testing. b Correlation between the presumed impacts of genetic variants on the protein and age of hearing loss onset.
In three of the seven families with COL4A5 variants (families 3, 6, and 8), the variants (c.866G > T, c.1634G > A, and c.647G > C), all of which affected glycine residues in the Gly-X-Y repeat of the collagenous domain, were predicted to have low impacts (Table 232,33,34,35,36,37,38. The onset of hearing loss in these probands varied, occurring at ages 8, 20, and 45 years (Fig. 4b).
In four families, the variants in COL4A5 were predicted to have high impacts (Table 232,33,34,35,36,37,38. Three male cases (families 2, 4, and 5) harbored a frameshift variant (c.2246_2255del), an exonic deletion (exon2_19del), and a nonsense variant (c.4291 C > T), respectively, while a female case (Family 7) harbored a nonsense variant (c.3841 A > T). All the three males had moderate level hearing loss at the time of genetic testing and developed hearing loss during childhood (6–11 years old) (Fig. 4a, b). The female patient also had moderate level hearing loss at the time of genetic testing and developed hearing loss at 28 years old. While hearing levels were similar in both males and females, the age of onset was higher in the female patient (Fig. 4a, b).
In cases with COL4A5 variants, the variants with high impacts tended to result in earlier onset hearing loss, with higher levels of hearing loss at the time of genetic testing. The variants with low impacts were associated with less pronounced hearing loss and later hearing loss onset. As the predicted impacts of variants on the protein increased, hearing ability tended to decline, suggesting a genotype–phenotype correlation in the COL4A5-related AS hearing phenotype. COL4A4 variant c.1323_1340del was predicted to have low impact and identified in a male proband (family 1). The proband presented with late onset hearing loss (age 50 years) and hearing loss at the time of genetic testing was moderate (Fig. 4a, b).
Discussion
We describe identification of eight causal variants in COL4A genes and evaluation of their relationships with hearing function in AS families. An autosomal dominant COL4A4 variant and seven different X-linked COL4A5 variants were detected in eight families. Mid-frequency dominant hearing loss was found in all families during the study period. The longest follow-up duration was 10.7 years (for Family 5, in which a COL4A5 variant was found in the proband), and all the patients maintained normal, mild or moderate hearing loss over follow-up, with hearing impairment progression mainly detected at high frequencies. Hence, identified hearing impairments were predominantly at mid-to-high frequency and the changes were more prominent at higher frequencies than those in mid-to-lower frequencies.
There are no previous reports of quantitative examination of the association between COL4A4 variants and hearing loss, presumably because of the limited number of cases with hearing loss carrying COL4A4 variants. In the present study, the patient with an autosomal dominant COL4A4 variant exhibited hearing characteristics similar to those of patients with COL4A5 variants.
Zhang et al. reported the hearing levels of 87 young male patients with AS, including seven adult cases, carrying COL4A5 variants and found that they had bilateral symmetric sensorineural hearing loss23. In that study, only one case was followed up for longer than 3 years, and hearing loss in the patient cohort was predominantly observed at mid-frequencies, with a gradual decline at high frequencies. Based on pure-tone threshold distributions, they speculated that hearing loss rate during the school years of study subjects would be approximately 5 dB HL/year. Our data, including long-term longitudinal follow-up of mainly adult cases, indicated mean long-term changes in hearing loss values of only 0.25, 0.29, 0.57, and 2.60 dB HL/year at 500, 1000, 4000, and 8000 Hz, respectively. Overall, hearing impairment was dominant at younger ages in AS and progression slowed in adulthood, regardless of genotype. Further, hearing level decline in patients with AS was predominantly at high frequencies.
A discrepancy between renal symptoms and hearing loss phenotype is established in AS, with renal symptoms generally preceding hearing loss in male patients with COL4A5 variants39. However, studies systematically examined the presence and onset of each phenotype are lacking. In the present study, we observed that all six male patients with COL4A5 variants first exhibited renal symptoms, followed by subsequent development of hearing loss. These results suggest that regular hearing tests should be considered for patients with AS once they present with renal symptoms.
Histological studies of the cochlea in individuals with AS have revealed degeneration of hair cells, stria vascularis40, spiral ligament41, and spiral ganglion cells. Loss of hair cells and spiral ganglion cells was observed only in the basal turn, which is responsible for high-frequency sounds9. Further, separation of the basilar and basement membranes has been specifically observed in patients with AS9. Conversely, type IV collagen α1, α3, and α5 chains are broadly distributed in the human cochlea, including in structural components, such as the basement membrane of the sensory epithelium, spiral ligament, spiral limbus, and Reissner’s membrane, as well as in other parts, such as Schwann cells and blood vessels8,9.
In our study, despite the detection of predominantly mid-frequency hearing loss at a younger age, hearing loss progression tended to more prominently affect high frequencies. According to previous histopathological findings in cases with AS, the most notable feature is detachment of the basilar membrane and basement membrane immediately under the organ of Corti, which is believed to be associated with chemical changes resulting from collagen abnormalities; however, this phenomenon is independent of cochlear position, which does not explain the mid-frequency deficits and progression of high-frequency hearing loss. Histologically, the characteristic tissue changes at the basal turn (the region associated with high-frequency hearing) in patients with AS are primarily loss of hair cells and spiral ganglion cells42. Zehnder et al. hypothesized that normal vibration of the organ of Corti contributes to potassium circulation and that impaired potassium recycling may be responsible for sensorineural hearing loss in AS8, although no physiological studies have been reported to support this concept. In a histopathological study of patients with AS conducted by Merchant et al., cellular infiltration was observed in Nuel’s space9. If these nonstructural pathologies precede threshold elevation and contribute to hearing loss progression in AS, they hold promise as targets for drug development. More detailed investigations using animal models are warranted to address these issues.
All missense variants of COL4A5 identified in this study involved glycine. Molecular studies have revealed that the collagen fibers consist of three chains wound together in a tight triple helix, where every third amino acid is glycine. The three chains are connected by hydrogen bonds, with donor hydrogens are in glycine residues and the acceptors in other amino acids; hence, the Gly-X-Y structure is fundamental to collagen structure. Our clinical findings indicate a strong relationship between hearing loss symptoms and variants affecting glycine, likely attributable to the molecular biology of the collagen structure.
The four variants predicted to have high impacts on the protein (three truncating variants and one large exonic deletion) were associated with ages of hearing loss onset of 6, 9, 11 years in male probands, and 28 years in a female, suggesting that variants with high impacts on the protein are associated with earlier onset hearing loss, similar to previous results from pediatric patients reported by Zhang et al.23. In the female patient who developed hearing loss at 28 years old, it is anticipated that functional collagen 4 α5-chain proteins would be produced from the unaffected allele, which could avoid X chromosome inactivation in females, resulting in comparatively milder symptoms in the affected individual than those observed in male patients. One possible underlying mechanism of various ages of onset is that collagen damage needs to progress to some degree before symptoms appear, even in the presence of the genetic variants, and that the rate of the damage progression may differ depending on the effect of genetic variants on the protein.
The findings of the current longitudinal follow-up study further support and provide evidence for the previous suggestion of genotype–phenotype correlations in AS. As shown in Fig. 4, our findings suggest a possible trend toward a relationship between hearing phenotypes and genotypes, in terms of both hearing levels and hearing loss onset; genetic variants with high impacts on the protein were associated with more severe hearing loss and earlier age of hearing loss onset. Moreover, our study provides additional data on hearing loss progression rate and frequency-specific hearing loss. Specifically, our findings demonstrate that hearing loss begins in the mid-to-high frequencies during childhood in individuals with AS and progresses, but in adulthood, high-frequency hearing loss advances more rapidly. While the sample size is limited, these findings are valuable for the clinical management of AS, providing insights into hearing loss progression that can guide diagnosis, monitoring, and intervention strategies.
The primary limitation of this study was sample size. Because of the extremely low incidence of AS cases with hearing loss, the number of patients included was insufficient to ensure comprehensive results. For example, the genotype–phenotype correlation data would be strengthened by statistical analysis, but the limited sample size made such analysis infeasible. In particular, hearing phenotypes associated with the rare variant in COL4A4 require confirmation in additional patients; although no reported cases of hearing loss have been directly associated with this variant, it is an established cause of autosomal dominant AS32,33,34,35,36,37. Furthermore, there was a report of cases with autosomal dominant AS presenting with hearing loss as the sole symptom24. It is also known that penetrance of renal symptoms is incomplete in cases with autosomal dominant AS39. These findings support that the hearing phenotype detected in the patient in the present study is associated with the COL4A4 variant. Future studies should aim to include a larger cohort to facilitate quantitative validation with statistical analysis, and to expand on our findings and eventually establish a global phenotype registry with broad applications for both research and clinical purposes.
In the present study, the long-term clinical course of hearing loss in patients diagnosed with AS was examined in relation to causal genetic variants. AS caused by COL4A4 and COL4A5 variants typically presented as a mid-frequency-dominant hearing loss. Most patients presented with moderate hearing loss at earlier ages, which then progressed more rapidly at higher frequencies. Given the hearing loss progression, implementation of auditory compensations, specifically adjustments to hearing aids, is necessary. Long-term, regular follow-up is important to manage the progression of hearing loss in patients with AS.
Methods
Ethics
This study was approved by the Ethics Committees of the National Hospital Organization (R3-NHOkankaku-02) and National Hospital Organization Tokyo Medical Center (Application No. R21-053), and Keio University Hospital (Application No. 2015-0235-2), and was conducted in accordance with the tenets of the Declaration of Helsinki. Genetic testing was performed after obtaining informed consent from the patients or, in the case of children, their parents.
Case enrolment
Individuals who underwent hearing loss gene testing at the Division of Hearing and Balance Research, National Institute of Sensory Organs, National Hospital Organization Tokyo Medical Center, Tokyo, Japan, between 2002 and 2018 were initially enrolled in the study. Eight probands and four of their family members with variants in either COL4A4 or COL4A5, who were diagnosed with AS, were further investigated by retrospective chart review.
Clinical evaluation
For clinical evaluation, a history of the present illness and a family history were obtained for each patient and their family members. Laboratory findings were obtained from medical records. Standard PTA was available for all patients, and speech intelligibility and DPOAE were available for a subset of cases. Hearing loss severity was determined according to the criteria proposed by the GENDEF study group43, with a pure-tone average over 0.5, 1, 2, and 4 kHz in the better-hearing ear defined as follows: < 20 dB HL normal, 20–40 dB HL mild hearing loss, 41–70 dB HL moderate hearing loss, 71–95 dB HL severe hearing loss, and > 95 dB HL profound hearing loss.
Genetic analysis
For genetic analysis, genomic DNA was extracted from peripheral blood leukocytes using the Genomix DNA extraction kit (Biologica Co., Nagoya, Japan). First, the GJB2 and mitochondrial m.1555 A > G and m.3243 A > G variants, which are common causative genes for hereditary hearing loss in the Japanese population, were analyzed according to previous reports44, and no variants were found. For genetic screening of COL4A5 in six probands (III-3 of Family 2, III-1 of Family 3, III-1 of Family 4, IV-2 of Family 5, IV-1 of Family 6, III-1 of Family 7), the coding region (exons 1–53, based on NM_0033380.3) was analyzed by Sanger sequencing using the polymerase chain reaction (PCR) primer sets described in Supplementary Table S1 because targeted resequencing of hearing loss genes was not available at the time of their genetic analysis. PCR conditions were as follows: denaturation at 95 °C for 15 min; 35 cycles each consisting of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s; followed by 10 min at 72 °C. In male proband of Family 4, no amplification of exons 2–19 was observed, whereas exons 1 and 20–53 were normally amplified, indicating a hemizygous deletion of exons 2–19 in COL4A5 which is located on the X chromosome and present as a single copy in males. Control amplifications for exons 2–19 were performed under the same condition.
For genetic analysis of the two remaining probands (III-1 of Family 1, proband of Family 1; III-2 of Family 8, proband of Family 8), genomic DNA was subjected to targeted resequencing of 154 hearing loss genes using NextSeq 500 (Illumina, CA, USA) against the human reference sequence (GRCh37.p13), and the resulting data analyzed as previously described45. For segregation analysis, genomic DNA of family members other than the probands were analyzed, if available, by Sanger sequencing after PCR for the corresponding exons of the variants that were found in the probands. The pathogenicity of each genetic variant was evaluated according to the ACMG/AMP 2015 Variant Interpretation Guidelines46. The population frequency of copy number variants was assessed using the Database of Genomic Variants47 and gnomAD SVs48. All variants in the study are described based on the canonical transcripts, NM_000495.5 and NM_000092.5 for COL4A5 and COL4A4, respectively. All analyses in the study were conducted using GRCh37 as a human genome reference sequence.
Data availability
All variant data have been deposited in MGeND ( [https://mgend.med.kyoto-u.ac.jp/](https:/mgend.med.kyoto-u.ac.jp) , accession number: MGS000093.1, open to public on November 7/2025) and are presented in the manuscript and Tables. The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Gubler, M. et al. Alport’s syndrome. A report of 58 cases and a review of the literature. Am. J. Med. 70 (3), 493–505 (1981).
Habib, R. et al. Alport’s syndrome: experience at Hôpital Necker. Kidney Int. Suppl. 11, S20–S28 (1982).
Hudson, B. G. The molecular basis of goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J. Am. Soc. Nephrol. 15 (10), 2514–2527 (2004).
Hudson, B. G., Reeders, S. T., Tryggvason, K. & Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of goodpasture and Alport syndromes and diffuse leiomyomatosis. Biol. Chem. 268 (35), 26033–26036 (1993).
Barker, D. F. et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248 (4960), 1224–1227 (1990).
Mochizuki, T. et al. Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat. Genet. 8 (1), 77–81 (1994).
Feingold, J. et al. Genetic heterogeneity of Alport syndrome. Kidney Int. 27 (4), 672–677 (1985).
Zehnder, A. F. et al. Distribution of type IV collagen in the cochlea in Alport syndrome. Arch. Otolaryngol. Head Neck Surg. 131 (11), 1007–1013 (2005).
Merchant, S. N. et al. Temporal bone histopathology in Alport syndrome. Laryngoscope 114 (9), 1609–1618 (2004).
Moon, I. S., Bang, M. Y., Shim, D. B., Shin, S. H. & Choi, J. Y. Severe to profound hearing loss in patients with progressed alport’s syndrome. Acta Otolaryngol. 129 (9), 982–987 (2009).
Kashtan, C. E. & Michael, A. F. Alport syndrome. Kidney Int. 50 (5), 1445–1463 (1996).
Jais, J. P. et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J. Am. Soc. Nephrol. 11 (4), 649–657 (2000).
Marcocci, E. et al. Autosomal dominant Alport syndrome: molecular analysis of the COL4A4 gene and clinical outcome. Nephrol. Dial Transpl. 24 (5), 1464–1471 (2009).
Storey, H., Savige, J., Sivakumar, V., Abbs, S. & Flinter, F. A. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J. Am. Soc. Nephrol. 24 (12), 1945–1954 (2013).
Heidet, L. et al. Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J. Am. Soc. Nephrol. 12 (1), 97–106 (2001).
Zhang, Y. et al. Genotype-phenotype correlations in 17 Chinese patients with autosomal recessive Alport syndrome. Am. J. Med. Genet. A. 158A (9), 2188–2193 (2012).
Bekheirnia, M. R. et al. Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 21 (5), 876–883 (2010).
Alves, F. R. A. & Ribeiro, F. A. Q. Clinical data and hearing of individuals with Alport syndrome. Braz J. Otorhinolaryngol. 74 (6), 807–814 (2008).
Dai, Y. et al. Clinical and genetic mapping of X chromosome in the X-linked dominant inherited alport’s syndrome. Saudi J. Kidney Dis. Transpl. 19 (5), 767–774 (2008).
Jais, J. P. et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a European community Alport syndrome concerted action study. J. Am. Soc. Nephrol. 14 (10), 2603–2610 (2003).
Gross, O., Netzer, K. O., Lambrecht, R., Seibold, S. & Weber, M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol. Dial Transpl. 17 (7), 1218–1227 (2002).
Nabais Sá, M. J. et al. Collagen type IV-related nephropathies in portugal: pathogenic COL4A5 mutations and clinical characterization of 22 families. Clin. Genet. 88 (5), 462–467 (2015).
Zhang, X. et al. X-linked Alport syndrome: pathogenic variant features and further auditory genotype-phenotype correlations in males. Orphanet J. Rare Dis. 13 (1), 229 (2018).
Rosado, C., Bueno, E., Fraile, P., García-Cosmes, P. & González-Sarmiento, R. A new mutation in the COL4A3 gene responsible for autosomal dominant Alport syndrome, which only generates hearing loss in some carriers. Eur. J. Med. Genet. 58 (1), 35–38 (2015).
Hertz, J. M., Thomassen, M., Storey, H. & Flinter, F. Clinical utility gene card for: Alport syndrome. Eur J. Hum. Genet ;20(6):. (2012).
Yamamura, T. et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. 98 (6), 1605–1614 (2020).
Gillion, V. et al. Genotype and outcome after kidney transplantation in Alport syndrome. Kidney Int. Rep. 3 (3), 652–660 (2018).
Kamura, M. et al. Trimerization and Genotype-Phenotype correlation of COL4A5 mutants in Alport syndrome. Kidney Int. Rep. 5 (5), 718–726 (2020).
Liapis, H. & Jain, S. The interface of genetics with pathology in Alport nephritis. J. Am. Soc. Nephrol. 24 (12), 1925–1927 (2013).
Savige, J. et al. X-Linked and autosomal recessive Alport syndrome: pathogenic variant features and further Genotype-Phenotype correlations. PLoS One. 11 (9), e0161802 (2016).
Shang, S. et al. Genotype-phenotype correlation and prognostic impact in Chinese patients with Alport syndrome. Mol. Genet. Genomic Med. 7 (7), e00741 (2019).
Boye, E. et al. Determination of the genomic structure of the COL4A4 gene and of novel mutations causing autosomal recessive Alport syndrome. Am. J. Hum. Genet. 63 (5), 1329–1340 (1998).
Nabais Sá, M. J. et al. Collagen type IV-related nephropathies in portugal: pathogenic COL4A3 and COL4A4 mutations and clinical characterization of 25 families. Clin. Gene. 88 (5), 456–461 (2015).
Kamiyoshi, N. et al. Genetic, Clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin. J. Am. Soc. Nephrol. 11 (8), 1441–1449 (2016).
Oh, J. et al. Clinical application of a phenotype-based NGS panel for differential diagnosis of inherited kidney disease and beyond. Clin. Genet. 99 (2), 236–224 (2021).
Zhu, F. et al. Identification of a novel COL4A4 variant in Compound-Heterozygous state in a patient with Alport syndrome and histological findings similar to focal segmental glomerulosclerosis (FSGS). Front Genet. 2019 Jan 28:9748 .
Park, E. et al. Genetic study in Korean pediatric patients with Steroid-Resistant nephrotic syndrome or focal segmental glomerulosclerosis. J. Clin. Med. 9 (6), 2013 (2020).
Guo, C. et al. Severe Alport phenotype in a woman with two missense mutations in the same COL4A5 gene and preponderant inactivation of the X chromosome carrying the normal allele. J. Clin. Invest. 95 (4), 1832–1837 (1995).
Nozu, K., Yamamura, T., Horinouchi, T. & Alport Syndrome Aug 28 [Updated 2025 Feb 27]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. (2001). Available from: https://www.ncbi.nlm.nih.gov/books/NBK1207/
Gregg, J. B. & Becker, S. F. Concomitant progressive deafness, chronic nephritis, and ocular lens disease. Arch. Ophthalmol. 69, 293–299 (1963).
Winter, L. E., Cram, B. M. & Banovetz, J. D. Hearing loss in hereditary renal disease. Arch. Otolaryngol. 88 (3), 238–241 (1968).
Ungar, O. J., Nadol, J. B. & Santos, F. Temporal bone histopathology of X-linked inherited Alport syndrome. Laryngoscope Investig Otolaryngol. 3 (4), 311–314 (2018).
Mazzoli, M. et al. Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment. Audiol. Med. 1, 148–150 (2003).
Yamamoto, N. et al. Prevalence of TECTA mutation in patients with mid-frequency sensorineural hearing loss. Orphanet J. Rare Dis. 12 (1), 157 (2017).
Mutai, H. et al. Whole exome analysis of patients in Japan with hearing loss reveals high heterogeneity among responsible and novel candidate genes. Orphanet J. Rare Dis. 17 (1), 114 (2022).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424 (2015).
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L. & Scherer, S. W. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42 (Database issue), D986–D992 (2014).
Collins, R. L. et al. A structural variation reference for medical and population genetics [published correction appears in Nature. ;590(7846):E55. (2021). https://doi.org/10.1038/s41586-020-03176-6]. Nature. 2020;581(7809):444–451.
Acknowledgements
We are grateful to all the study participants. This research was supported by a Grant-in-Aid for Clinical Research from the National Hospital Organization of Japan (R3-NHO (kankakuki)-02) to TM, Japan Agency for Medical Research and Development (AMED) under Grant Numbers 23gn010063h0002 and 23ek0109634h0001 to TM, and Japan Society for the Promotion of Science (KAKENHI) to MF (24H00648), and SM (21K09660). The Funders played no role in the research.
Author information
Authors and Affiliations
Contributions
TM designed the study. HM, KN and TM contributed to genomic sequencing analysis and validation of variants. HM, KN, and TM contributed to variant interpretation. SK, HS, MH, NW, NA, SA, MF, and TM obtained clinical data and samples. SM and TM analyzed data. SM, MF and TM drafted the manuscript. MF and TM finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Matsuzaki, S., Nara, K., Mutai, H. et al. Clinical features of hearing loss and genotype–phenotype correlations in Alport syndrome caused by COL4A4 or COL4A5 variants. Sci Rep 15, 43803 (2025). https://doi.org/10.1038/s41598-025-27772-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-27772-6
