
Abstract
The palm Euterpe precatoria (Mart.), one of the most widely distributed species in the Amazon basin, plays a crucial role for traditional riverine communities. It thrives in nutrient-poor, well-drained soils, important traits for forest restoration. Additionally, the species is extensively exploited for açaí extraction. We sampled 23 natural geographic populations in the Brazilian states of Amazonas and Rondônia, characterizing them using single nucleotide polymorphism (SNP) markers. We divided the dataset into putatively neutral and outlier SNPs (under selection) to estimate genomic diversity and structure. To identify potential causes of selective pressure, we explored the factors that may have contributed to the differences and similarities between the two datasets. Our results suggest that anthropogenic pressure may be impacting the species’ genomic diversity. We also found significant population structure, likely shaped by limited gene flow influenced by river flow and anthropogenic activity. Based on these patterns, we propose that sampling at least 35 individuals per site across 18 localities to capture the species’ genomic diversity. The genomic groups identified here should guide future conservation actions, such as germplasm banking, and restoration efforts.
Similar content being viewed by others

Introduction
The Brazilian Amazônia has the highest diversity of palms (148) in the region1. Among the palm species that occur in there, açaí-do-amazonas or açaí-solitário (Euterpe precatoria Mart.) stands out for its wide distribution and extensive use. The species occurs from Central America to northern South America, with especially high abundance in the central and western Amazônia2. It is one of the most hyperdominant plant species in the bioregion and has the highest levels of relative abundance across different forest types3. Euterpe precatoria also thrives in nutrient-poor and well-drained latosols and ultisols, which makes it an important alternative for forest restoration in degraded areas of the Amazônia4.
Traditionally, the species E. precatoria has been used in construction, utensils, and medicine5, but it is especially important as a food resource, for fruits and heart-of-palm. In the last 10 years, açaí fruit production in Brazil has grown by 80% (Instituto Brasileiro de Geografia e Estatística)6, with 31% of the production occurring in the state of Amazonas7,8. This palm plays an essential role for traditional riverine human populations, contributing to the economic, social, and cultural subsistence of 6 million families9. Assessing the genomic diversity patterns and population structure of E. precatoria at the Amazônia scale can help us understand the factors that influence the dynamics of the species, such as selective pressure, gene flow, and inbreeding. With this information, we were able to identify populations and sites with specific genomic variation to be used in restoration efforts within the Amazônia bioregion and in the creation of germplasm banks for their conservation.
Population genomics provides comprehensive molecular insights into species diversity and population structure, as next-generation sequencing (NGS) enables the identification of millions of single nucleotide polymorphisms (SNPs), even in non-model organisms. Although genetic diversity and population structure in E. precatoria have been previously investigated using microsatellites7, SNP markers offer higher resolution for detecting population structure due to their abundance and broader genomic coverage10,11. Population genomics is based on two main principles. First, neutral loci across the genome are similarly affected by demography and evolutionary history, as with microsatellite markers, and can therefore be used to estimate genomic diversity and structure. Second, it enables the identification of loci under selection that show outlier patterns, and the reliable detection of these markers may help uncover strong signals of population differentiation12,13.
Outlier loci may present different estimates of diversity and genomic structure in natural populations, as they may indicate putative adaptation or functionality. In addition to changes in population estimates, genomic sequences with outlier SNPs can be annotated according to gene ontology terms. This can allow us to determine cellular components, molecular functions, and biological processes in which each gene product is involved14, providing insights into traits under selection in the species. By combining neutral and under selection loci, it is possible to obtain a clearer picture of the current state of natural populations and how selection is acting on them, helping determine which populations should be used in conservation and restoration programs for the species15,16,17. In this study, we applied a genotyping-by-sequencing (GBS) approach to identify SNP markers, using restriction enzyme digestion followed by Illumina sequencing. This reduced-representation method allows cost-effective SNP discovery in non-model species.
The main goal of this research is to analyze the genomic diversity and population structure of E. precatoria using putatively neutral and outlier SNPs. The specific objectives are: (1) To identify the putative outlier SNPs and define gene ontology terms associated with the sequences in which they occur. Our hypothesis is that the growing and continuous extraction of açaí is creating selective pressures on the sampled populations, and that the gene ontology terms indicate functions related to productivity. (2) To estimate genomic diversity and population structure based on both SNP datasets, identify their differences and similarities, and explore potential factors underlying these patterns. Our hypothesis is that, since outlier markers are subject to selective pressures, the differences between outlier and neutral markers may indicate the adaptation the species is undergoing. (3) To establish minimum sample sizes and define factors for identifying areas to be used for the restoration and conservation of the species. The use of two SNP datasets may improve the identification of priority sites for initiatives involving the species.
Results
SNP discovery
A total 1,077,552 loci were genotyped in 309 individuals from 23 localities (Fig. 1) in the Brazilian Amazônia. The mean coverage per sample was 12.6× (SD = 2.8×, min = 7.1×, max = 28.3×), with a mean of 101.0 sites per locus. After applying the filters in the populations module, 17,514 high-quality SNPs were retained.

Sampling locations. Localities were named according to the nearest municipality. Map colors represent predominant Vegetation Denomination/Natural Area or Anthropic Area according to the Instituto Brasileiro de Geografia e Estatística74. Base map from IBGE (https://geoftp.ibge.gov.br/informacoes_ambientais/vegetacao/vetores/escala_250_mil/versao_2021/). Map created by A.F.F. in QGIS version 3.34.10-Prizren (QGIS Development Team, https://qgis.org).
Identification of putative outlier SNPs
The pcadapt approach, which uses principal components analysis (PCA) to identify SNPs associated with population structure, identified 414 loci putatively under selection (2.36%; Supplementary Fig. S1a; Supplementary Fig. S2a–c online). In contrast, fsthet and BayeScan methods, both based on FST outlier detection, identifying loci with higher-than-expected genetic differentiation under neutrality18,19 detected 1,049 (5.99%; Supplementary Fig. S1b online) and 2,421 (13.82%; Supplementary Fig. S1c online) SNPs, respectively. Between pcadapt and fsthet, only one locus was found to be putatively under selection. In contrast, 357 SNPs were identified as outliers between pcadapt and BayeScan. Fsthet and BayeScan identified 499 SNPs in common. Among the three methods, only three SNPs were identified as outliers (see Supplementary Fig. S1d online). These discrepancies likely reflect differences in model assumptions and the stringency of outlier classification. To minimize false positives and account for method-specific biases, 860 SNPs (4.91%) were considered putative outliers, while 16,654 SNPs (95.09%) were classified as neutral, forming the two analysis datasets. Using multiple complementary approaches enhances the reliability of outlier detection by reducing Type I error19.
Among the 552 sequences generated by Stacks that contained outlier SNPs – sequences in which at least two of the three methods (pcadapt, fsthet, or BayeScan) identified an outlier SNP – 173 showed BLAST hits, 156 were successfully mapped, and 132 were functionally annotated (Supplementary Table S1; Supplementary Fig. S3a online). The species with the highest number of top hits with E. precatoria were the palms Elaeis guineensis Jacq. (54 hits), Cocos nucifera L. (40 hits), and Phoenix dactylifera L. (27 hits; Supplementary Fig. S3b online). A total of 307 genes were identified within these sequences. Based on GO terms, 111 are linked to molecular function, 104 to biological processes, and 92 to cellular components (Supplementary Fig. S3c online). Genes associated with binding and catalytic activity were prominent in the molecular function category, while those involved in cellular and metabolic processes stood out in the biological process category. In the cellular component category, genes related to cellular and membrane structures were the the predominant group.
Genomic diversity of Euterpe precatoria based on SNP markers
Based on 16,654 putatively neutral loci, the highest levels of genomic diversity, measured by observed heterozygosity (HO), gene diversity (HS), and allelic richness (AR), were found in the localities along the Alto Solimões River, São Paulo de Olivença, and Santo Antônio de Iça. In contrast, localities along the Uatumã and Amazonas River, such as São Sebastião do Uatumã and Parintins, consistently exhibited lower diversity estimates (HO, HS, and A). Private alleles (Ap) were more frequent in Porto Velho and Manicoré. Fixation indices were generally low or zero across localities (Table 1), indicating an excess of heterozygotes relative to Hardy-Weinberg expectations.
Resampling based on minimum sample size using the SaSii approach indicated stability in the estimates with 60 individuals (Supplementary Fig. S4). According to the results, 15 individuals per site were sufficient to capture 90% of the genomic diversity estimates for E. precatoria. This suggests that our sampling reflects the actual species composition at most sites, except for four sites in the Alto Solimões (Table 1), where sampling was lower. To capture all possible rare and common alleles, a sample size of 35 individuals was the ideal value (Supplementary Fig. S4).
When analyzing the 860 putative outlier SNPs, an overall increase in diversity metrics was observed, with the same localities from Alto Solimões maintaining the highest HO, HS, and AR values. Conversely, the lowest diversity estimates shifted to Urucurá (Uatumã River) and localities from Rondônia. Fixation indices remained negative across all sites (Table 2).
Population structure of E. precatoria based on SNP markers
The analysis of 16,654 putatively neutral SNPs indicated a clear population divergence, as reflected by FST values, which measure genetic divergence among localities. The low but positive FIS, contributing alongside FST to a positive FIT, indicates a slight heterozygote deficit within populations, possibly due to mild deviations from random mating. In contrast, the 860 outlier SNPs identified population subdivisions as the primary structuring factor, with positive FST and negative FIS values (Table 3).
AMOVA results showed that variation was predominantly within populations (58.81%). However, when considering outlier SNPs, within-population variation increased to 66%. The AMOVA-based analogue of FST (ՓST) was higher for neutral markers (0.412, p < 0.001) compared to outlier SNPs (0.342, p < 0.001), indicating stronger population differentiation with neutral SNPs (Table 3).
Pairwise FST calculated from neutral SNPs revealed the greatest genomic divergence between Atalaia do Norte (Alto Solimões River) and Parintins (Amazonas River, FST = 0.612), two sites located at opposite ends of the state of Amazonas, near the borders with the state of Pará and Peru (Fig. 2a). The lowest divergence was observed between Presidente Figueiredo (Uatumã River) and Anori (Solimões River, FST = 0.011). Comparing neutral and outlier markers, a general trend toward reduced divergence was observed, although Atalaia do Norte, Benjamin Constant, and Tabatinga showed increased FST values in the outlier dataset (Fig. 2b). The order of the sites was similar in the heatmaps for both datasets, a pattern further reinforced by NJ dendrograms, which grouped populations along the Uatumã and Amazon Rivers, followed by sites from the Madeira and Solimões Rivers, with a distinct cluster formed by Alto Solimões River (Fig. 2c,d).

Heatmaps of pairwise FST values and neighbour-joining dendrograms for 309 açaí palms (E. precatoria) sampled across 23 localities in the states of Amazonas and Rondônia. (a) Genomic divergence estimated from 16,654 putatively neutral SNPs; (b) Genomic divergence from 860 putatively outlier SNPs; (c) Dendrogram based on Nei’s distance and the agglomerative neighbor-joining method using neutral SNPs; (d) Dendrogram based on outlier SNPs. Consensus topologies were generated from 1,000 bootstraps with the Cavalli‐Sforza and Edwards (1967) chord distance.
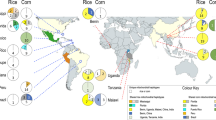
In the map of Fig. 3a, we observe the results of the sNMF analysis based on neutral SNPs, which identified 16 genomic groups. This number of clusters was defined using the cross-entropy criterion, selecting the value of K that minimized model error (see Supplementary Fig. S5 online). The analysis revealed a high degree of genetic structuring among localities and captured signals of introgression between groups. Nearby sites tended to share similar group compositions, as observed in the Alto Solimões River region, where Atalaia do Norte, Benjamin Constant, and Tabatinga all shared genomic Group 9. However, some sampling sites formed exclusive clusters, such as those located along the Solimões River.

Genetic structure of 309 E. precatoria palms based on 16,654 putatively neutral SNPs and 860 outlier SNPs. (a)Geographic Distribution of the genomic groups defined by DAPC with K-means clustering in the states of Amazonas and Rondônia, with K = 12 (neutral SNPs) and K = 9 (outlier SNPs); (b) Bar plot of sNMF ancestry coefficients for K = 16, based on neutral SNPs; (c) Bar plot of sNMF ancestry coefficients for K = 8, based on outlier SNPs. Localities selected to represent the main genomic clusters (see in Discussion) are highlighted with a rectangle around their names in panel a and c. Base map from DIVA-GIS repository (https://diva-gis.org/data.html; Brazil, Inland Water). Map created by A.F.F. in QGIS version 3.34.10-Prizren (QGIS Development Team, https://qgis.org).
In the DAPC analysis by K-means, also based on neutral SNPs, 12 genomic groups were identified, based on the lowest Bayesian Information Criterion (BIC) (Fig. 3b; Supplementary Fig. S6a–d online). Similarly, nearby localities generally shared group compositions, but some differences emerged. Atalaia do Norte, in the Alto Solimões River, formed an exclusive cluster (Group 3). Codajás, Anori, and Presidente Figueiredo were grouped together in a single cluster (Group 2), while Manicoré, Novo Airão, Manacapuru, and Borba shared Group 7. The combination of Groups 2 and 7 in this region suggests distinct and localized patterns of gene flow compared to other areas.
In the map of Fig. 3c, we observe the results of the sNMF analysis based on 860 outlier SNPs, which identified eight genomic groups (Supplementary Fig. S7 online). Compared to the 16 groups detected with neutral SNPs, the number of groups was reduced by half. Several previously exclusive clusters became shared among localities. For instance, sampling sites at the beginning of the Uatumã River and along the Amazon River now clustered together in Group 4. Overall, the sNMF results with outlier SNPs revealed broader groupings, possibly reflecting selection-driven convergence across distant sites.
In the DAPC analysis by K-means, the number of genomic groups also decreased, from 12 (neutral markers) to nine (Fig. 3d; Supplementary Fig. S8a–d online). Atalaia do Norte joined other Alto Solimões River localities in Group 3, while Urucurá and Parintins clustered with Amazon River sites (Group 9). Porto Velho grouped with other sites from Rondônia in Group 4. Additionally, localities that previously formed a single group (Group 7) with neutral SNPs—such as Manicoré, Novo Airão, Manacapuru, and Borba—were now split into two distinct groups: Manicoré and Novo Airão in Group 5, and Manacapuru and Borba in Group 2. Compared to sNMF, K-means identified one additional group, although the general patterns of regional clustering remained consistent.
Finally, using both neutral and outlier SNPs, a significant correlation was found between genetic and geographic distance, according to Mantel’s test. For the neutral SNPs, the test resulted in a p-value = 0.0001 and r = 0.906; for outlier SNPs, p-value = 0.0001 and r = 0.729. Thus, regardless of selection, geographically closer individuals were also genetically closer.
Discussion
As E. precatoria is one of the most hyperdominant species in the Amazônia region3, and since 17% of the Amazônia’s territory has already been lost since pre-Columbian times20, we aimed to provide applied results from population genomics that can contribute to its use in the restoration of the biome. This palm has also become the main harvested species for NTFP in recent years21, making the results applicable to future conservation programs. For this reason, efforts have been made to re-evaluate and expand the sampling areas of E. precatoria in the Brazilian Amazônia. GO terms were identified in the DNA sequences with outlier SNPs, and genomic diversity and population structure were estimated using both neutral and outlier markers.
The identification of putatively outlier SNPs revealed loci associated with a range of adaptive responses, suggesting that selective pressures are acting on multiple functional traits across the studied populations. Although the methods applied rely on distinct mechanisms and assumptions and detected distinct sets of markers22, a consistent set of outlier SNPs was recovered, supporting the hypothesis that human-driven extraction may be influencing genetic variation. Despite the limited proportion of annotated sequences – partly due to the absence of a reference genome23,24 and the BLASTX program’s focus on coding regions25– the identified GO terms offer important insights into ongoing selection processes.
Notably, we found loci associated with heat stress tolerance and enhanced photosynthetic efficiency (Loci 1507, 14540 and 28678), as well as resistance to fungal infection and response to jasmonic acid signaling (Loci 7191 and 10984). Genes involved in the hormonal regulation of root growth, such as auxin signaling and root morphogenesis, were also detected (Loci 2728, 515 and 3018; Supplementary Table S1 online). These traits may reflect selective pressures favoring increased stress tolerance and photosynthetic efficiency, ultimately contributing to greater energetic efficiency and fruit production. Additionally, these functions point to broader adaptive responses, likely driven by habitat disturbance and climate-related stress. Given that this is the first genome-wide scan for E. precatoria, we prioritized a broad perspective to capture candidate loci potentially relevant across multiple environmental and anthropogenic contexts. Future studies could build on these findings by testing hypotheses of local adaptation under specific land-use regimes.
Neutral markers are shaped by mutations, recombination, genetic drift, and gene flow. In contrast, outlier SNPs are also influenced by selective forces15 and may provide insights into adaptive advantages17,26,27. Consequently, the evolutionary processes acting on these two sets of markers differ, and the genomic patterns defined by outlier SNPs may not mirror those revealed by neutral markers15,27.
In our study, we observed that the population structure analysis based on outlier SNPs identified fewer genomic groups compared to analysis with neutral markers (Fig. 3). Although this may initially seem contradictory, such a pattern is expected when directional selection acts similarly across geographically distinct populations, favoring shared adaptive traits and reducing genomic divergence at loci under selection. This could explain why localities that were initially structured into distinct genomic groups based on neutral SNPs became clustered when outlier SNPs were considered. The functional annotation of outlier loci supports this interpretation, revealing associations with GO terms related to traits that may confer adaptive advantages potentially enhance fruit yield (Supplementary Table S1 online). While we hypothesize that such traits are under selection due to high demand for açaí fruits28, our current approach does not account for environmental predictors or phenotypic validation. Therefore, further studies integrating genotype–phenotype associations and ecological data are needed to confirm this relationship.
The study by Ramos et al.7, which analyzed 19 populations of E. precatoria also included in our sampling, identified high genetic diversity across all localities, providing important information on the species’ conservation status. By expanding the geographic coverage and analyzing both neutral and outlier SNPs, our study provides additional insights into how ongoing NTFP extractivism may be influencing this status. This interpretation is supported by the consistency observed between the results obtained from neutral and outlier markers. The localities along the Uatumã and Amazon Rivers, where lower genomic diversity was detected, are more anthropized and located closer to the capital city of Manaus (Fig. 1). In contrast, populations from the Alto Solimões region, which exhibited higher diversity, are more geographically isolated and subject to lower extraction rates (Produção da extração vegetal e da silvicultura6; see Supplementary Table S2). Although we did not perform a formal correlation analysis, this spatial pattern suggests a possible link between anthropogenic pressure and reduced genomic diversity. This may be associated with the logistical advantages of harvesting closer to urban centers, where the perishability of the fruit and reduced transportation times encourage higher extraction volumes8. Further analyses integrating extraction intensity, landscape features, and genomic data could help clarify this relationship.
Although the higher genomic diversity of the populations in the Alto Solimões River may result from lower extractive pressure, isolation by distance (IBD), driven by pollination and seed dispersal, may also affect the species’ diversity and structure patterns. Pollination of E. precatoria is primarily carried out by beetles from Staphylinidae, Chrysomelidae, and Curculionidae families, as well as bees from the Halictidae family29, with an average distance of 447 m4. Dispersal can range from 4 to 1400 m, with an average of 400 m, and the majority of seeds are deposited close to the parent palm. This behavior characterizes the species’ primary dispersal syndrome, which occurs through precipitation4. Additionally, there are reports of dispersal occurring via rivers30. As pointed out by Ramos et al.7 pollination and dispersal syndromes can limit gene flow and promote IBD. The fact that diversity was mostly concentrated in one region highlights how geographic constraints can influence the genomic diversity of E. precatoria. The influence of IBD was also observed in population structuring, as subdivisions and differences within localities were the main factors identified by overall FST and AMOVA (Table 3), in both SNP datasets. The results of Mantel’s tests, supported by pairwise FST and NJ dendrograms (Fig. 2) indicated that the gene flow was proportional to geographic distances, with the species being dispersed mainly across nearby regions7.
Reinforcing the importance of rivers for gene flow (Fig. 3), we found that the distribution of genomic groups often aligned with the directionality of river flow, forming a geographic gradient. Nearby localities tended to share genomic clusters with neighboring sites, as previously observed with microsatellite markers7. Like E. precatoria, the palm Mauritia flexuosa L.f., also hyperdominant in the Amazônia3, exhibited an IBD pattern in population structure when analyzed with microsatellite markers31,32. Population plots indicated that genetic structure followed watersheds boundaries, with populations in the same basin being genetically closer. In contrast, another Amazonian palm, Astrocaryum aculeatum G.Mey., did not exhibit IBD in the 15 populations analyzed using microsatellites. However, genetic groups were still formed according to the hydrographic basin in which the populations were located32. According to the authors, one of the main factors responsible for this structuring was the human-mediated transport of products along the rivers32. Along with short-distance dispersal by rodents and birds33, human transport has been identified as a secondary dispersal mechanism for E. precatoria, carried out by local harvesters, who help disperse genetic material over long distances34.
This dispersal can occur due to various factors, including preferences for fruit quality. Consequently, sites that did not show genomic structures consistent with their river patterns may be experiencing anthropogenic dispersal. This was the case, for example, with the genomic groups from Presidente Figueiredo, identified by DAPC with K-means on both types of SNPs. The management of E. precatoria by humans may have intensified in recent decades6. However, there is evidence that the species has been managed for centuries by Amerindian populations35. One of the main indications is its abundance and richness, which are likely the result of transformation caused by pre-Columbian societies36,37. This long and continuous process has shaped gene flow patterns, creating a genomic structure distinct from that expected based solely on river dynamics. While our results point to the importance of both natural and anthropogenic factors in shaping gene flow, future studies integrating extraction records and hydrological connectivity could provide a more detailed understanding of these dynamics.
In recent years, the highest levels of deforestation in the Amazônia occurred in the states of Pará, Mato Grosso, Rondônia, and Amazonas20, with the latter two included in this study. Promoting the sustainable use of biodiversity and monetizing ecosystem services are among the strategies that may help reverse this trend1. Due to its hyperdominance across various forest types, as terra-firme (upland areas), non-floodable areas, and várzeas (lowland areas)38, and soil conditions within the Amazônia bioregion3,4, E. precatoria stands out as a valuable resource for restoration programs. Moreover, as shown in this study, extractive pressure driven by high demand for açaí fruits may be contributing to a decline in the species’ genomic diversity in certain regions of Amazonas.
A comprehensive understanding of the factors influencing genomic diversity and population structure is crucial for making informed decisions about the use of species. Funk et al.15 suggested that combining data from neutral and outlier markers improves restoration and conservation strategies, as these markers provide complementary insights. Therefore, the information presented here can support the identification of locally adapted genotypes and the genetic similarities among them, ultimately contributing to the preservation of the species’ gene pool and enhancing its resilience in ongoing and future efforts17. Analyses of population structure and individual assignment can be used to estimate gene dispersal, detect immigrants, and evaluate the influence of natural or anthropogenic barriers on connectivity39. Notably, we observed a greater number of genomic groups when using neutral SNPs compared to outliers. This likely reflects the influence of directional selection on outlier loci, which can reduce variability within groups, whereas neutral markers better capture population structure.
Given the pronounced genomic structuring observed among the sampled localities – likely driven by IBD and limited gene flow – we identified specific sites that serve as source regions representing the main genomic clusters of E. precatoria, primarily guided by neutral SNPs and further confirmed by outlier SNPs. When multiple localities belonged to the same genomic group, we prioritized those with the highest genomic diversity, primarily based on observed heterozygosity. We opted for observed heterozygosity (HO) as it directly reflects realized genetic variation within populations and is not dependent on assumptions of Hardy-Weinberg equilibrium. Additionally, we avoided metrics such as number of alleles and private alleles, which are highly sensitive to sample size and thus less reliable for meaningful comparisons across populations40.
Thus, the genomic clusters identified here not only reflect evolutionary processes but also offer practical guidance for conserving E. precatoria genetic diversity across its distribution range. Accordingly, Atalaia do Norte, Tabatinga, São Paulo de Olivença, and Fonte Boa were selected to represent the diversity of the Alto Solimões River region. Coari, Codajás, Anori, and Manacapuru captured the genomic variation of the Solimões River basin. In the Amazon basin, Itapiranga and Parintins showed distinct genomic profiles. In the Negro River basin, Novo Airão was the only sampled site and represented that region. In the Uatumã River region, all three localities – São Sebastião do Uatumã, Urucurá, and Presidente Figueiredo – exhibited distinct genomic groups. Finally, in the Madeira basin, Borba and Manicoré (Amazonas state), along with Porto Velho and Guajará-Mirim (Rondônia state), stood out due to their unique genomic structure. In Fig. 3, these selected localities are highlighted with a rectangle around their names to facilitate identification. Nevertheless, we acknowledge that additional sampling across other regions of the species’ distribution, particularly in the western and non-Brazilian portions of its range, could further refine the genomic landscape and conservation recommendations.
To translate these genomic insights into practical guidelines for restoration and conservation, it is important to consider both the distance between locations and the number of individuals per site when planning such initiatives41. To minimize the chances of collecting genetically related seeds, a safe distance for collection should be greater than 470 m4. A sampling size of 35 individuals was found to be sufficient to capture a representative set of both common and rare alleles in the populations. Euterpe precatoria has a predominantly allogamous mating system with cross-pollination42,43. Previous studies have shown that for cross-pollination, collecting 30 unrelated individuals will capture 95% of the diversity, although it may not capture the rarest alleles in the populations. This percentage can be increased to 99% by collecting 45 individuals44,45,46,47,48. We recommend including 35 to 45 individuals per location, especially considering the spatial genetic structure influenced by pollination and primary seed dispersal4.
Conclusion
This study provides a comprehensive overview of the genomic diversity and population structure of E. precatoria. The analysis of outlier SNPs revealed loci associated with traits that may reflect selective pressures related to fruit productivity, as well as broader adaptive responses to environmental stress. The comparison between neutral and outlier SNPs identified divergences and similarities. Regarding divergence, fewer genomic groups were identified with outlier SNPs, which may reflect directional selection. This interpretation is supported by functional annotation, particularly of GO terms related to traits enhancing fruit productivity. However, further studies incorporating genotype–phenotype associations are needed to confirm this link. As for similarities, both SNP datasets revealed areas of lower genomic diversity, especially in more anthropized regions. Although no formal correlation was done, this spatial pattern suggests association between anthropogenic pressure and reduced diversity. Moreover, gene flow appears to be shaped by the species’ syndromes, along with river flow and human-mediated movement, explaining the highly structured genomic groups across the species’ distribution. This strong structuration poses a challenge for sampling: at least 18 of the 23 localities and 35 individuals per site are needed to fully capture the species’ diversity. Proper sampling strategies are therefore essential to ensure the representation of this diversity in conservation and restoration programs. In the future, modeling efforts based on the SNPs characterized here — or using whole genome sequencing —will be important to identify individuals with greater resilience to critical climate change scenarios.
Materials and methods
Sampling
We collected adult leaves from 309 individuals of E. precatoria across 23 localities between 2011 and 2015: 20 in the state of Amazonas and three in the state of Rondônia, with 4 to 15 individuals sampled per site (Fig. 1). The sampled regions are located within the basins of the Alto Solimões/Solimões, Amazonas, Negro, Madeira, and Uatumã Rivers. It is important to note that the individuals sampled belong to E. precatoria var. precatoria, as all sampled areas fall within the distribution range of this variety, corresponding to Central and Western Amazônia. The other recognized variety, E. precatoria var. longevaginata, occurs in the Andean region49. The appropriate permissions for the collection of plant material were taken, and the collections were registered according to the National System for the Management of Genetic Heritage and Associated Traditional Knowledge (SISGEN), as stated by Brazilian Decree No. 8,772 (May 11, 2016) and regulated by Brazilian Law No. 13,123 (May 20, 2015; SISGEN numbers: AA9537B and A411583, Brazil). The plant material was taxonomically identified by specialists of the genus Euterpe at the Universidade Federal do Amazonas (UFAM), Dr. Santiago L. F. Ramos and Dr. Doriane Picanço-Rodrigues, who are co-authors of this study.
DNA extraction and genotyping-by-sequencing library
The DNA was extracted from adult plant leaves using 2% CTAB50. DNA quantification was performed by electrophoresis on a 1% agarose gel with GelRedTM (Biotium, Hayward) and by using a Qubit fluorometer (Thermo Fisher Scientific, US).
We applied the genotyping-by-sequencing (GBS) approach to obtain SNP markers. This technique uses restriction enzymes to reduce DNA complexity, followed by sequence identification via adapters (barcodes), enabling multiplex libraries for NGS51. Specifically, we used the restriction enzymes MseI and PstI for double digestion of the samples, as described by Poland et al.52. Barcodes were added to the digested fragments for individual identification. Polymerase chain reaction (PCR) was used to enrich the barcoded fragments, with primers complementary to the adapters in the GBS library. Sequencing of PCR products was performed on the Illumina NextSeq 2000 platform.
SNP identification
FastQC53 was used to verify the overall quality of the raw reads. The Stacks 2.62 pipeline54 was applied in several steps of the SNP calling process: sample demultiplexing, filtering, catalog building, and SNP markers identification.
The process_radtags module was used to demultiplex all individuals according to their barcodes and quality filters were applied, including quality scores (-q) and reads with non-called bases. Correction of over-represented sequences was also performed. FastQC was again used to check the quality of the sequences for each individual, and the results were summarized by MultiQC55. Due to the unavailability of a reference genome for Euterpe spp., the ustacks processing was done de novo. For the minimum depth of reads to form a stack, the parameter m = 4 was considered. The number of nucleotide variations accepted for stack alignment was M = 3, and the number of secondary reads considered was N = 5. The catalog was built using cstacks, based on the identification of consensus sequences for each SNP locus. The maximum number of mismatches allowed between loci (n) was set to the same value as the M parameter used in ustacks. Population-level filters were applied using the populations module, considering the minor allele frequency (min-maf) equal to 1%, the occurrence of the alleles (r) in 75% of the individuals in the population, and the occurrence of the alleles in all but one population.
Identification of putative loci under selection
We applied three different methods (pcadapt, fsthet, and BayeScan) to detect outlier SNPs putatively under selection. Following the recommendations of Luikart et al.10, who suggest using multiple complementary statistical tests to confirm outlier behavior and minimize false positives, we considered SNPs to be outliers if they were simultaneously identified by at least two of the three methods. This conservative approach increases confidence in the identified outliers by reducing method-specific biases and avoiding misleading results caused by demographic effects or technical issues. The overlap of outlier SNPs detected by different methods was visualized using a Venn diagram, generated with the R package VennDiagram 1.7.356 in R 4.2.157. After detecting the outlier SNPs, two datasets were retained for analysis of genomic diversity and population structure: one consisting of putatively neutral SNPs and the another of outlier SNPs.
The R package pcadapt58 is based on principal components analysis (PCA) and determines vectors with z-scores, measuring the relationship between the SNPs and the first principal component58. Using the Mahalanobis distance59, each SNP was analyzed according to the z-scores to identify those that did not follow the majority point distribution58, which were considered outliers. We applied pcadapt 4.0.358 and tested K principal components (with K ranging from 1 to 20). After computing the p-values, we applied a false discovery rate (FDR) threshold of 10% (q < 0.1). Loci with q-values below this threshold were considered outliers.
Both the R package fsthet60 and BayeScan18 apply Wright’s fixation index, FST. Following the island migration model61, fsthet quantified the expected FST-HE ratio and identified the SNPs with excessively high or low FST61. The fsthet 1.0.160 was used to calculate the FST and HE values based on allele frequency variation, with 500 bootstrap replications generating confidence intervals. Candidate outlier SNPs were identified at both extremes (lower and upper) of the distribution, using an alpha level of 0.05.
Finally, BayeScan18 decomposed FST into beta (population-specific) and alpha (locus-specific) components, according to a logistic regression. For each locus, the posterior probability of including or not the alpha component in the selection model was estimated. This was determined using reversible-jump Markov chain Monte Carlo (RJ-MCMC), considering the migration island model and allele frequency uncertainty18. For this, BayeScan 2.118 generated 20 pilot runs, each with 5,000 RJ-MCMC iterations, followed by a burn-in of 50,000 iterations, and then 150,000 RJ-MCMC additional iterations with a thinning interval of 10. Outlier SNPs were considered those with an FDR threshold of 5% (q ≤ 0.05), the default recommend by BayeScan.
After identifying the outlier SNPs that were detected by at least two methods, a BLASTX search was performed. Using blast2GO62, genomic data were analyzed against the NCBI database, and the species with the highest number of annotation-associated top hits were identified. Sequences with significant BLASTX results had their associated functional annotation mapped using the gene ontology terms which summarize information about cellular components, molecular functions, and biological processes in which each gene product was involved. The results were compiled with the use of the online tool WEGO63.
Analysis of genomic diversity and minimum sample size
Genomic diversity analysis was estimated for putatively neutral and outlier SNPs. To define the estimates of observed heterozygosity and gene diversity (HO and HS), allelic richness (AR), fixation index (f), and their confidence intervals (determined with 1,000 bootstraps), we used the R package hierfstat 0.5.1164. The number of alleles (A) and private alleles (AP) were estimated using the R package adegenet 2.1.165 and the R package poppr 2.9.666.
Using only the putatively neutral SNPs, the minimum sample size was defined with Sample Size Impact, SaSii 1.067. Neutral loci were selected for this analysis because they reflect background levels of genetic variation, not directly influenced by selection, and provide accurate estimates of genetic diversity, which are appropriate for sampling strategies. SaSii is an R script used to infer the effect of sample size on genetic diversity. The program randomizes subsamples from an original dataset and estimates genetic parameters for each subsample, such as the average number of alleles, allele frequencies, expected and observed heterozygosities, as well as the genetic distance (FST) between the subsample and the original dataset67. SaSii was used with two objectives: (1) to detect whether the original sampling size was sufficient to obtain the genetic parameters, and (2) to define the ideal sampling size for conservation and restoration projects with E. precatoria.
The parameters used were “sample_size” = 5, “resampling” = 20, “min_frequency” = 0.05 (minimum allele frequency), and “max_indv_pop” = 100 (individuals used in each population; https://sasii.readthedocs.io/en/latest/index.htm). For the last parameter, however, the analysis was carried out until the graphs stabilized.
Population structure
Population structure analyses were conducted separately on both putatively neutral and outlier SNP datasets. First, the overall Wright’s F-statistics (FST, FIT, FIS) were calculated, as well as the pairwise FST between the 23 localities, using the R package hierfstat64. Heatmaps of the pairwise FST results were created using the R package heatmaply 1.5.068.
We assessed the genomic variation within and between groups using 20,000 permutation-based Molecular Analyses of Variance (“locus-by-locus” AMOVA), performed with Arlequin 3.5.2.269. Agglomerative analysis was conducted using Nei’s genetic distance70 and the Neighbor-Joining (NJ) clustering method. For this, we used the R package poppr66. Dendrograms were generated with confidence intervals based on 1,000 bootstrap replications.
To investigate the patterns of genomic structure, we used two methods: Discriminant Analysis of Principal Components (DAPC) and the sparse non-negative matrix factorization (sNMF). DAPC analyses were performed using the R package adegenet65. We applied DAPC with the K-means clustering to identify the optimal number of genomic groups (K), increasing K in successive runs. The optimal K was selected based on the Bayesian Information Criterion (BIC)71. We then used the α-score function to determine the optimal number of principal components (PCs) to retain, and generated scatter and bar plots.
The sNMF method assumes that the genomic population data result from an admixture of K parental lineages, estimating the ancestry coefficients from multilocus genotypes. We tested values of K from 1 to 23, corresponding to the 23 sampled localities, using 200,000 iterations and ten repetitions for each K. To visualize the results of the simulations, we used the cross-entropy criterion. This analysis was performed with the R package LEA 3.16.072.
Additionally, we assessed the correlation between Cavalli-Sforza and Edwards’ genetic distances and geographical distances (in km) among localities using the Mantel test73, implemented in the R package adegenet65.
Data availability
The datasets generated during and/or analysed during the current study are available in the NCBI repository,The datasets generated during and/or analysed during the current study are available in the NCBI repository, PRJNA1289687. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1289687.
References
Kikuchi, T. Y. P. & Callado, C. H. Brazilian Amazonian palm-stem types and uses: a review. Acta Amazon. 51, 334–346 (2021).
Henderson, A., Galeano, G., Euterpe & Prestoea (eds) Neonicholsonia (Palmae). 72 (Published for Organization for Flora Neotropica by the New York Botanical Gard, 1996).
Ter Steege, H. et al. Hyperdominance in the Amazonian tree flora. Science (1979) 342, (2013).
Ramos, S. L. F. et al. Paternity analysis, pollen flow, and Spatial genetic structure of a natural population of Euterpe precatoria in the Brazilian Amazon. Ecol. Evol. 8, 11143–11157 (2018).
Bussmann, R. W. & Zambrana, N. Y. P. Facing global markets - usage changes in Western Amazonian plants: The example of Euterpe precatoria Mart. and E. oleracea Mart. Acta Societatis Botanicorum Poloniae. 81, 257–261. (2012).
IBGE. Produção da Extração Vegetal e da Silvicultura. https://sidra.ibge.gov.br/tabela/289 (2021).
Ramos, S. L. F. et al. Genetic structure in populations of Euterpe precatoria Mart. In the Brazilian Amazon. Front. Ecol. Evol. 8, (2021).
Melo, G. S., Costa, F. S. & Silva, L. C. Da. O cenário da produção do açaí (Euterpe spp.) no Estado do Amazonas/ the production scenario of açaí (Euterpe spp.) in the state of Amazonas. Brazilian J. Dev. 7, 71536–71549 (2021).
Lopes, E. et al. Mapping the socio-ecology of Non timber forest products (NTFP) extraction in the Brazilian amazon: the case of açaí (Euterpe precatoria Mart) in acre. Landsc. Urban Plan. 188, 110–117 (2019).
Luikart, G., England, P. R., Tallmon, D., Jordan, S. & Taberlet, P. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 4, 981–994 (2003).
Helyar, S. J. et al. Application of SNPs for population genetics of nonmodel organisms: new opportunities and challenges. Mol. Ecol. Resour. 11, 123–136 (2011).
Etter, P. D. & Johnson, E. RAD paired-end sequencing for local de novo assembly and SNP discovery in non-model organisms. In Data Production and Analysis in Population Genomics: Methods and Protocols (ed. Bonin, F. P. and A.) 888 135–151. (Springer Science + Business Media New, 2012).
Zucchi, M. I. et al. Patterns of genome-wide variation, population differentiation and SNP discovery of the red banded stink bug (Piezodorus guildinii). Sci. Rep. 9, 1–11 (2019).
Blake, J. A. Ten quick tips for using the gene ontology. PLoS Comput. Biol. 9, 9–10 (2013).
Funk, W. C., McKay, J. K., Hohenlohe, P. A. & Allendorf, F. W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 27, 489–496 (2012).
Luikart, G. et al. Population genomics: Advancing understanding of nature. In Population Genomics: Concepts, Approaches and Applications (ed. Om P. Rajora) 3–79. https://doi.org/10.1007/13836_2018_60 (2019).
Breed, M. F. et al. The potential of genomics for restoring ecosystems and biodiversity. Nat. Rev. Genet. 20, 615–628 (2019).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A bayesian perspective. Genetics 180, 977–993 (2008).
Vaux, F., Dutoit, L., Fraser, C. I. & Waters, J. M. Genotyping-by-sequencing for biogeography. J. Biogeogr. 50 262–281. https://doi.org/10.1111/jbi.14516 (2023).
Albert, J. S. et al. Human impacts outpace natural processes in the Amazon. Science (1979) 379, (2023).
Lopes, R. et al. Seasonality of fruit production of Euterpe Oleracea and E. precatoria açaí palm trees cultivated in the metropolitan region of Manaus (AM). Revista Agro@Mbiente on-Line. 16, 1–14 (2022).
Alves-Pereira, A. et al. A population genomics appraisal suggests independent dispersals for bitter and sweet Manioc in Brazilian Amazonia. Evol. Appl. 13, 342–361 (2020).
Costa, M. F., Francisconi, A. F. & Vancine, M. H. Zucchi, M. I. Climate change impacts on the Copernicia Alba and Copernicia prunifera (Arecaceae) distribution in South America. Revista Brasileira De Bot. 45, 807–818 (2022).
Morais, J. G. S. et al. Genomic population structure of Parkia platycephala Benth. (Leguminosae) from Northeastern Brazil. Genet. Resour. Crop Evol. 70, 251–261 (2023).
Gish, W. & States, D. J. Identification of protein coding regions by database similarity search. Nat. Genet. 3, 266–272 (1993).
Allendorf, F. W., Hohenlohe, P. A. & Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709 (2010).
Benestan, L. M. et al. Conservation genomics of natural and managed populations: Building a conceptual and practical framework. Mol. Ecol. 25, 2967–2977 (2016).
Cartaxo, C. B., Vasconcelos, C., de Papa, M. A. M., de Gonzaga, D. & de Álvares, V. da, D. S. de O. M. S. Euterpe Precatoria Mart.: Boas Práticas de Produção Na Coleta e Pós-Coleta de Açaí-Solteiro. (2020).
Küchmeister, H., Silberbauer-Gottsberger, I. & Gottsberger, G. Flowering, pollination, nectar standing crop, and nectaries of Euterpe precatoria (Arecaceae), an Amazonian rain forest palm. Plant Syst. Evol. 206, 71–97 (1997).
Bovi, M. L. A. & de Castro, A. Assaí. in Selected Species and Strategies To Enhance Income Generation from Amazonian Forests (eds Clay, J. W. & Clement, R. C.) 58–67 (FAO, Rome, Italy, (1993).
Melo, W. A., Freitas, C. G., Bacon, C. D. & Collevatti, R. G. The road to evolutionary success: insights from the demographic history of an Amazonian palm. Heredity (Edinb). 121, 183–195 (2018).
Ramos, S. L. F. et al. Natural populations of Astrocaryum aculeatum Meyer in amazonia: genetic diversity and conservation. Plants 11, 1–16 (2022).
Zona, S. & Henderson, A. A review of animal-mediated seed dispersal of palms. Selbyana 11, 6–21 (1989).
Smith, N. Palms and People in the Amazon. Palms and People in the Amazon (Springer, 2015).
Clement, C. R. Melhoramento De Espécies Nativas. Recursos genéticos & melhoramento-plantas (Fundação De Apoio à Pesquisa (Agropecuária de Mato Grosso-Fundação MT, 2001).
Levis, C. et al. How people domesticated Amazonian forests. Front. Ecol. Evol. 5, (2018).
Clement, C. R. et al. Disentangling domestication from food production systems in the neotropics. Quaternary 4. https://doi.org/10.3390/quat4010004 (2021).
Bernardes Filho, L. A. Perfil econômico E análise Da Qualidade Do Fruto E Polpa De açaí (Euterpe Precatoria Mart.) Comercializados Em Manaus, Amazonas (Instituto Nacional de Pesquisas da Amzônia, 2021).
Konrad, H., George, J. P. & Szukala, A. The state of forest genetic diversity: Anthropogenic impacts and conservation initiatives. In Ecological Connectivity of Forest Ecosystems 55–89. https://doi.org/10.1007/978-3-031-82206-3_4 (Springer Nature Switzerland, 2025).
Allendorf, F. W., Luikart, G. H. & Aitken, S. N. Conservation and the Genetics of Populations (Wiley, 2012).
Bocanegra-González, K. T. & Guillemin, M. L. Guidelines for the restoration of the tropical timber tree Anacardium excelsum: first input from genetic data. Tree Genet. Genomes 14, (2018).
Ramos, S. L. F. et al. Mating system analysis of Açaí-do-Amazonas (Euterpe precatoria Mart.) using molecular markers. Crop Breed. Appl. Biotechnol. 19, 126–130 (2019).
Perrut-Lima, P., Sebbenn, A. M., Francisconi, A. F., Picanço-Rodrigues, D. & Clement, C. R. Genetic diversity and mating system of Euterpe precatoria in three localities along the lower Solimões river in central Amazonia. Silvae Genet. 72, 81–91 (2023).
Lawrence, M. J., Marshall, D. F. & Davies, P. Genetics of genetic conservation. I. Sample size when collecting germplasm. Euphytica 84, 89–99 (1995).
Crossa, J. & Vencovsky, R. Basic sampling strategies: Theory and practice. Collect. Plant Genet. Div. Tech. Guidel. 2011 Update 1–28 (2011).
Hale, M. L., Burg, T. M. & Steeves, T. E. Sampling for microsatellite-based population genetic studies: 25 to 30 individuals per population is enough to accurately estimate allele frequencies. PLoS ONE 7, (2012).
Hoban, S. & Schlarbaum, S. Optimal sampling of seeds from plant populations for ex-situ conservation of genetic biodiversity, considering realistic population structure. Biol. Conserv. 177, 90–99 (2014).
Basey, A. C., Fant, J. B. & Kramer, A. T. Producing native plant materials for restoration: 10 rules to collect and maintain genetic diversity. Native Plants J. 16, 37–53 (2015).
Teixeira, B. et al. A review of the genus euterpe: botanical and genetic aspects of açai, the purple gold of the Amazon. Bot. J. Linn. Soc. https://doi.org/10.1093/botlinnean/boae060 (2024).
Doyle, J. J. & Doyle, J. L. Genomic plant DNA Preparation from fresh tissue-CTAB. Phytochemical Bull. 19, 11–15 (1987).
Poland, J. A. & Rife, T. W. Genotyping-by‐sequencing for plant breeding and genetics. Plant Genome 5, (2012).
Poland, J. A., Brown, P. J., Sorrells, M. E. & Jannink, J. Development of high-density genetic maps for barley and wheat using a novel two-enzyme Genotyping-by-Sequencing approach. PLoS One. 7, e32253 (2012).
Andrews, S. FastQC: A quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Catchen, J., Hohenlohe, P. A., Bassham, S. & Amores, A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Ewels, P., Lundin, S. & Max, K. Data and text mining multiqc: summarize analysis results for multiple tools and samples in a single report. 32, 3047–3048 (2016).
Chen, H. & Boutros, P. C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 35, 7 (2011).
R Core Team. R: A language and environment for statistical computing. https://www.R-project.org/ (R Foundation for Statistical Computing, 2022).
Luu, K., Bazin, E. & Blum, M. G. B. Pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 17, 67–77 (2017).
Maronna, R. A. & Zamar, R. H. Robust estimates of location and dispersion for high-dimensional datasets. Technometrics 44, 307–317 (2002).
Flanagan, S. P. & Jones, A. G. Constraints on the FST-heterozygosity outlier approach. J. Hered. 108, 561–573 (2017).
Wright, S. The genetical structure of populations. Ann. Eugen. 15, 323–354 (1949).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 (2008).
Ye, J. et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 46, W71–W75 (2018).
Goudet, J. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes. 5, 184–186 (2005).
Jombart, T. & Ahmed, I. Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281 (2014).
Scaketti, M. et al. Sample size impact (SaSii): an R script for estimating optimal sample sizes in population genetics and population genomics studies. PLoS One. 20, e0316634 (2025).
Galili, T., O’Callaghan, A., Sidi, J., Sievert, C. & Heatmaply An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 34, 1600–1602 (2018).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Nei, M. Genetic distance between populations. In Molecular Evolutionary Genetics 208–253 (Columbia University Press, 1987).
Jombart, T. et al. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94 (2010).
Frichot, E. & François, O. L. E. A. An R package for landscape and ecological association studies. Methods Ecol. Evol. 6, 925–929 (2015).
Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220 (1967).
IBGE. Geociências - Vegetação. https://geoftp.ibge.gov.br/informacoes_ambientais/vegetacao/vetores/escala_250_mil/versao_2021/ (2021).
Acknowledgements
We thank Chico Mendes Institute for Biodiversity Conservation (ICMBio) and SISGEN for granting sampling and research permits. We are also greatful to Dr. Charles R. Clement of the National Institute for Amazon Research (INPA), who coordinated CT-Amazônia/CNPq project 575588/08-0, responsible for the 2011 sampling. This study was supported by the National Council for Scientific and Technological Development (CNPq—313417/2023-7).
Author information
Authors and Affiliations
Contributions
A.F.F. conducted the research, extracted and analyzed data, interpreted the results, and wrote and reviewed the manuscript. M.S. extracted and analyzed data. J.A.M.M. and I.A.S.C. prepared the genomic library, while G.O.F. contributed to data extraction and M.S.M. contributed to the structural analysis. F.M.C. and D.M.M. assisted in interpreting the results, and R.O.P. interpreted the GO terms. S.L.F.R. and D.P.R. collected the samples. M.T.G.L. provided financial support for the collections, and M.I.Z. designed the experiment and secured funding. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Francisconi, A.F., Scaketti, M., Morales-Marroquín, J.A. et al. High genomic structuring in Euterpe precatoria in the Brazilian Amazônia and new implications for restoration and conservation efforts. Sci Rep 16, 1525 (2026). https://doi.org/10.1038/s41598-025-31277-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-31277-7
