
Abstract
Yaks have successfully adapted to the extreme conditions of the Qinghai-Tibetan Plateau, although the mechanisms underlying their hepatic energy utilization remain poorly understood. Using histological, transcriptomic, and metabolomic approaches, this study compared liver characteristics between yaks and cattle at similar altitudes, as well as between yaks at different altitudes. Results revealed that yaks possess smaller hepatic sinusoids and greater glycogen storage capacity than cattle. Transcriptomic analysis showed that differentially expressed genes (DEGs) in yaks are predominantly involved in carbohydrate and lipid metabolism, with significant up-regulation of key genes such as FBP1 (gluconeogenesis), ACAA1 and ACOX1 (lipolysis), and ACO1 and MDH1 (TCA cycle). High-altitude yaks exhibited even narrower sinusoids and higher glycogen levels, along with DEGs enriched in metabolic pathways including acetyl-CoA synthesis and the TCA cycle. Specifically, G6PC1 was up-regulated, while ACOX1, ACADS, and ACO1 were down-regulated in high-altitude yaks. These findings suggest that yaks maintain energy homeostasis through enhanced lipolytic metabolism and increased TCA cycle activity. Compared to low-altitude yaks, those at high elevations appear to rely more on oxidative phosphorylation for energy production, highlighting key metabolic adaptations that support survival in hypoxic environments.
Similar content being viewed by others

Introduction
The Qinghai-Tibetan Plateau, one of the highest and harshest geographical regions globally, features extreme ecological conditions such as hypoxia, low temperatures, intense UV radiation, and nutrient scarcity1. Long-term natural selection has driven indigenous species to evolve a series of adaptive phenotypes and molecular mechanisms to sustain their survival and reproduction in high-altitude environments2. For example, plateau pika (Ochotona curzoniae) significantly reduces hypoxia-induced myocardial injury by upregulating the key gene EPAS1 in the hypoxia-inducible factor pathway, thereby enhancing its hypoxic tolerance3. In high-altitude mammals like Myospalacinae, positive selection acts on genes involved in energy transport and enzyme catalytic functions, significantly optimizing their energy metabolism efficiency to cope with the high-energy-demanding survival environment4. Furthermore, in human populations residing at high altitudes, HIF-2α has been found to exhibit strong signals of positive selection within the HIF signaling pathway, highlighting its critical role in hypoxia adaptation5.Yaks (Bos grunniens) have developed multifaceted adaptive changes over time to survive in the Qinghai-Tibetan Plateau environment. Studies have shown that several key genes related to hypoxic response, such as HIF1A, VEGFA, and EPAS1, are significantly upregulated in the tissues of plateau yaks, which helps regulate angiogenesis, oxygen transport, and cellular metabolic reprogramming, thus maintaining their physiological homeostasis under hypoxic conditions6,7. Moreover, yak colostrum is rich in active glycometabolites such as UDP-galactose, N-acetylglucosamine, and UDP-glucose, which not only provide energy support for newborns but also participate in hypoxia adaptation and immune regulation through glycosylation modifications8. Recent studies have also found that under high-altitude hypoxic conditions, the expression of mitochondrial dynamics-related gene FIS1 is significantly upregulated in yak kidneys, suggesting that it promotes mitochondrial fission to maintain mitochondrial network dynamic balance and functional integrity, thereby enhancing cellular tolerance to hypoxic stress9.
The liver is the core organ of energy metabolism, primarily responsible for glucose, fatty acid, and protein metabolism processes, playing a crucial role in maintaining peripheral blood nutrient homeostasis and systemic energy balance10,11. Previous studies have shown that the liver plays an important role in adapting to the plateau environment. Liver TCA cycle, gluconeogenesis, glycogenesis, fatty acid oxidation and synthesis were enhanced during acute plateau exposure in mice, in addition, carbohydrates are the main energy substances and fatty acids play an important supplementary12. Moreover, various species have developed distinct liver adaptation strategies. Myospalax baileyi enhances the malate-aspartate shuttle (MA) activity to facilitate cytosolic NADH transport into mitochondria, thus improving energy supply efficiency; Ochotona curzoniae depends on enhanced hepatic gluconeogenesis to provide energy substrates necessary for locomotion13. Through long-term natural and artificial selection, the yak has evolved not only distinctive morphological14,15,16, physiological17,18,19 and genetic traits that set it apart from other bovine species20,21 but also a liver-centered adaptive strategy: this organ fine-tunes gene expression and key metabolic enzyme activities, thereby providing an additional, unique mechanism for thriving in the extreme plateau environment22,23.Recent studies have shown that lipid catabolism, fatty acid synthesis, glucose uptake, and fatty acid oxidation are regulated in yak during the cold seasons24. However, the energy metabolism network is highly complex, and the specific metabolic regulatory mechanisms in the yak liver underlying adaptation to the plateau hypoxic environment remain unclear.
This study integrates histology, transcriptomics, metabolomics, and molecular biology techniques to systematically investigate the energy metabolism adaptation mechanisms of yak liver under plateau hypoxia and low temperatures, aiming to reveal the physiological and molecular basis of efficient energy utilization from a multi-omics perspective.
Materials and methods
Animals and sample collection
All the animal studies were performed based on the guidelines of the Institutional Animal Care and Use of Laboratory Animals and were approved by the Animal Welfare and Ethics Committee at the Qinghai University. To investigate hepatic adaptation differences in yaks from different altitudes, liver tissue samples were collected from adult male yaks at local slaughterhouses, five adult male yaks (3–5 years old) were selected from Qumalai County (altitude: 4500 m) (QML-Y), and five adult male yaks (XH-Y) along with five adult male cattle (XH-C) of the same age range were selected from Xunhua County (altitude: 2600 m). To meet statistical requirements, all omics data analyses in this study achieved the necessary minimum number of biological replicates. Specifically, transcriptome sequencing and quantitative real-time PCR validation experiments were conducted with three replicates per group (n = 3). Whereas metabolomic analysis was performed with five replicates per group (n = 5). Liver tissues were rapidly dissected from the deep parenchyma near the central vein (approximately 1 cm³ in size), samples intended for histological staining were stored at room temperature in centrifuge tubes containing fixative, while the remaining samples were aliquoted into cryovials, rapidly frozen in liquid nitrogen, and transported under cryogenic conditions for subsequent analyses.
Histological study
Fresh samples were fixed and dehydrated The fresh tissue samples were embedded in paraffin, and the embedded blocks were sectioned into 4 μm thick slices., followed by hematoxylin-eosin (HE) and periodic acid-Schiff (PAS) staining of the paraffin-embedded Sect25. The HE staining procedure was to deparaffinize the paraffin sections sequentially by xylene I and II (20 min each), hydrate twice with anhydrous ethanol (5 min each) and rinse with running water. After hematoxylin staining for 3–5 min, they were washed in water and differentiated with a weak acidic solution. After washing with distilled water, it was sequentially dehydrated with 85% and 95% ethanol (5–10 min each) and retained with eosin for 5 min. Finally, it was thoroughly dehydrated by anhydrous ethanol three times (5 min each), transparent with xylene I and II (5 min each), and sealed with neutral gum. The PAS staining procedure was to dewax the paraffin sections until hydration, then stain them first in PAS dye solution B for 10–15 min, and wash them with water to remove excess dye; then transfer them to act in PAS dye solution A for 25–30 min, rinse them with running water for 5 min, and treat them with dye solution C for 30 s. After completion of staining, the sections were rinsed in tap water, differentiated in hydrochloric acid solution, and then washed thoroughly in water. To enhance contrast, sections were blued in ammonia and washed in water. Sections were finally dehydrated in gradient alcohol, transparent in xylene, and sealed in neutral gum.
Observe with Nikcon (DS-Ri2) optical microscope and image acquisition. The related data were directly measured using Image-Pro Plus 6.0 Chinese image analysis system, and the individual hepatocyte area, hepatic sinus area, and hepatic glycogen content per unit area were measured. Experimental data were analyzed using IBM SPSS Statistics 27 and differences between groups were assessed by independent samples t-test. The results were expressed as “mean ± standard deviation” (\(\:\stackrel{-}{x}\)±SD) for comparing significant differences in each structural parameter between XH-C and XH-Y at the same altitude and between XH-Y and QML-Y.
Transcriptome sequencing
The bioinformatics pipeline for RNA sequencing data analysis comprised five key steps: initial quality assessment, sequence alignment, transcript quantification, statistical evaluation of differential expression, and functional enrichment analysis. The collected liver tissue samples of cattle and yaks at same altitude and yaks at different altitudes were sent to Tianjin Novozymes Bioinformatics Co., Ltd. Website (https://cn.novogene.com/). In this analysis, the experimental design incorporated three biological replicates per group (n = 3) to ensure the robustness of statistical analysis and the reproducibility of results. Total RNA was extracted from samples stored in dry ice using the RNA Extraction Kit (Tiangen, DP419), and RNA integrity was accurately assessed using the Agilent 2100 Bioanalyzer. The mRNA was enriched using magnetic bead-based isolation, followed by double-stranded cDNA synthesis catalyzed by M-MuLV reverse transcriptase. The purified cDNA underwent end repair, A-tailing, and adapter ligation. Fragments of 250–300 bp were selectively isolated using AMPure XP beads, with subsequent PCR amplification and final purification to complete library preparation. The transcriptome was subjected to paired-end sequencing using the Illumina HiSeq 2500 platform, generating 150 bp paired-end reads. To ensure the quality and reliability of the data analysis, the raw data were filtered using Fastp software to remove splices, N-containing and low-quality read lengths (Qphred ≤ 5 bases, more than 50% of the total read length). (Fastp v0.23.4 software source: https://github.com/OpenGene/fastp). The clean reads were mapped to the reference genome (ncbi_bos_mutus_gcf_000298355_1_bosgru_v2_0) of Bos mutus using HISAT 2.2.1 software. ༈HISAT 2.2.1 software source༚http://daehwankimlab.github.io/hisat2/download/༉FPKM was used to measure the transcript or gene expression level. Differential expression genes (DEGs) were identified between yaks and cattle at same altitude and yaks at different altitudes using the DESeq2 based on the negative binomial distribution. The P values were adjusted for controlling the false discovery rate. Genes with |log2(Fold Change)| ≥ 1 and FDR value < 0.05 were assigned as DEGs. The differentially expressed gene sets obtained from screening were subjected to GO functional enrichment analysis and KEGG pathway enrichment analysis using the cluster Profiler (v3.8.1) software26,27.
Metabolomic profiling
Metabolomics sample preparation: Precisely 50 mg of liver tissue was weighed and transferred to a 2 mL centrifuge tube. Then, 500 µL of pre-cooled extraction solvent (methanol: chloroform = 3:1, v/v) was added, along with the internal standard 2-chloro-L-phenylalanine at a final concentration of 1 mg/mL. After vortex mixing for 30 s, stainless steel grinding beads were added, and the sample was homogenized at 35 Hz for 4 min, followed by ultrasonic treatment in an ice-water bath for 5 min. This cycle was repeated three times to ensure thorough tissue lysis and efficient metabolite extraction. After centrifugation (12,000 rpm, 15 min), the supernatant was collected. An aliquot of 80 µL from each sample was pooled to create a quality control (QC) sample. All extracts were dried using a vacuum concentrator. Then, 60 µL of methoxyamine hydrochloride solution was added, and after mixing, the samples were incubated in an 80 °C water bath for 30 min. Subsequently, 80 µL of BSTFA reagent was added, and incubation was carried out at 70 °C for 1.5 h. After derivatization was complete, the samples were cooled to room temperature, and 5 µL of FAMEs was added as a retention index standard for subsequent chromatographic retention time calibration. GC-TOF-MS analysis conditions: Analysis was performed using a Gas Chromatography-Time-of-Flight Mass Spectrometry (GC-TOF-MS) system. Chromatographic separation was achieved using a DB-5MS quartz capillary column (30 m × 250 μm × 0.25 μm). The injection volume was 1 µL, employing a splitless injection mode. The column temperature program was set as follows: initial temperature 50 °C, held for 1 min, then ramped at 10 °C/min to 310 °C, and maintained for 8 min. The carrier gas was high-purity helium, with a constant flow rate of 1.0 mL/min. The mass spectrometer ion source temperature was set at 250 °C, with electron impact (EI) ionization energy of 70 eV. The scan range was m/z 50–500, with a solvent delay time of 6.27 min. 2-Chloro-L-phenylalanine was used as the internal standard and added at the beginning of sample preparation to monitor and correct for variations in extraction efficiency, instrument response, and matrix effects throughout the analytical process. This approach ensures accurate and reliable quantification of metabolite levels across all samples.
Principal component analysis (PCA) of experimental samples was performed using the software SIMCA (V14.1) with the parameter setting: auto-fit. Raw data were filtered using the Perl 6.0 program to remove data with no identifiable substance name and no spectral similarity (samples with more than 50% missing substances were filtered directly, and less than 50% were simulated using R (V3.6.2)). (Perl 6.0 software source: https://www.perl.org/). The DMwR package K Nearest Neighbor (KNN) algorithm was used for missing value simulation, and the preprocessed data were annotated with the HMDB database (V4.0) and the KEGG COMPOUND database (https://www.kegg.jp/kegg/compound/). Metabolites were identified using perl 6.0 software with P-value < 0.05 and VIP > 1 (with replicates) or p-value < 0.05 and FC > 1.2 | FC < 0.83 (without replicates) were considered differential metabolites. The raw data were pre-processed and 364 Peak were retained for multivariate analysis of liver metabolites. Pathway function analysis of differential metabolites was performed using the KEGG PATHWAY database28,29 (https://www.kegg.jp/kegg/pathway.html).
Quantitative real-time PCR
RNA extraction for RT-qPCR validation was performed using the Trizol method. Approximately 20 mg of liver tissue was homogenized in 1 mL of Trizol reagent, followed by phase separation with chloroform, isopropanol precipitation, 75% ethanol washing, and final dissolution in RNase-free water. Based on the results of transcriptome DEGs analysis, we designed primers using Oligo 7 software and performed real-time PCR using SYBR® Premix Ex Taq™II (TaRaKa, Dalian, and China) in order to detect the expression levels of energy metabolism-related gene mRNAs (The primer information is shown in Table 1). The β-actin was used as a reference gene to normalize gene expression30. The quantitative real-time PCR amplification protocol was set as follows: initial denaturation at 95 °C for 30 s, followed by 40 cycles of amplification (95 °C for 15 s denaturation, and 60 °C for 30 s combined annealing/extension). Finally, melting curve analysis was performed to verify amplification specificity by gradually increasing the temperature from 65 °C to 95 °C with fluorescence acquisition at 0.5 °C increments. The expression of each candidate gene was calculated using the formula 2−ΔΔCt31 and the significance of the data was tested by independent samples t-test.
Results
Morphological adaptation of Yak to plateau environment
Histological studies were carried out on XH-C, XH-Y and QML-Y (Fig. 1). HE and PAS staining results are shown in Fig. 1a. Compared with the XH-C, the hepatic sinus of XH-Y was significantly reduced (P<0.01) (Fig. 1b). The hepatic glycogen of XH-Y content was significantly increased (P<0.05) (Fig. 1c). Similarly, compare with the XH-Y, the hepatic sinus of QML-Y were significantly reduced (P<0.01) (Fig. 1b) and the hepatic glycogen content of QML-Y was significantly increased (P<0.01) (Fig. 1c). In addition, the number of hepatocytes was significantly lower in QML-Y than in XH-Y (P<0.01) (Fig. 1d).

Basic structure and quantitative measurement of liver of XH-C, XH-Y and QML-Y. note: (a) HE staining and PAS staining. (b) relative area of hepatic sinus per unit area. (c) relative content of glycogen per unit area. (d) relative area of hepatocytes. ** indicates that the difference is extremely significant, P < 0.01; *Indicates significant difference, P < 0.05. Hepatic sinusoids in HE-stained sections are indicated by arrows. Glycogen in PAS-stained sections is labeled with a triangle.
Adaptation of Yak to Highland hypoxic environment at transcriptional level
Transcriptome profile of liver tissues
Quality control analysis of the transcriptome sequencing data demonstrated excellent performance across all key metrics: The clean reads ranged from 6.68 Gb to 8.57 Gb, with Q20 and Q30 scores reaching 98.29%−98.59% and 95.39%−96.01% respectively, while the GC content remained stable at 48.55%−50.13%. These robust quality indicators confirm that our transcriptomic data meet stringent sequencing quality standards, ensuring their reliability for subsequent bioinformatics processing and functional genomic studies. Data is available at NCBI SRA, accession numbers: PRJNA1162664.
Differential gene expression analysis
We used DESeq2 software to identify the genes that were DEGs, and |log2(Fold Change)| ≥ 1 and FDR value < 0.05 as the screening criteria (Fig. 2) (DESeq2 software source: https://bioconductor.org/packages/devel/bioc/html/DESeq2.html). We identified 1,546 DEGs between XH-C and XH-Y, in which 741 were up-regulated and 805 were down-regulated (Fig. 2a), 564 DEGs between XH-Y and QML-Y, of these DEGs, 307 were up-regulated and 257 were down-regulated (Fig. 2b).

Volcano plots of DEGs. note: (a) XH-C vs. XH-Y (b) QML-Y vs. XH-Y. The red and green dots represent genes up-regulated and down-regulated.
GO enrichment analysis of DEGs
To further investigate the biological functions and regulatory mechanisms underlying the DEGs in the liver of XH-C versus XH-Y, as well as XH-Y versus QML-Y, we carried out a GO analysis focusing on biological processes. All the DEGs were annotated in the GO database and were categorized into 691 functional groups between XH-C and XH-Y, 407 functional groups between XH-Y and QML-Y. In XH-C and XH-Y, GO biological processes such as oxidation-reduction process, fatty acid metabolic process, and lipid metabolic process were significantly enriched. Similarly, between XH-Y and QML-Y, processes including oxidation-reduction process, lipid transport, and protein dephosphorylation showed notable enrichment (Table 2).
KEGG enrichment analysis of DEGs
The results of KEGG pathway enrichment analysis using DEGs are shown in Fig. 3. The most enriched pathways between XH-C and XH-Y included Pyruvate metabolism, Biosynthesis of unsaturated fatty acids, Fatty acid metabolism, Bile secretion, PPAR signaling pathway, Cholesterol metabolism and Steroid hormone biosynthesis (Fig. 3a). Multiple key genes exhibited significant upregulation across various metabolic pathways. Specifically, genes such as COX1, COX2, COX3, along with ND1, ND2, ND3, ND4, ND5 and ND6, were enriched in reactive oxygen species-related pathways. The genes ELOVL7, FADS2 and ACOX1 were involved in fatty acid metabolic pathways, while SCD and FADS1 were enriched in fatty acid biosynthetic processes. Additionally, ACACB and ACSS2 genes participated in pyruvate metabolic pathways. All these genes showed a significant upregulation trend in yaks (Table 3).

KEGG enrichment analysis. note: (a)XH-C vs. XH-Y (b) QML-Y vs. XH-Y. The horizontal coordinate is the significance level of pathway enrichment and the vertical coordinate is the KEGG pathway.
By contrast, the most enriched pathways in XH-Y and QML-Y included steroid biosynthesis, steroid hormone biosynthesis, PPAR signaling pathway, bile secretion and cholesterol metabolism (Fig. 3b). In high-altitude yaks, multiple key metabolic genes exhibited significant downregulation. Specifically, the CPX1, GGT1 and GSTA4 genes are primarily involved in glutathione metabolism pathways, while the PCK1, RXRG, APOA5 and ACSL6 genes were enriched in the PPAR signaling pathway (Table 4).
Metabolomic profiling
Multivariate statistical analysis
Principal component analysis (PCA) was performed on the collected data (Fig. 4). From the results of PCA score map, it could be seen that all the samples were within the 95% confidence interval (Hotelling’s T-squared ellipse), indicating the high reliability of the data (Fig. 4a). Next, the data were analyzed by the Partial Least Squares-Discriminant Analysis (PLS-DA) method. These multivariate statistical results indicate good intra-group reproducibility and significant inter-group separation across all comparison groups (interspecies comparisons at same altitude, intraspecies comparisons across altitudes) (Fig. 4b).

Multivariate statistical analysis. note: (a) XH-C vs. XH-Y PCA score plots (b) XH-Y vs. QML-Y PCA score plots (c) XH-C vs. XH-Y PLS-DA score plots (d) XH-Y vs. QML-Y PLS-DA score plots.
Analysis of key differential metabolites
A total of 319 and 324 metabolites were identified in XH-C vs. XH-Y and XH-Y vs. QML-Y. Importantly, the results of the study showed that only one differential metabolite (xanthosine) was detected between cattle and yaks; whereas, a total of 59 significantly different metabolites were identified between yaks at different altitudes, including L-Malic acid, Fumaric acid, D-galacturonic acid and phosphoric acid (VIP > 1, P < 0.05). The differential metabolites of yak at different altitudes were further analyzed (Fig. 5).

Hepatic metabolic profiles between yaks at different altitudes. note: (a). Heat map of differential metabolites. The abscissa indicates the sample name, and the ordinate indicates the differential metabolite. (b). KEGG Pathway Enrichment Analysis Diagram for Differentially Expressed Genes Between XH-Y and QML-Y. The x-axis represents enrichment factors; higher enrichment factors indicate more significant accumulation of differentially expressed metabolites within the pathway. Point colors denote p-values, while bubble sizes reflect the number of annotated metabolites exhibiting differential expression within the pathway.
We performed hierarchical clustering on all significantly different metabolites (P < 0.05) and visualized the differential expression level of metabolites based on VIP value. Significant differences in metabolites were noted between the XH-Y and QML-Y (Fig. 5a). KEGG analysis of metabolites in the liver of XH-Y and QML-Y revealed that the metabolites were significantly enriched in pathways Pyruvate metabolism, Pentose and glucuronate interconversions, Oxidative phosphorylation, and TCA cycle (Fig. 5b). Furthermore, key metabolites including fumarate, succinate, malate, glycerol, and octadecenoic acid were significantly enriched in pathways associated with energy metabolism.
Quantitative real-time PCR validation of functional gene expression
To verify the reliability of gene expression in the RNA-Seq data, we selected three key genes related to gluconeogenesis (G6PC1、PCK1、FBP1), three genes involved in fatty acid catabolism (ACAA1、ACOX1、ACADS), and five genes related to the TCA cycle (IDH1、ACO1、SDHA、MDH1、FH) for further validation analysis based on the results of the RNA-Seq analysis (Fig. 6). The original RNA concentration ranged from 4807 to 5646 ng/µL, with A260/A280 ratios between 2.004 and 2.01. These data indicate that the extracted RNA has high purity and good quality. The results showed that XH-Y liver tissues exhibited unique adaptive strategies compared with those of XH-C living at the same altitude. XH-Y liver gluconeogenesis-related genePCK1 and FBP1 was up-regulated, lipid oxidation-related genes ACOX1 were significantly up-regulated, and tricarboxylic acid cycle-related genes FH and MDH1 were significantly up-regulated in yak liver (Fig. 6a). The quantitative real-time PCR results revealed enhanced lipid oxidation and TCA cycle activity in yaks compared to cattle.

Relative expression of genes related to energy metabolism. note: (a) XH-C vs. XH-Y. (b) XH-Y vs. QML-Y. ** indicates that the difference is extremely significant, P < 0.01; *Indicates significant difference, p < 0.05.
compared with XH-Y, the QML-Y gluconeogenesis-related gene G6PC1 was significantly up-regulated, lipid oxidation-related genes ACOX1 and ACADS were significantly down-regulated, and the tricarboxylic acid cycle-related gene IDH1、ACO1 and SDHA was significantly down-regulated (Fig. 6b). The quantitative real-time PCR analysis demonstrated attenuated lipid oxidation and TCA cycle activity in QML-Y compared to their XH-Y counterparts. The results of the quantitative real-time PCR data were in general agreement with the transcriptome results.
Mechanisms of energy metabolism in cattle versus Yaks and across Yak altitudes
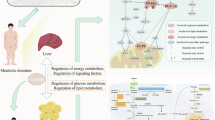
In conclusion, our results suggest that XH-Y have an enhanced capacity for material and energy exchange through a narrower hepatic sinusoidal structure, which represents an important physiological adaptive feature when compared to common XH-C. This adaptive process was accompanied by an increase in glycogen accumulation, as well as up-regulation of the gluconeogenesis-related gene FBP1 and PCK1. At the same time, the expression levels of lipolysis-related genes ACOX1, as well as TCA cycle-related genes FH and MDH1, were also significantly increased. When comparing QML-Y with XH-Y, we found that the hepatic sinusoidal structure was narrower in QML-Y. The differential metabolites (fumarate, succinate, malate, glycerol and octadecenoic acid) exhibited significant enrichment in pathways associated with energy metabolism. In addition, glycogen accumulation was higher in QML-Y, and the expression of the glycogen-related gene G6PC1 was up-regulated. However, IDH1、ACO1 and SDHA, a gene involved in the TCA cycle, and the fatty acid degradation-related genes ACOX1 and ACADS were down-regulated in QML-Y (Fig. 7).

The regulation of the yak under long-term nutritional stress. note: red: up-regulation of genes expression or enhanced the pathways; green: down-regulation of genes expression or diminished the pathways. →: promote or result in. ⊥: the genes expression or metabolic pathways were inhibited. Square: gene; Oval: metabolic process; Hexagon: metabolite.
Discussions
As an important metabolic organ, the liver plays an important role in highland hypoxic environment. Previous studies have shown that Tibetan sheep and other species can meet the high-energy demand of the Qinghai-Tibetan Plateau by regulating the lipid metabolism, glucose metabolism and amino acid metabolism of the liver32,33,34. However, studies on the mechanisms underlying the adaptation of yak liver to high-altitude environments remain limited. To address this issue, the present study employed a combination of histological, transcriptomic, metabolomic, and molecular biological approaches to systematically elucidate the adaptive mechanisms by which the yak liver maintains energy metabolic homeostasis under hypoxic high-altitude conditions.
Our study found that XH-Y hepatic sinus were significantly smaller than XH-C, and the hepatic sinus of QML-Y were significantly smaller than XH-Y. Liver glycogen content was higher in XH-Y than in XH-C, and higher in QML-Y than XH-Y. It was found that rats exposed to a hypoxic conditions exhibited increased hepatic glycogen accumulation35. Similarly, hepatic glycogen content was significantly increased in mice under simulated high-altitude conditions36.Therefore, we speculate that XH-Y may adapt to the energy demands of the high-altitude hypoxic environment through a narrower hepatic sinusoidal structure and enhanced glycogen storage. Furthermore, compared with XH-Y, QML-Y appears to exhibit additional adaptive modifications, including smaller hepatocytes, a narrower hepatic sinusoidal structure, and greater glycogen accumulation.
Transcriptomic analysis revealed that differentially expressed genes (DEGs) identified in the comparison between XH-C and XH-Y were significantly enriched in pathways such as pyruvate metabolism, biosynthesis of unsaturated fatty acids, and fatty acid metabolism; whereas in the XH-Y versus QML-Y comparison, DEGs were primarily enriched in pathways related to bile secretion and cholesterol metabolism. Moreover, metabolomic profiling revealed that, in the XH-Y versus QML-Y comparison, critical TCA cycle intermediates—including fumarate37, succinate38, and malate39—along with glycerol40 and octadecenoic acid41 (key metabolites linked to fatty acid metabolism), were significantly enriched in pathways involved in energy metabolism. These metabolomic findings are highly consistent with the transcriptomic results, collectively indicating a clear adaptive divergence in energy metabolism regulation among yak populations inhabiting different altitudes. To further clarify their molecular regulatory basis, we conducted an in-depth analysis of key genes involved in energy metabolism.
Quantitative real-time PCR results showed that, compared to XH-C, XH-Y livers exhibit significantly up-regulated expression of the gluconeogenic gene PCK1 and FBP1, lipid oxidation-related genes ACOX1, and elevated levels of TCA cycle genes FH and MDH1.The gluconeogenic genes PCK1, FBP1, and G6PC1 regulate the conversion of oxaloacetate to phosphoenolpyruvate, fructose-1,6-bisphosphate to fructose-6-phosphate, and glucose-6-phosphate, respectively42,43,44. Additionally, lipid catabolism-related genes ACOX1, ACADS, and ACAA1 exhibit significant activity in triglyceride hydrolysis and fatty acid β-oxidation processes45,46,47. Key enzymes in the TCA cycle, such as IDH1, ACO148, SDHA49, MDH150, and FH51, catalyze steps including isocitrate dehydrogenation, succinate oxidation, and the conversion of fumarate to L-malate, which are crucial for mitochondrial function52. Studies have shown that increased glycogen storage may result from reduced glycolysis or enhanced gluconeogenesis32. Under acute hypoxic conditions, gluconeogenesis in mouse liver is rapidly enhanced53,54; high-altitude adapted rats increase hepatic glycogen storage by enhancing gluconeogenesis12,55. Furthermore, research on lipid metabolism has found that high-altitude hypoxia enhances fatty acid oxidation in the mouse liver36. Lipids stored in lipid droplets are hydrolyzed to release energy during periods of energy shortage56,57. Chronic hypoxia also induces a significant increase in TCA cycle metabolites in cardiac tissue32, and pancreatic cancer cells under hypoxic conditions show up-regulated expression of pyruvate dehydrogenase genes associated with the TCA cycle58. Our transcriptomic study comparing XH-C and XH-Y revealed significant differences in pyruvate metabolism, unsaturated fatty acid biosynthesis, fatty acid metabolism, and cholesterol metabolism pathways. Based on these findings, we speculate that yak livers may enhance hepatic glycogen synthesis and storage by up-regulating PCK1 and FBP1 expression and produce ATP through increased lipid hydrolysis and TCA cycle activity, thereby meeting their energy demands at high altitudes.
In our transcriptomic and metabolomic analyses, we found that KEGG pathways related to pyruvate metabolism, oxidative phosphorylation, and the TCA cycle were significantly enriched in QML-Y compared to XH-Y. Quantitative real-time PCR results further demonstrated that the expression of the gluconeogenesis-related gene G6PC1 was significantly upregulated in QML-Y, while the expression levels of lipid oxidation-related genes ACOX1 and ACADS were markedly downregulated. Additionally, the expression of IDH1、ACO1 and SDHA, a key gene involved in the TCA cycle, was also significantly reduced in QML-Y. Studies have shown that the reduction in lipid hydrolysis may be associated with a negative feedback regulatory mechanism triggered by increased intestinal lipid absorption, thereby suppressing hepatic lipid synthesis59. Oxidative phosphorylation (OXPHOS) represents one of the primary mechanisms through which cells generate large amounts of ATP via energy transfer processes60. Accumulating evidence suggests that, within the cytoplasm, oxidative phosphorylation collaborates with glycolysis to maintain energy homeostasis in energy-intensive cells such as hepatocellular carcinoma cells61. Moreover, under hypoxic conditions, human lung cancer cells can upregulate OXPHOS levels to produce more ATP and meet their elevated energy demands62. These findings indicate that oxidative phosphorylation plays a crucial role in energy supply under both high-energy-demand and hypoxic conditions. Based on these findings, we hypothesize that the pronounced upregulation of G6PC1 may be a key contributor to glycogen accumulation in the livers of QML-Y. Metabolomic analysis also revealed a significant increase in metabolites associated with oxidative phosphorylation in QML-Y, suggesting that these animals may enhance the OXPHOS process to more efficiently generate energy under hypoxic conditions. However, this adaptive mechanism requires further investigation to fully elucidate its functional significance.
Furthermore, we candidly acknowledge the following specific limitations in this study: both the transcriptome sequencing and subsequent RT-qPCR validation employed only three biological replicates per group (n = 3), which, while meeting the minimum recommended standards for high-throughput research, still constitutes a small sample size design. Although we have minimized false positive risks through rigorous experimental controls and statistical methods, the limited sample size may affect statistical power. Secondly, all samples were obtained from a single geographical region (Qinghai Province), potentially subject to regional environmental or genetic influences. To strengthen conclusion universality, future research will broaden geographical sampling to include yaks and cattle from diverse high-altitude regions, complemented by multi-omics approaches incorporating proteomic and epigenomic dimensions for deeper mechanistic insights into high-altitude adaptation.
Conclusions
In conclusion, the present study demonstrates that yaks are more efficient in meeting the metabolic demands of highland hypoxic environment through a narrower hepatic sinusoidal structure and significant hepatic glycogen accumulation compared to cattle. As well as significantly increased lipolytic metabolism and tricarboxylic acid (TCA) cycle activity, yaks produce energy more efficiently to meet the metabolic demands of highland hypoxic environment. High-altitude yaks exhibited smaller hepatocytes, narrower hepatic sinusoids and significant glycogen storage compared to low-altitude yaks. In addition, it is possible that high-altitude yaks maintain their energy levels by enhancing the efficiency of the oxidative phosphorylation pathway. However, further research is needed to fully understand the exact mechanism behind this adaptation.
Data availability
Sequence data that support the findings of this study have been deposited in the European Nucleotide Archive with the primary accession code PRJNA1162664.
References
Hu, R. et al. Nutritional interventions improved rumen functions and promoted compensatory growth of growth-retarded Yaks as revealed by integrated transcripts and Microbiome analyses. Front. Microbiol. 10, 318 (2019).
Burtscher, M., Gatterer, H., Burtscher, J. & Mairbaurl, H. Extreme terrestrial environments: life in thermal stress and hypoxia. A narrative review. Front. Physiol. 9, 572 (2018).
Liu, N. et al. A highland-adaptation mutation of the Epas1 protein increases its stability and disrupts the circadian clock in the plateau Pika. Cell. Rep. 39, 110816 (2022).
YuanTing, G. et al. Phylogenomic relationships and molecular convergences to subterranean life in rodent family Spalacidae. Zool. Res. 42(5), 671–674 (2021).
Lee, F. S. Hypoxia inducible factor pathway proteins in high-altitude mammals. Trends Biochem. Sci. 49, 79–92 (2024).
Tiwari, M. et al. Hypoxia related genes modulate in similar fashion in skin fibroblast cells of Yak (Bos grunniens) adapted to high altitude and native cows (Bos indicus) adapted to tropical climate during hypoxia stress. Int. J. Biometeorol. 68, 1675–1687 (2024).
Tiwari, M. et al. Selection signatures for high altitude adaptation in livestock: A review. Gene 927, 148757 (2024).
Amarjeet et al. Characterizing metabolome signature of colostrum, transition and mature milk of Indigenous cows (Bos indicus) adapted to high altitude environment of Leh-Ladakh. Food Chem. 464, 141767 (2025).
Yuan, D. et al. Characterization of gene expression related to mitochondrial morphology and function in Yak kidney at different altitudes. Chin. J. Anim. Sci. 60, 262–267 (2024).
Jing, X. et al. Comparison between Tibetan and Small-tailed Han sheep in adipocyte phenotype, lipid metabolism and energy homeostasis regulation of adipose tissues when consuming diets of different energy levels. Br. J. Nutr. 124, 668–680 (2020).
Alexandre, P. A. et al. Liver transcriptomic networks reveal main biological processes associated with feed efficiency in beef cattle. BMC Genom. 16, 1073 (2015).
Hong, J. Y. et al. Metabolic programming from neonate to adulthood in rats with maternal hypoxia. Int. J. Clin. Exp. Med. 9(2), 2921–2928 (2016).
Zhu, R. et al. Functional difference of malate-aspartate shuttle system in liver between plateau Zokor (Myospalax baileyi) and plateau Pika (Ochotona curzoniae). Acta Physiol. Sin. 177, 186 (2012).
Guan, J. et al. Comparative analysis of the MicroRNA transcriptome between Yak and cattle provides insight into high-altitude adaptation. PeerJ 5, e3959 (2017).
Shao, B. et al. Morphological adaptations of Yak (Bos grunniens) tongue to the foraging environment of the Qinghai-Tibetan plateau. J. Anim. Sci. 88, 2594–2603 (2010).
Zou, H. et al. Effects of nutritional deprivation and re-alimentation on the feed efficiency, blood biochemistry, and rumen microflora in Yaks (Bos grunniens). Animals 9, 10 (2019).
Chen, Y. B. et al. Single-cell transcriptomic survey of cell diversity and functional changes in Yak hearts at different altitude. Proteomics 23, 2200345 (2023).
Wan, R. et al. Characteristics of pulmonary microvascular structure in postnatal Yaks. Sci. Rep. 11, 18265 (2021).
Meng, X. Q. et al. UPLC-QTOF/MS-based metabolomic analysis of plasma reveals altitude effects on Yaks (Bos grunniens). Thai J. Vet. Med. 51, 551–559 (2012).
Li, J. Y. et al. Changes in the expression levels of elastic fibres in Yak lungs at different growth stages. BMC Dev. Biol. 21, 11 (2021).
Meng, X. Q. et al. Changes of related to energy metabolism genes in the Yak lung tissue after birth. Basic. Clin. Pharmacol. Toxicol. 126, 421–422 (2020).
Jing, X. P. et al. Energy substrate metabolism in skeletal muscle and liver when consuming diets of different energy levels: comparison between Tibetan and Small-tailed Han sheep. Animal 15, 100162 (2021).
Rui, L. Energy metabolism in the liver. Compr. Physiol. 4, 177–197 (2014).
Xiong, L. et al. The study of the response of fat metabolism to long-term energy stress based on serum, fatty acid and transcriptome profiles in Yaks. Animals 10, 1150 (2020).
Nettleton, G. S. & Carpenter, A. M. Studies of the mechanism of the periodic acid-Schiff histochemical reaction for glycogen using infrared spectroscopy and model chemical compounds. Stain Technol. 52, 63–77 (1977).
Kanehisa, M. et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012).
Kanehisa, M., S, Goto & KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Science: Publication Protein Soc. 28, 11: 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Tiwari, M. et al. Selection of species specific panel of reference genes in peripheral blood mononuclear cells of native livestock species adapted to trans-Himalayan region of Leh-Ladakh. Sci. Rep. 12, 18473 (2022).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Midha, A. D. et al. Organ-specific fuel rewiring in acute and chronic hypoxia redistributes glucose and fatty acid metabolism. Cell. Metab. 35, 504–516 (2023).
Li, D. et al. Liver transcriptome shows differences between acute hypoxia-tolerant and intolerant individuals of greater Amberjack (Seriola dumerili). Animals 13, 2717 (2023).
Chen, Q. et al. A study on the differences in rumen microbiota-liver gluconeogenesis-mitochondrial interaction between Tibetan sheep and Hu sheep in the Qinghai-Tibet plateau. Animals 15, 1603 (2025).
Kong, C., Chen, B. & Zhao, H. The effects of intermittent hypoxia on hepatic glycogen in rats and the intervention role of tempol. Biotech. World. 8, 62–63 (2014).
Liu, G. et al. Energy metabolic mechanisms for high altitude sickness: downregulation of Glycolysis and upregulation of the lactic acid/amino acid-pyruvate-TCA pathways and fatty acid oxidation. Sci. Total Environ. 894, 164998 (2023).
Zhong, X. et al. Complex metabolic interactions between ovary, plasma, urine, and hair in ovarian cancer. Front. Oncol. Vol. 12, 916375 (2022).
Liu, X. et al. Biosynthetic pathway and metabolic engineering of succinic acid. Front. Bioeng. Biotechnol. Vol. 10, 843887 (2022).
Wei, Z. et al. Microbial biosynthesis of L-Malic acid and related metabolic engineering strategies: advances and prospects. Front. Bioeng. Biotechnol. Vol. 9, 765685 (2021).
Hui, S. et al. Quantitative fluxomics of Circulating metabolites. Cell. Metabolism Vol. 32, 4 (2020).
Ikeguchi, S. et al. Inhibitory effect of the gut microbial Linoleic acid metabolites, 10-oxo-trans-11-octadecenoic acid and 10-hydroxy-cis-12-octadecenoic acid, on BV-2 microglial cell activation. J. Pharmacol. Sci. Vol. 138, 1 (2018).
Lamont, B. J. et al. Expression of human fructose-1,6-bisphosphatase in the liver of Transgenic mice results in increased glycerol gluconeogenesis. Endocrinology 147, 2764–2772 (2006).
Weston, B. W. et al. Glucose-6-phosphatase mutation G188R confers an atypical glycogen storage disease type 1b phenotype. Pediatr. Res. 48, 329–334 (2000).
Tang, X. et al. G6PC1 expression as a prognostic biomarker associated with metabolic reprogramming and tumor microenvironment in hepatocellular carcinoma. Front. Immunol. 16, 1623315 (2025).
Morita, A. et al. Novel ACOX1 mutations in two siblings with peroxisomal acyl-CoA oxidase deficiency. Brain Dev. 43, 475–481 (2021).
He, M. et al. Identification and characterization of new long chain acyl-CoA dehydrogenases. Mol. Genet. Metab. 102, 418–429 (2011).
Ferdinandusse, S. et al. Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J. Lipid Res. 42, 1987–1995 (2001).
Geisbrecht, B. V. & Gould, S. J. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J. Biol. Chem. 274, 30527–30533 (1999).
Renkema, G. H. et al. SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors. Eur. J. Hum. Genet. 23, 202–209 (2015).
Shi, C. et al. The acetylation of MDH1 and IDH1 is associated with energy metabolism in acute liver failure. iScience 27, 109678 (2024).
Ajalla Aleixo, M. A. et al. Structural, biochemical and biophysical characterization of Recombinant human fumarate hydratase. FEBS J. 286, 1925–1940 (2019).
Wang, C. et al. The tricarboxylic acid cycle is inhibited under acute stress from carbonate alkalinity in the gills of Eriocheir sinensis. Comp. Biochem. Physiol. Part D Genomics Proteomics 51, 101245 (2024).
Li, C. et al. The IGF axis in baboon pregnancy: placental and systemic responses to feeding 70% global ad libitum diet. Placenta 28, 1200–1210 (2007).
Li, W., Jiang, T. & Zhao, C. Changes of hepatic ACC1 and CPT1A and their effects on glucose and lipid metabolism in male mice at medium and high altitude. J. Chin. High. Alt Med. Biol. 45, 200–205 (2024).
She, X. et al. Research advances on alterations of hepatic glucose and lipid metabolism under hypobaric hypoxia at high altitudes. China J. Mod. Med. 35, 37–45 (2025).
Chao, M. K., Yi, X. D. & Pang, W. J. Regulation of the mTOR signaling pathway on lipogenesis. China J. Biochem. Mol. Biol. 38, 1477–1485 (2022).
Son, S. H. et al. Perilipin 2 (PLIN2)-deficiency does not increase cholesterol-induced toxicity in macrophages. PLoS ONE. 7, e33063 (2012).
Obeidat, N. M. et al. Effects of Cyclic acute and chronic hypoxia on the expression levels of metabolism related genes in a pancreatic cancer cell line. Biomed. Rep. 17, 81 (2022).
Zhao, S. et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579(7800), 586–591 (2020).
Kowaltowski, A. J. & Abdulkader, F. Textbook oxidative phosphorylation needs to be rewritten. Trends Biochem. Sci. 50, 87–88 (2025).
Zhang, Y. et al. Multifaceted roles of aerobic Glycolysis and oxidative phosphorylation in hepatocellular carcinoma. PeerJ 11, e14797 (2023).
Öğünç Keçeci, Y. & İncesu, Z. Mitochondrial oxidative phosphorylation became functional under aglycemic hypoxia conditions in A549 cells. Mol. Biol. Rep. 49, 8219–8228 (2022).
Acknowledgements
We are grateful to our colleagues in the laboratory for their assistance in sample collection.
Funding
This study was supported by the Second Tibetan Plateau Scientific Expedition and Research Program (grant number: 2019QZKK0501) and Qinghai Basic Research Project (grant number: 2023-ZJ-708) in analysis and interpretation of date.
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology, Jiarui Chen and Qing Wei; software, Shuwen Wang, Zhanlei Rong, Nating Huang and Jingyi Li; formal analysis, Jingqing Ma and Na Song; investigation, Jingqing Ma; resources, Jiarui Chen and Qing Wei; data curation, Jingqing Ma, Zhanlei Rong, Xun Zhang, Nating Huang, Shuwen Wang and Jingyi Li; writing—original draft preparation, Jingqing Ma and Na Song; writing—review and editing, Jiarui Chen and Qing Wei; project administration, Jiarui Chen and Qing Wei; funding acquisition, Jiarui Chen. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All of the animal studies were performed based on the guidelines of the Institutional Animal Care and Use of Laboratory Animals and were approved by the Animal Welfare and Ethics Committee at the Qinghai University.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, J., Song, N., Huang, N. et al. Effective energy harvesting of Yak by regulating the glucose and lipid metabolism in liver to adapt to the plateau environment. Sci Rep 16, 944 (2026). https://doi.org/10.1038/s41598-025-30526-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-30526-z
