
Abstract
The species is often described as the basic unit of biology, yet defining what constitutes a species has been a long-standing challenge. The advent of modern molecular techniques in conjunction with advanced analytical approaches now provide new opportunities for the robust and repeatable delineation of taxa from previously difficult to assess species complexes. Using marine mussels of the genus Mytilus (smooth-shelled blue mussels of the globally distributed Mytilus edulis species complex) we tested regionally differentiated putative taxa from the Northern and Southern hemispheres using a Bayesian species delimitation model that infers species trees. Using a multilocus panel of 54 single nucleotide polymorphic loci (SNPs) we tested four alternative hypotheses against the hypothesis of the currently recognised taxonomy to better understand the evolutionary history and the contemporary species of this complex. Only one model provided a better fit than the contemporary taxonomy model: this best fit model included the three reference Northern hemisphere taxa (M. edulis, M. galloprovincialis, M. trossulus) plus M. chilensis (Chile), M. platensis (Argentina), M. planulatus (Australia) and the newly recognised M. aoteanus (New Zealand). Phylogenetic reconstruction based on our nuclear DNA-based SNP data suggests that M. trossulus is the oldest of the modern smooth-shelled blue mussels, that a first migration event from the north to the south occurred that gave rise to M. platensis and M. chilensis in South America, subsequently that M. edulis and M. galloprovincialis diverged in the Northern hemisphere, and that subsequently again there was a second migration event from the north that gave rise to M. planulatus in Australia and M. aoteanus in New Zealand. Our findings provide very strong support for earlier mitochondrial DNA-based phylogenetic findings for globally distributed blue mussels and also help to clarify uncertainty about the number of north-to-south migration events that gave rise to important Mytilus speciation events in South America and in Australasia.
Similar content being viewed by others

Introduction
Species delimitation is a crucial task in taxonomic studies that involves identifying and defining distinct species within a taxonomic group1. The process of species delimitation plays a critical role in resolving taxonomic problems and understanding the biodiversity and genetic diversity within species complexes2,3. One such taxonomic problem is the delimitation of species within the global complex of marine smooth-shelled blue mussels (family Mytilidae), specifically the Mytilus edulis species complex4,5. The existence of this mussel complex has long been recognised in the Northern hemisphere6,7,8,9, but the taxonomic status of a number of different regionally distributed blue mussels in the Southern hemisphere is much less clear and requires further attention4.
Smooth-shelled blue mussels of the genus Mytilus are anti-tropically distributed bivalve molluscs9 that hold significant ecological10,11 and economic importance12,13,14. However, whilst the distribution of smooth-shelled blue mussels throughout the world at regional scales is well established (reviewed by Gardner et al.4) the taxonomic classification of the Southern hemisphere mussels (e.g., Chile, Argentina, New Zealand, Australia, Southern Ocean islands including the Falkland Islands, Kerguelen Islands, Campbell Islands, Auckland Islands) is still not entirely clear, and at times has been hotly debated4,15. The application of species delimitation methods to the analysis of genetic data has proven invaluable in resolving taxonomic issues within the Mytilus edulis species complex. By using both nuclear and mitochondrial genetic data, researchers have been able to assess species boundaries and recognise distinct species within the complex5. Studies conducted during the last 20 years that have focussed on the description and/or delimitation of mussels in South America5,15,16,17, Australia18,19,20, New Zealand21 and islands of the Southern Ocean22,23 have contributed to our understanding of the taxonomic diversity and genetic integrity of native populations, as well as the threats posed by climate change, introduced species and hybridisation4,16,21,24,25,26.
Single nucleotide polymorphisms (SNPs) have emerged as powerful markers for species delimitation in recent years27. SNPs are highly informative genetic markers that can provide valuable insights into genetic diversity and population structure28,29. They are advantageous because they can be easily identified and genotyped with high throughput technologies30. Furthermore, the use of large sets of informative markers, such as SNPs, allows for accurate and robust delimitation of closely related species27. SNPs have been applied in species delimitation studies, particularly in taxonomically challenging groups that exhibit cryptic speciation processes. For example, species delimitation analyses using genome-wide SNPs have been conducted for various taxa, including turtles27, plants31, fishes32, sponges33, butterflies34, rodents35, and frogs36. These studies have demonstrated the utility and effectiveness of SNP-based approaches for resolving taxonomic problems and improving the understanding of species boundaries.
Whilst it is difficult to estimate the species tree for a group of organisms that has recently diverged, such as the Mytilus edulis complex, the use of a coalescent model is the most informative and robust methodological approach37,38. Coalescence analyses have been implemented in various software packages and can use different types of input data (sequence data, polymorphic sites, etc.). However, only a small number of software packages allow estimates of species trees directly from SNP data. This approach has the advantage that there is no recombination within a locus because each locus is a single SNP. In addition, SNP data are the most robust markers of the genome, therefore they represent particularly well the genetic differences between species39. However, it would be extremely demanding, from a computational point of view, to estimate the individual genealogy of each SNP locus, and even if it could be done, such locus-specific results would be of low resolution. To address this, some computational tools (e.g. SNAPP) have adopted a strategy that mathematically integrates all possible genealogies for each SNP locus. This method allows for estimating the probability of allele frequency changes between ancestor-descendant nodes40, making it a highly efficient approach—surpassing other methods—for species delimitation.
Leaché et al.41 addressed the problem of species delimitation using estimation of marginal probabilities. This method requires the a priori specification of models that incorporate the candidate species to be evaluated, allowing for the estimation of each model’s marginal likelihood. Bayes factors are then employed to compare alternative species delimitation models that differ in the number of inferred species—an approach referred to as Bayes Factor Delimitation (BFD). BFD relies on path sampling to estimate the marginal likelihood (ML) of population divergence models directly from SNP data, without the need to integrate over gene trees. This approach circumvents the requirement of specifying a guide tree to represent the true species relationships, a step that often introduces potential sources of error42.
The objective of the present paper was therefore to test for SNP-based global species boundaries in regional representatives of the Mytilus edulis species complex from the Southern hemisphere in the context of accepted reference species from the Northern hemisphere. This approach utilises an hypothesis testing framework that permits the testing of multiple different options (putative species designations or groupings) based on geographically differentiated taxa - that is, putatively different species.
Materials and methods
Samples
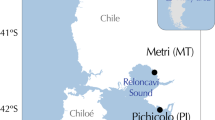
A total of 106 mussels was randomly collected from 21 sampling sites across 12 countries (Table 1; Fig. 1). Details of the collection methods, dates of collection, storage methods and the sampling sites are described for mussels from Argentina by Zbawicka et al.17, from Australia by Zbawicka et al.20,22, from Chile by Larraín et al.15, and from New Zealand by Gardner et al.21. Reference Northern hemisphere mussels (M. edulis, M. galloprovincialis, M. trossulus) were collected from Canada, France, Italy, the Netherlands, Northern Ireland (UK), Ukraine, Spain and the United States. The reference Northern hemisphere mussels were collected from localities at which earlier work had shown ‘pure’ reference species to exist (reviewed in Gardner et al.4). Mussels from reference populations of each pure Northern Hemisphere species were included to encompass the greatest possible intraspecific variation. All these mussels, from both the Northern and Southern hemispheres, have previously been used in regional analyses investigating genetic structure based on SNP marker variation15,17,20,21,22.

Map of the location of the reference and sample sites. The colour of the codes represents the current taxonomy according to WoRMS of the species of the Mytilus edulis species complex (red = M. galloprovincialis; black = M. trossulus, purple = M. edulis, light green = M. chilensis; dark green = M. platensis, light blue = M. planulatus). The map was created using the rworldxtra v1.0 package running on the R project43.
DNA extraction and SNP calling
DNA extraction and quantification methods have been described by Gardner et al.21, Larraín et al.15, and Zbawicka et al.17,20,22. Samples were genotyped using the Sequenom MassARRAY iPLEX platform44. In total, 54 independent nuclear SNP loci were identified that were taxonomically informative at the global scale15,17,20,21,22, with sequences deposited in GenBank (KT713378–82; HQ257471; KJ871039–57; KT713368–74). Linkage disequilibrium and assignment to chromosomes was checked and reported in previous studies21,45,46. It is these 54 SNPs for the 106 mussels from 12 countries and 21 sampling sites that are employed in the analyses described below.
Bayes factor delimitation (BFD)
We performed the BFD analysis using SNAPP40, implemented as a plugin in BEAST v.2.447. To define the candidate species models, previously published information was used (e.g. 5,15,17,20,21,22). We tested six different models, including the currently accepted taxonomy (model A) as obtained from the World Register of Marine Species (WoRMS - https://www.marinespecies.org/), which was contrasted with five alternative models (models B-E) (Table 2). All six models included the accepted Northern hemisphere species including their intraspecific variability (M. edulis, M. galloprovincialis, M. trossulus) and models B to E had, variously, 1, 2, 3 or 4 additional Southern hemisphere putative species combinations that were selected to reflect possible different speciation scenarios (reviewed by Gardner et al.4). We estimated the marginal likelihood of each model by running path sampling with 24 steps (15,000 MCMC steps, 1500 pre-burn in steps). The Bayes Factor (BF) test statistic (2*ln(BF)) was then used to compare the strength of support according to the framework of Kass & Raftery48. A positive BF test statistic (2 × loge) reflects evidence in favour of model 1 (the first or null model being tested), whereas a negative BF value is considered as evidence favouring model 2 (the alternative model). The BF scale is as follows: 0 < 2×logeBF<2 is not worth more than a bare mention, 2 < 2 × loge BF < 6 is positive evidence, 6 < 2 × loge BF < 10 is strong support, and 2 × loge BF > 10 is decisive.
Species trees
Species tree reconstruction was performed using the Bayesian package SNAPP, implemented in BEAST v2.447. This method is specifically designed to infer species trees directly from biallelic SNP data, without the need to reconstruct gene trees for individual loci. Instead, SNAPP estimates the species tree by mathematically integrating over all possible genealogies for each SNP49. The tree was reconstructed based on the species’ assignments defined by the selected species group (model B). Independent runs were conducted using a Markov Chain Monte Carlo (MCMC) chain length of 4,000,000 iterations, with samples taken every 100 steps, resulting in a total of 40,000 samples per analysis. The first 20% of the samples (8,000) were discarded as burn-in. Convergence of Bayesian analyses was verified in Tracer v1.5 by examining trace plots and ensuring effective sample size (ESS) values > 200 for all estimated parameters (posterior, likelihood, θ, λ, tree height). Two independent SNAPP runs yielded consistent posterior estimates, indicating convergence. The output files were compared and combined using LogCombiner v1.8.1. The remaining trees were summarised using TreeAnnotator (included with the BEAST package) to obtain a consensus species tree with posterior probabilities assigned to each node. The tree was rooted according to the Yule speciation model, taking into account the ancestry of Mytilus trossulus9,50. All analyses were carried out on the CIPRES Science Gateway (https://www.phylo.org), which facilitated the computational demands of the SNAPP model—particularly important given the high resource requirements associated with large SNP datasets.
Results
In total, five different models covering a range of possible speciation scenarios were tested. The first model (model A) represented the current taxonomic situation as described by WoRMS, with the three reference Northern hemisphere species plus three putative Southern hemisphere species. Subsequent models (B to E) tested various combinations of putative Southern hemisphere species in conjunction with the three reference Northern hemisphere species. The Bayes Factor (BF) score for each model was compared to the BF score for model A. Three of the alternative models (C, D and E) had BF scores larger than that of model A, meaning that they were less well supported than model A. However, model B [the three Northern hemisphere reference species plus M. chilensis (Chile), M. platensis (Argentina), M. planulatus (Australia) and M. aoteanus (New Zealand)] had a lower BF score than model A, making model B the best fit model overall (Table 3). The model with the lowest level of support was that for the three reference Northern hemisphere species plus all Southern hemisphere mussels in one group (i.e., model E with four species).
BEAST was used to construct the species tree of the best supported model (Fig. 2). This tree revealed the early origin of M. trossulus, followed by the origin of the South American mussels (M. chilensis and M. platensis), then the separation of North Atlantic M. edulis from Mediterranean Sea M. galloprovincialis, and finally the divergence between the Australasian blue mussels (M. planulatus in Australia and M. aoteanus in New Zealand). Bootstrap support values for all of these branching events were 100%, except for the split between M. edulis and M. galloprovincialis, which was 81% (Fig. 2).

Species tree based on the best fit model (model B) from the Mytilus edulis species complex estimated with 54 SNP loci. Posterior probabilities are shown on branches. FigTree v1.4.2 software51 was used to visualize the tree.
Discussion
The taxonomy of the smooth shelled blue mussels of the Mytilus edulis species complex has been uncertain for decades52,53,54 and has been revised following the recognition of a new species or as the status of, for example, a subspecies is no longer accepted (e.g. 4,5,55). Recently, modern genetic markers such as single nucleotide polymorphisms (SNPs) and advances in statistical analyses have helped resolve longstanding questions about species boundaries within various species complexes. Here, we apply a Bayesian framework to test hypotheses about species boundaries of putative smooth-shelled blue mussel species at the global level, and then we examine the phylogenetic history of these mussels. We find evidence of the need to recognise a new species and also our nuclear DNA-based testing confirms earlier mitochondrial DNA-based methods that examined the evolutionary history of these mussels, including the number of migration events from north to south, and the relative timing of these events.
How many species of smooth-shelled blue mussels are there?
At the time of writing, the World Register of Marine Species (WoRMS -https://www.marinespecies.org/) recognises six species of smooth-shelled blue mussels: Mytilus chilensis Hupé, 1854, Mytilus edulis Linnaeus, 1758, Mytilus galloprovincialis Lamarck, 1819, Mytilus planulatus Lamarck, 1819, Mytilus platensis A. d’Orbigny, 1842, and Mytilus trossulus A. Gould, 1850. Using our SNP panel and taking the accepted WoRMS taxonomy as our starting (null) hypothesis we tested four alternative hypotheses with various combinations of species and/or geographic groupings of mussels. Only one alternative hypothesis provided a better fit than the current taxonomy. This best fit model involved the separation of native Australian mussels from native New Zealand mussels, with all other aspects of the currently recognised taxonomy being retained. It was Powell56 who first described M. aoteanus, collected from Wellington Harbour, based on shell morphometric differences when compared to native Australian mussels. These differences included ‘…. the dorsal slope (longer in New Zealand shells), hinge teeth (fewer in New Zealand shells), shape of the posterior retractor scar (narrower in New Zealand shells) and the anterior adductor scar (larger in New Zealand shells)’ (Gardner et al.4, p. 167). However, very soon after, the species status of M. aoteanus was changed by Fleming57 to subspecies status in keeping with the globally recognised M. edulis complex (e.g. 58) – thus, M. edulis aoteanus57. Since this time, various authors have employed the subspecies or full species forms of nomenclature for native New Zealand mussels (reviewed by Gardner et al.4), although, as noted above, neither the binomial nor the trinomial form is recognised by WoRMS. Nonetheless, we now know that there are clear shell morphometric56,59, shell shape59, genetic20,22,60 and species delimitation (present paper) differences between native Australian (the distribution of M. planulatus is centred around the island of Tasmania) and native New Zealand mussels, and these are, we suggest, sufficient to recognise the full specific status of M. aoteanus when applied to native New Zealand mussels. This would mean that there are, in fact, seven species of smooth-shelled blue mussel at the global level.
The evolutionary history of the Mytilus edulis species complex
Based on our panel of taxonomically informative SNP loci and the best fit species delimitation model, we used BEAST to construct a phylogenetic tree that best reflects the evolutionary history of the seven species of smooth-shelled blue mussels. Consistent with earlier interpretations, M. trossulus (which is thought to have a North Pacific Ocean origin – 9,50,60) was recognised as the oldest species. The next oldest branch on the tree gives rise to the two South American taxa, M. chilensis on the Pacific Ocean coast and M. platensis on the Atlantic Ocean coast, which are thought to be derived from Northern hemisphere mussels. This finding supports the conclusion of Gérard et al.60 based on COI and 16 S mitochondrial DNA sequencing, who highlighted pronounced genetic differentiation between Northern and Southern hemisphere blue mussels and who suggested that this split pre-dated the divergence between Northern hemisphere M. edulis and M. galloprovincialis. Our nuclear DNA SNP-based results confirm this interpretation. Gérard et al.60 dated the Northern versus Southern hemisphere split to be between 0.5 M and 1.3 M ybp (average 0.84 M ybp), an estimate that places the event during the Pleistocene (0.01–1.8 M ybp) and which is reasonably consistent with the earlier estimate produced by Hilbish et al.9 of 1.2 M ybp based on sequence variation of the 16 S gene. The next bifurcation in our tree is for the Northern hemisphere M. edulis (Atlantic Ocean) and M. galloprovincialis (Mediterranean Sea), followed by a second, quite recent, migration event from the Northern to the Southern hemisphere, to give rise to M. planulatus in Australia and M. aoteanus in New Zealand. Hilbish et al.9 reported evidence of this second north to south migration event, while Gérard et al.60 noted that the evidence supported two migration events (with differential barriers to nuclear and mitochondrial gene flow) or a single migration event and ‘a view of the composition of the nuclear genome biased by taxonomic preconception’ (Gérard et al. 60, p. 84). In other words, Gérard et al.60 are saying that if one views the Australasian mussels as part of the Mytilus edulis species complex and not as taxa distinct from it then the interpretation is one historical migration event (to South America only) but if one views the Australasian mussels as being distinct (i.e., as M. planulatus in Australasia or as M. planulatus in Australia and as M. aoteanus in New Zealand) then the interpretation is two events (to South America and then to Australasia). However, it is possible that the first migration event occurred earlier than either molecular study suggests, given that the fossil record indicates the presence of Mytilus deposits along both coasts of South America dating back to the Miocene epoch (~ 5.3–23 million years before present), which contain native blue mussels (Kiel and Nielsen61, as reviewed in Gardner et al.4). Most recently, Popovic et al.26 estimated that Australian M. planulatus diverged in allopatry from Northern hemisphere M. galloprovincialis between 0.1 and 0.6 M ybp (their work did not include native New Zealand mussels) thereby strongly supporting the hypothesis of a second, recent, migration event. Based on our current taxonomic understanding of M. planulatus in Australia and M. aoteanus in New Zealand as separate species then the two-event interpretation (as proposed by Hilbish et al. 9 and as discussed by Gérard et al.60) is strongly supported by our SDM findings.
Our nuclear DNA SNPs-based findings are in full agreement with the mitochondrial DNA-based results of Hilbish et al.9 and Gérard et al.60 and the SNPs-based work of Popovic et al.26, and provide strong support for the origin of two South American species before the origin of M. edulis and M. galloprovincialis in the Northern hemisphere, and also for the origin of two Australasian species after the northern origin of M. edulis and M. galloprovincialis. At the moment, the timing of the split between M. platensis (Atlantic Ocean) and M. chilensis (Pacific Ocean) in South America, and the timing of the split between M. planulatus (Australia) and M. aoteanus (New Zealand) in Australasia cannot be determined. The main limitation in estimating divergence times lies in the lack of reliable fossil records that would allow for a robust molecular clock calibration for the Southern Hemisphere Mytilus lineages. However, based on the most likely scenario of a North Atlantic to South Atlantic Ocean route of colonisation9, it seems likely that in the first migration event smooth-shelled blue mussels colonised the Atlantic Ocean (modern Argentina) and then the Pacific Ocean (modern Chile) coastlines, and for the second migration event, that mussels colonised Australia and then New Zealand (reviewed by Gardner et al. 4). These hypotheses need further testing to help us understand both the routes of colonisation and the timings of these two separate events.
Conclusions
We validated seven species within the Mytilus edulis complex by analysing specimens collected across their global distribution, while also integrating evidence from previous studies, including both molecular-based species delimitation and traditional taxonomic approaches. This comprehensive framework enabled us to confirm that mussels native to New Zealand and Australia represent two distinct species, and that South American blue mussels diverged independently from the other lineages within the complex.
Numerous unresolved phylogeographic and biogeographic questions still remain regarding the evolutionary history and distribution of the group. Addressing these issues will be essential for advancing our understanding of the complex spatial and evolutionary dynamics that have shaped the distribution of these taxa.
Accurate species delimitation is a central component of taxonomic research, as it plays a critical role in resolving systematic uncertainties, enhancing our understanding of biodiversity, and informing the development of effective conservation strategies. Furthermore, the implications of these results extend beyond evolutionary biology, offering valuable insights for applied contexts such as aquaculture, food provenance certification, and biosecurity management.
Data availability
All data analysed during this study are included in this published article. Additionally, the DNA sequences are stored in GenBank (KT713378–82; HQ257471; KJ871039–57; KT713368–74).
References
Sites, J. W. & Marshall, J. C. Delimiting species: A renaissance issue in systematic biology. Trends Ecol. Evol. 18 (9), 462–470 (2003).
Camargo, A. & Sites, J. W. Species delimitation: A decade after the renaissance. In The Species Problem –Ongoing Issues, (ed Pavlinov, I. Y.) Rijeka: InTech, 225–247 (2013).
Yang, Z. & Rannala, B. Unguided species delimitation using DNA sequence data from multiple loci. Mol. Biol. Evol. 31 (12), 3125–3135 (2014).
Gardner, J. P. A., Oyarzún, P. A., Toro, J. E., Wenne, R. & Zbawicka, M. Phylogeography of Southern hemisphere blue mussels of the genus Mytilus: evolution, biosecurity, aquaculture and food labelling. Oceanogr. Mar. Biol. Annu. Rev. 59, 139–232 (2021).
Oyarzún, P. A., Toro, J. E., Nuñez, J. J., Suarez-Villota, E. & Gardner, J. P. A. Blue mussels of the Mytilus Edulis species complex from South america: the application of species delimitation models to DNA sequence variation. PLoS ONE. 16 (9). https://doi.org/10.1371/journal.pone.0256961 (2021). e0256961.
Koehn, R. K., Hall, J. G., Innes, D. J. & Zera, A. J. Genetic differentiation of Mytilus Edulis in Eastern North America. Mar. Biol. 79, 117–126 (1984).
McDonald, J. H., Seed, R. & Koehn, R. K. Allozymes and morphometric characters of three species of Mytilus in the Northern and Southern hemispheres. Mar. Biol. 111, 323–333 (1991).
Gosling, E. M. Systematics and geographic distribution of Mytilus. In The Mussel Mytilus: Ecology, Physiology, Genetics and Culture; (ed Gosling, E. M.) Elsevier: Amsterdam, The Netherlands, 1–20 (1992).
Hilbish, T. J. et al. Origin of the Antitropical distribution pattern in marine mussels (Mytilus spp.): routes and timing of transequatorial migration. Mar. Biol. 136, 69–77 (2000).
Crooks, J. A. Characterizing ecosystem-level consequences of biological invasions: the role of ecosystem engineers. Oikos 97, 153–151 (2002).
Borthagaray, A. I. & Carranza, A. Mussels as ecosystem engineers: their contribution to species richness in a Rocky Littoral community. Acta Oecol. 31, 243–250 (2007).
FAO. The State of World Fisheries and Aquaculture 2024 – Blue Transformation in action. Rome. (2024).
Michalek, K., Ventura, A. & Sanders, T. Mytilus hybridisation and impact on aquaculture: A minireview. Mar. Genomics. 27, 3–7 (2016).
González-Poblete, E., Hurtado, F. C., Rojo, C. & Norambuena, R. Blue mussel aquaculture in chile: small or large scale industry? Aquaculture 493, 113–122 (2018).
Larraín, M. A., Zbawicka, M., Araneda, C., Gardner, J. P. A. & Wenne, R. Native and invasive taxa on the Pacific Coast of South america: impacts on aquaculture, traceability and biodiversity of blue mussels (Mytilus spp). Evol. Appl. 11, 298–311 (2018).
Oyarzún, P. A., Toro, J. E., Cañete, J. I. & Gardner, J. P. A. Bioinvasion threatens the genetic integrity of native diversity and a natural hybrid zone: smooth-shelled blue mussels (Mytilus spp.) in the Strait of Magellan. Biol. J. Linn. Soc. 117, 574–585 (2016).
Zbawicka, M., Trucco, M. I. & Wenne, R. Single nucleotide polymorphisms in native South American Atlantic Coast populations of smooth shelled mussels: hybridization with invasive European Mytilus galloprovincialis. Genet. Sel. Evol. 50, 5 (2018).
Dias, P. J., Fotedar, S. & Snow, M. Characterisation of mussel (Mytilus sp.) populations in Western Australia and evaluation of potential genetic impacts of mussel Spat translocation from interstate. Mar. Freshw. Res. 65, 486–496 (2014).
Ab Rahim, E. S. et al. Species composition and hybridisation of mussel species (Bivalvia: Mytilidae) in Australia. Mar. Freshw. Res. 67, 1955–1963 (2016).
Zbawicka, M., Wenne, R., Dias, P. J. & Gardner, J. P. A. Combined threats to native smooth-shelled mussels (genus Mytilus) in australia: bioinvasions and hybridisation. Zoo J. Linn. Soc. 194, 1194–1211 (2022).
Gardner, J. P. A., Wenne, R., Westfall, K. R. & Zbawicka, M. Invasive mussels threaten regional scale genetic diversity in Mainland and remote offshore locations: the need for baseline data and enhanced protection in the Southern ocean. Glob Change Biol. 22, 3182–3195 (2016).
Zbawicka, M., Gardner, J. P. A. & Wenne, R. Cryptic diversity in smooth-shelled mussels on Southern ocean islands: Connectivity, hybridisation and a marine invasion. Front. Zool. 16, 32 (2019).
Fraïsse, C. et al. Fine-grained habitat associated genetic connectivity in an admixed population of mussels in the small isolated kerguelen Islands. Peer Community J. 1, e10 (2021).
Westfall, K. M. & Gardner, J. P. A. Genetic diversity of Southern hemisphere blue mussels of the genus Mytilus (Mytilidae; Bivalvia) and the identification of non-indigenous taxa. Biol. J. Linn. Soc. 101, 898–909 (2010).
Westfall, K. M. & Gardner, J. P. A. Interlineage Mytilus galloprovincialis Lmk. 1819 hybridisation yields inconsistent genetic outcomes in three Southern hemisphere regions. Biol. Invasions. 15, 1493–1506 (2013).
Popovic, I., Matias, A. M. A., Bierne, N. & Riginos, C. Twin introductions by independent invader mussel lineages are both associated with recent admixture with a native congener in Australia. Evol. Appl. 13, 515–532 (2020).
Shaffer, H. & Thomson, R. Delimiting species in recent radiations. Syst. Biol. 6 (56), 896–906 (2007).
Shin, J., Kwon, S., Lee, J., Min, H. & Kim, N. Genetic diversity of maize kernel starch-synthesis genes with snaps. Genome 10 (49), 1287–1296 (2006).
Wenne, R. Single nucleotide polymorphism markers with applications in conservation and exploitation of aquatic natural populations. Animals 13 (6), 1089 (2023).
Gabur, I. et al. Finding invisible quantitative trait loci with missing data. Plant. Biotechnol. J. 12 (16), 2102–2112 (2018).
Meudt, H., Lockhart, P. & Bryant, D. Species delimitation and phylogeny of a new Zealand plant species radiation. BMC Ecol. Evol. 1 (9), 111 (2009).
Wanghe, C. et al. Phylogenetic relationship and taxonomic status of Gymnocypris eckloni (Schizothoracinae) based on specific locus amplified fragments sequencing. Front. Ecol. Evol. 10, 933632 (2022).
Kelly, J. & Thacker, R. New shallow water species of Caribbean Ircinia Nardo, 1833 (Porifera: Irciniidae). Zootaxa 4 (5072), 301–323 (2021).
Campbell, E., Gage, E., Gage, R. & Sperling, F. Single nucleotide polymorphism-based species phylogeny of greater fritillary butterflies (Lepidoptera: nymphalidae: Speyeria) demonstrates widespread mitonuclear discordance. Syst. Entomol. 2 (45), 269–280 (2019).
Martínez-Borrego, D., Arellano, E., González-Cózatl, F. & Rogers, D. Species delimitation and integrative taxonomy of the Reithrodontomys Mexicanus (Rodentia: Cricetidae) cryptic complex. Ecol. Evol. 13(8), e10355 (2023).
Yuan, Z. et al. Historical mitochondrial genome introgression confounds species delimitation: evidence from phylogenetic inference in the Odorrana Grahami species complex. Curr. Zool. 1 (69), 82–90 (2023).
Fujita, M. K., Leaché, A. D., Burbrink, F. T., McGuire, J. A. & Moritz, C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 27 (9), 480–488 (2012).
Mason, N. A., Fletcher, N. K., Gill, B. A., Funk, W. C. & Zamudio, K. R. Coalescent-based species delimitation is sensitive to geographic sampling and isolation by distance. Syst. Biodiv. 18 (3), 269–280 (2020).
Kornai, D., Jiao, X., Ji, J., Flouri, T. & Yang, Z. Hierarchical heuristic species delimitation under the multispecies coalescent model with migration. Syst. Biol. 73 (6), 1015–1037 (2024).
Bryant, D., Bouckaert, R., Felsenstein, J., Rosenberg, N. A. & RoyChoudhury, A. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29, 1917–1932 (2012).
Leaché, A. D., Fujita, M. K., Minin, V. N. & Bouckaert, R. R. Species delimitation using Genome-Wide SNP data. Syst. Biol. 63 (4), 534–542 (2014).
Leaché, A. D. & Fujita, M. K. Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). Proc. R Soc. B Biol. Sci. 277, 3071–3077 (2010).
South, A. & rworldxtra Country boundaries at high resolution. R package version 1.01. (2023). https://CRAN.R-project.org/package=rworldxtra
Gabriel, S., Ziaugra, L. & Tabbaa, D. SNP genotyping using the sequenom massarray iPLEX platform. Curr Protoc. Hum. Genet Chapter 2: (2009). Unit 2.12.
Wenne, R., Bach, L., Zbawicka, M., Strand, J. & McDonald, J. H. A first report on coexistence and hybridization of Mytilus trossulus and M. edulis mussels in Greenland. Polar Biol. 39 (2), 343–355 (2016).
Zbawicka, M. et al. Genetic differentiation in Mytilus populations across climatic zones in Greenland. J Biogeogr. e70067 (2025).
Bouckaert, R. et al. BEAST 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 10 (4), e1003537 (2014).
Kass, R. E. & Raftery, A. E. Bayes factors. J. Am. Stat. Assoc. 90, 773–795 (1995).
Matschiner, M. Species tree inference with SNP data. In: (eds Pereira-Santana, A., Gamboa-Tuz, S. D. & Rodríguez-Zapata, L. C.) Plant Comparative Genomics. Methods in Molecular Biology, vol 2512. Humana, New York (2022).
Riginos, C. & Cunningham, C. W. Local adaptation and species segregation in two mussel (Mytilus Edulis × Mytilus trossulus) hybrid zones. Mol. Ecol. 14, 381–400 (2005).
Rambaut, A. & FigTree (Version 1.4.2) [Software]. Institute of Evolutionary Biology, University of Edinburgh. (2018). Retrieved from http://tree.bio.ed.ac.uk/software/figtree/
Koehn, R. K. The genetics and taxonomy of species in the genus Mytilus. Aquaculture 94 (2–3), 125–145 (1991).
Gardner, J. P. A. Mytilus galloprovincialis (Lmk) (Bivalvia, Mollusca): the taxonomic status of the mediterranean mussel. Ophelia 35 (3), 219–243 (1992).
Seed, R. Systematics, evolution and distribution of mussels belonging to the genus Mytilus: an over-view. Am. Malacol Bull. 9, 123–137 (1992).
Illesca, A., Oyarzún, P. A., Toro, J. E. & Gardner, J. P. A. Morphometric variability of smooth-shelled blue mussels from the Pacific Coast of South America. Biol. J. Linn. Soc. 125, 194–209 (2018).
Powell, A. W. B. New Zealand molluscan systematics with descriptions of new species, part 3. Records Auckl. Inst. Museum. 5, 87–91 (1958).
Fleming, C. A. Notes on new Zealand recent and tertiary mussels (Mytilidae). Trans. R Soc. N Z. 87, 165–178 (1959).
Soot-Ryen, T. A report of the family mytilidae (Pelecypoda). Allan Hancock Pacifc Expedition. 20, 1–75 (1955).
Ravalo, L. G. O. Native and invasive smooth shelled blue mussels Mytilus spp in New Zealand: Evolutionary Biosecurity and Aquaculture Implications. PhD tesis, Victoria University of Wellington, New Zealand Available at - (2024). https://openaccess.wgtn.ac.nz/articles/thesis/Native_and_Invasive_Smooth_Shelled_Blue_Mussels_Mytilus_spp_in_New_Zealand_Evolutionary_Biosecurity_and_Aquaculture_Implications/25989046
Gérard, K., Bierne, N., Borsa, P., Chenuil, A. & Feral, J. P. Pleistocene separation of mitochondrial lineages of Mytilus spp. Mussels from Northern and Southern hemispheres and strong genetic differentiation among Southern populations. Mol. Phylogenet Evol. 49, 84–91 (2008).
Kiel, S. & Nielsen, S. N. Quaternary origin of the inverse latitudinal diversity gradient among Southern Chilean mollusks. Geology 38, 955–958 (2010).
Acknowledgements
We thank the CIPRES Science Gateway for providing access to its platform, which facilitated several of our analyses.
Funding
This work was funded by the projects FONDECYT 1230212 (JT) and FONDECYT 1251077 (POC).
Author information
Authors and Affiliations
Contributions
P.A.O.: Conceptualisation, methodology, and writing—review and editing. J.E.T.: Investigation and validation. M.Z.: Investigation and validation. R.W.: Investigation and validation. J.P.A.G.: Conceptualisation, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oyarzún, P.A., Toro, J.E., Zbawicka, M. et al. Global species delimitation of smooth-shelled blue mussels in the Mytilus edulis complex. Sci Rep 15, 43860 (2025). https://doi.org/10.1038/s41598-025-29759-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-29759-9
