
Abstract
The impact of brewing materials on the spatial distribution of eukaryotic microbiota in pit muds (PMs) of Luzhou-flavor liquor was investigated using high-throughput sequencing of 18 S rDNA amplicons to compare the differences in eukaryotic microbiota composition between PMs derived from three grains and five grains. Our findings revealed contrasting trends in the spatial distribution of microbiota structure between the two types of PMs, with the Chao1 index of eukaryotic microbiota in three-grain PMs being higher than that of five-grain PMs. Significant differences in relative abundance were observed among the various samples, including Brettanomyces custersianus, Debaryomyces sp., Pichia fementans, Pichia sp., Naganishia sp., Saccharomyces selenospora, Saccharomyces cerevisiae, and Saccharomycopsis fibuligera in yeast, and Aspergillus fumatus, Aspergillus sp., and Penicillium chrysogenum in mold. Linear discriminant analysis based on 14 dominant operational taxonomic units accurately classified all samples, indicating that our discriminant model can be utilized for quality control of microbiota in PMs. These results provide a foundation for precisely regulating the spatial distribution of fungal community structure and quality control by adjusting the composition of brewing raw materials.
Similar content being viewed by others


Introduction
Baijiu is one of the six most popular distilled liquors worldwide. It has a unique flavor owing to variations in brewing materials, geographical locations, and brewing techniques1. This traditional Chinese liquor has been brewed for thousands of years using grains as the main ingredient. Saccharification involves the use of Daqu, Xiaoqu, or bran koji as starters, followed by bilateral or solid fermentation and distillation. The final product is then aged and blended to achieve a strong aroma2. During fermentation, unique geographical and climatic conditions play a crucial role in the production of aroma compounds by microorganisms in Daqu and pit mud (PM). This results in various flavor types, including Luzhou-flavor, Qing-flavor, Maotai-flavor, and other special flavor3. Among these, Luzhou-flavor liquor (LFL) is the most popular in the Chinese market, accounting for over 70% of the market share4. The primary aroma component of LFL is ethyl hexanoate, which is predominantly produced by caproic acid-producing bacteria present in PM. Other esters are formed through the complex metabolism and morphological changes of solid, liquid, and gas substances by distiller’s yeast and PM microorganisms during fermentation5,6. Consequently, the metabolic activity and community structure of microorganisms, particularly aroma-producing microorganisms, are crucial in the LFL brewing process. Understanding the relationship between microbial structure, interactions, and their products during liquor fermentation is a key theoretical issue in liquor research because of the variety of brewing microorganisms involved.
Microorganisms determine the flavor and quality of LFL and also play key roles in the efficiency and outcomes of chemical reactions during fermentation. The dominant microbiota dynamically change across different fermentation stages. The production of LFL involves three basic processes: saccharification, alcoholic fermentation, and formation of flavor compounds. Saccharification is accomplished by extracellular enzymes secreted by microorganisms. In addition to prokaryotes like Bacillus and Staphylococcus, yeasts and molds also play important roles in saccharification7. Molds are aerobic fungi that mainly exist during the initial fermentation stages and include Rhizopus, Aspergillus, Mucor, and Monascus. Molds play a crucial role in the production of liquor as a starter, providing many enzymes, including amylase, glucoamylase, protease, cellulase, lipase, and pectinase. They serve as the main source of glucoamylase and acid proteases. These enzymes can decompose most of the nutrients in raw materials and lay the foundation for the growth and metabolism of other microorganisms. Additionally, some molds can produce citric acid, succinic acid, acetic acid, and other organic acids8. Yeasts in the PM of LFL primarily include Saccharomyces cerevisiae, Pichia kudriavzevii, Kazachstania exigua, Geotrichum silvicola, Hansenula spp., Torulopsis spp., and Candida spp. These yeasts are obligate or facultative anaerobes that are crucial in alcohol fermentation and the formation of flavor compounds in LFL. The proportions of these yeasts vary across different fermentation stages7,9. S. cerevisiae and K. exigua play major roles in ethanol production, whereas P. kudriavzevii and G. silvicola assist in ethanol production during continuous fermentation9,10.
The formation mechanisms of specific flavor components in liquors have always been a focus of scientific research. Yeasts produce various enzymes, including alcoholases, esterases, fuselases, and lipases. Under anaerobic conditions, yeast ferments glucose into alcohol and carbon dioxide via glycolysis and acetone anaerobic degradation. Additionally, yeast synthesizes organic acids, higher alcohols, and glycerol. Lipids produced by biochemical reactions in yeast are a key flavor source in liquor. For example, although methyl mercaptan is an important aroma component in liquor, its biotransformation mechanism remains unclear. However, researchers have found that S. cerevisiae J14, a yeast isolated and identified from Daqu, which plays an important role in liquor fermentation, can synthesize benzyl mercaptan. Yeast can produce this aroma substance under certain conditions11,12. Similarly, P. kudriavzevii YF1702 has been shown to produce high levels of 2-phenylethanol under optimized fermentation conditions, significantly enhancing liquor aroma13. Furthermore, the selection of specific ester-producing yeasts can enhance the liquor-brewing process and significantly increase the proportion of certain aroma components. For instance, S. cerevisiae CM15 can significantly increase the content of ethyl acetate and acetate lactate, thereby enhancing the overall aroma profile of the liquor14,15. Further studies have shown that different microbiota exist in different regions and types of liquors produced during fermentation. Although the brewing process involves rich genetic diversity, relatively little genetic diversity has been studied for certain key microorganisms, such as Saccharomycopsis fibuligera16. This gap highlights the need for in-depth studies of these critical microbial populations.
Although numerous studies have investigated the diversity and structure of the microbiota and their impact on flavor substances during LFL fermentation, there is still a lack of sufficient data. For instance, the differences in microbiota at various fermentation depths and in different fermentation vessels, as well as the spatial distribution and influencing factors of fungal communities in PM, have not been fully elucidated17,18,19. Furthermore, microorganisms and their volatile components in different PMs and fermented grains significantly influence the quality and flavor of compound-flavor Baijiu20,21. In the liquor industry, “sorghum fragrance, corn sweet, rice net” clearly explain the relationship between liquor quality and raw materials8. Studies on various fermentation environments and raw materials have demonstrated that pretreating husks and glutinous millet can significantly alter the core flavor composition of light-flavor Baijiu. The relative abundance of microorganisms at different fermentation stages also significantly affects the final product’s quality22,23. Therefore, microorganisms play a crucial role in liquor fermentation through metabolism and reproduction, and also influence flavor compound production through complex interaction networks24,25. Based on existing research, further exploration and optimization of microbial interactions during fermentation are essential for improving liquor production efficiency and final product quality. To clarify the effect of fermentation materials on the spatial distribution of eukaryotic microbiota in the PM of LFL, this study compared the differences in eukaryotic microbiota composition in the upper, middle, and bottom layers of PMs using three- and five-grain mixtures. The findings provide a foundation for accurately controlling the spatial distribution of fungal community structures and ensuring the quality control of eukaryotic communities by adjusting the composition of raw materials.
Materials and methods
Sample collection
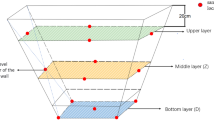
PMs from Baimai Spring Liquor Co., Ltd. (Jinan, China) were collected from the pits of LFL made with either three or five grains as the raw material. The three-grain liquor (M3G) was composed of sorghum, rice, and corn. The five-grain liquor (M5G) was composed of sorghum, rice, corn, wheat, and glutinous rice. The dimensions of the pit were 3.7 × 1.6 × 2.0 m. The sampling and processing methods were based on a previously published study26. Briefly, fifteen samples were collected form the upper layer (UP, n = 5), the middle layer (MP, n = 5), and the bottom (BP, n = 5) of the pits. The upper and middle layers were set at 0.5 and 1.3 m below the cellar surface, respectively. The pit mud was randomly collected from three points in the same layer and mixed as a sample26.
DNA extraction and high-throughput sequencing
The DNA were extracted from the PMs using a SPINeasy™ DNA pro kit for soil (MP Biomedicals, Eschwege, Germany). The V4 region of the small subunit rRNA gene was amplified using the eukaryotic primer pair TAReuk454FWD1 (5′–CCAGCASCYGCGGTAATTCC–3′) and TAReukREV3 (5′–ACTTTCGTTCTTGATYRA–3′), as previously described27,28. The 5’ end of the TAReuk454FWD1 primer was linked to a 12-nucleotide barcode sequence specific to each sample, as described in our previous study26. The polymerase chain reaction (PCR) production was detected using 1.5% agarose gel electrophoresis and purified using am AxyPrep™ DNA gel extraction kit (Axygen, Suzhou, China). Subsequently, the purified DNA was sequenced at Guangdong Meilikang Bio-Science Ltd. (Foshan, China) using the HiSeq platform (Illumina, CA, USA) with 2 × 250 bp kits following the manufacturer’s instructions. The raw reads were merged to tags using FLASH 1.2.8 and filtered using QIIME 1.9.0 to remove low-quality tags as our previously described26. Chimeric tags were removed using UCHIME 4.2. Subsequently, the remaining high-quality tags were clustered into operational taxonomic units (OTUs) using USEARCH v9 with 97% sequence similarity. α-Diversity indexes were calculated, and principal coordinates analysis (PCoA) based on weighted UniFrac distances was conducted using QIIME 1.9.0. The OTUs were annotated to taxa using the Ribosomal Database Project Classifier with the SILVA 138.2 databases.
Data analyses
Data are presented as mean ± standard error. The Kruskal–Wallis rank sum test with Dunn’s post-hoc test was performed using R 4.2.3, with the FSA package. A permutational multivariate analysis of variance was performed using the vegan package in R 4.2.3. A heatmap profile was created using the pheatmap package in R, version 4.2.3. For linear discriminant analysis, significantly different dominant operational taxonomic units (OTUs) were identified using the Kruskal–Wallis H-test. Extremely collinear dominant OTUs were filtered out based on their Spearman correlation coefficients (> 0.9 or < − 0.9) using the R psych and reshape2 packages. The samples were then randomly divided into training and test sets in an 8:2 ratio, and linear discriminant analysis was conducted using the MASS package in R. Correlation bubble diagrams were generated using the corrplot package in R. Statistical significance was set at p < 0.05.
Results
α-Diversity of eukaryotic microbiota in PM
The V4 hypervariable region of the 18 S rRNA gene was sequenced in the PM microbiomes of LFL brewed with different materials. Thirty PM samples, derived from two different brewing materials, were analyzed. To ensure accuracy, after quality control, 3,123 high-quality sequences were randomly extracted from each sample for further analysis. Principal coordinates analysis results based on the weighted UniFrac distance revealed contrasting spatial distribution trends in microbiota structure between the PM of M3G and M5G (Fig. 1A). The results of the α-diversity analysis showed that the observed OTU number and the Shannon and Chao1 indices for eukaryotic microorganisms were significantly lower in the upper and bottom layers of the PM than those in the middle layer (Fig. 1B–D, p < 0.05). Although the statistical test results were not significant, the Shannon index of eukaryotic microorganisms in the upper and middle layers of the PM of M3G showed trend toward being higher than that of M5G, whereas the Shannon index in the bottom layer of M3G was lower than that of M5G (Fig. 1C). The Chao1 index value of eukaryotes in the upper, middle, and bottom layers of the PM of M3G were higher than those of M5G (Fig. 1D).
Phylum level analysis of microbiota composition in PM
Ten eukaryotic microbial phyla were identified in this study, including SAR, Fungi, Holozoa, Aphelidea, Rigifillida, Breviatea, Excavata, Cryptophyta, Archaeplastida, and Amoebozoa (Fig. 1E). Moreover, Zoopagomycota, Mucoromycota, Dikarya, Cryptomycota, and Chytridiomycota were the major Fungi subgroup, and Stramenopiles, Rhizaria, and Alveolata were the major SAR subgroup (Fig. 1F).

Effects of different brewing materials and spatial locations on eukaryotic community composition in pit mud. (A) PCoA profile; (B) OTU number; (C) Shannon index; (D) Chao index; (E) Composition of the dominant phyla; (F) Major subgroups of SAR and Fungi. M3GUL, the pit mud sample was collected at the upper layer (0.5 m below the cellar surface) of the three-grain Baijiu brewing pit. M3GML, the pit mud sample was collected at the middle layer (1.3 m below the cellar surface) of the three-grain Baijiu brewing pit. M3GBL, the pit mud sample was collected at the bottom of the three-grain Baijiu brewing pit. M5GUL, the pit mud sample was collected at the upper layer of the five-grain Baijiu brewing pit. M5GML, the pit mud sample was collected at the middle layer of the five-grain Baijiu brewing pit. M5GBL, the pit mud sample was collected at the bottom of the five-grain Baijiu brewing pit. * P < 0.05; ** P < 0.01; *** P < 0.001.
Genus level analysis of microbiota composition in PM
At the genus level, 59 dominant eukaryotic microorganisms with a relative abundance of > 1% in at least one sample were detected. Some variations were observed among different samples. The dominant genera in these samples were analyzed; 11 dominant genera with significant differences were identified (Fig. 2A). Among these, the yeasts included Brettanomyces custersianus, Debaryomyces sp., Pichia fementans, Pichia sp., Naganishia sp., Saccharomyces selenospora, S. cerevisiae, and S. fibuligera. The molds included Aspergillus fumigatus, Aspergillus sp., and Penicillum chrysogenum. Further analysis showed that, except for Debaryomyces sp. and Pichia fementans, the relative abundance of the other nine eukaryotic microorganisms in the upper PM of M3G were significantly higher than those in the middle and bottom (Fig. 2B-L, p<0.05). Within the same pit position, the relative abundance of A. fumigatus in the middle and lower PM of M3G was significantly lower than that of M5G (Fig. 2B, p < 0.05).
In the upper PM of M3G, the relative abundances of B. custersianus and Penicillium chrysogenum were significantly higher than those in M5G (Fig. 2D and G, p < 0.05). Conversely, the middle PM of M3G showed significantly lower levels of Debaryomyces sp. than those of M5G (Fig. 2E, p < 0.05). The relative abundance of P. fermentans in the upper PM of M3G was significantly lower than that in M5G (Fig. 2H, p < 0.05). The relative abundance of Pichia sp. was significantly higher in the upper PM of M3G, but the opposite trend was observed in the lower PM (Fig. 2I, p < 0.05). The relative abundance of Naganishia sp. was notably lower in the PM of M3G than in the PM of M5G (Fig. 2F, p < 0.05). Additionally, the relative abundance of S. cerevisiae in the middle PM of the M3G was significantly lower than that of the M5G (Fig. 2K, p < 0.05). Similarly, S. fibuligera was significantly less abundant in the middle and lower layers of M3G PM than in those of M5G PM (Fig. 2L, p < 0.05).

Effects of different brewing materials and spatial locations on the dominant genera of eukaryotic microbiota in pit mud. (A) Heatmap profile shows the relative abundance of dominant genera and difference among groups; (B) Boxplot shows the difference of relative abundance of Aspergillus fumigatus; (C) Boxplot shows the difference of relative abundance of Aspergillus sp.; (D) Boxplot shows the difference of relative abundance of Brettanomyces custersianus; (E) Boxplot shows the difference of relative abundance of Debaryomyces sp.; (F) Boxplot shows the difference of relative abundance of Naganishia sp.; (G) Boxplot shows the difference of relative abundance of Penicillium chrysogenum; (H) Boxplot shows the difference of relative abundance of Pichia fermentans; (I) Boxplot shows the difference of relative abundance of Pichia sp.; (J) Boxplot shows the difference of relative abundance of Saccharomycopsis selenospora; (K) Boxplot shows the difference of relative abundance of Saccharomyces cerevisiae; (L) Boxplot shows the difference of relative abundance of Saccharomycopsis fibuligera. M3GUL, the pit mud sample was collected at the upper layer (0.5 m below the cellar surface) of the three-grain Baijiu brewing pit. M3GML, the pit mud sample was collected at the middle layer (1.3 m below the cellar surface) of the three-grain Baijiu brewing pit. M3GBL, the pit mud sample was collected at the bottom of the three-grain Baijiu brewing pit. M5GUL, the pit mud sample was collected at the upper layer of the five-grain Baijiu brewing pit. M5GML, the pit mud sample was collected at the middle layer of the five-grain Baijiu brewing pit. M5GBL, the pit mud sample was collected at the bottom of the five-grain Baijiu brewing pit. * p < 0.05; ** p < 0.01; *** p < 0.001.
The combination of different grains significantly influenced the co-occurrence patterns of eukaryotic microorganisms in the PM. In M3G, the highest levels were observed in P. chrysogenum (12), S. cerevisiae (11), S. fibuligera (11), B. custersianus (11), S. selenospora (10), Aspergillus sp. (10), and Naganishia sp. (10) (Fig. 3A). In M5G, the highest levels were found in P. fermentans (9), S. cerevisiae (8), Debaryomyces sp. (6), P. kudriavzevii (6), and W. anomalus (6) (Fig. 3B). Fusarium oxysporum was positively correlated with S. cerevisiae, Sporobolus stapfianus, and Arthrinium sp. in M3G (Spearman correlation coefficient > 0.6, p < 0.05; Fig. 3A), and with S. cerevisiae, P. kudriavzevii, P. fermentans, Debaryomyces sp., and Moiassezia sp. in M5G (Spearman correlation coefficient > 0.6, p < 0.05; Fig. 3B). Aspergillus sp. showed a significant positive correlation with B. custersianus, Naganishia sp., P. chrysogenum, Pichia sp., Pichia jarconii, S. cerevisiae, S. fibuligera, S. selenospora, W. anomalus, P. kudriavzevii, and Aspergillus fumigatus in M3G (Spearman correlation coefficient > 0.6, p < 0.05; Fig. 3A), whereas it was only significantly positively correlated with P. fermentans and Malassezia sp. in M5G (Spearman correlation coefficient > 0.6, p < 0.05; Fig. 3B).
B. custersianus showed a significant positive correlation with Naganishia sp., P. chrysogenum, Pichia sp., Pichia jaroonii, S. cerevisiae, S. fibuligera, S. selenospora, W. anomalus, P. kudriavzevii, and (A) fumigatus (Spearman correlation coefficient > 0.6, p < 0.05; Fig. 3A). However, Naganishia sp. and Pichia sp. were only significantly positively correlated with (B) custersianus in M5G (Spearman’s correlation coefficient > 0.6, p < 0.05; Fig. 3B). Furthermore, no significantly negatively correlated eukaryotic microorganisms were found in M3G, but a significant negative correlation was observed between B. custersianus and Alternaria alternata in M5G (Spearman correlation coefficient < − 0.6, p < 0.05; Fig. 3B).

Co-occurrence networks showed the co-occurrence relationships between each dominant eukaryotic species. (A) The three-grain liquor composed of sorghum, rice, and corn; (B) The five-grain liquor composed of sorghum, rice, corn, wheat, and glutinous rice. Spearman correlation coefficient > 0.6 or < − 0.6, and p < 0.05 was considered significant. The red and blue edges indicate positive and negative correlations, respectively.
Considering that these patterns may be crucial for maintaining the quality of M3G and M5G, analyzing the recognition patterns of these representative differential microbial compositions could offer a novel approach for quality control. Fifty-one significantly different dominant OTUs were selected from the dominant OTUs using the Kruskal–Wallis H-test (p < 0.05; Fig. 4A). Next, we filtered out the extremely convergent dominant OTUs based on Spearman’s correlation coefficients, resulting in only 14 dominant OTUs (Fig. 4B). Based on linear discriminant analysis of these 14 dominant OTUs, the constructed discriminant formula (Table 1) accurately classified all samples (Fig. 4C; Table 2). The findings suggest that the discriminant formula can be used to monitor and maintain the quality of microbiota in PM. Analysis of these 14 dominant OTUs allowed for precise classification of the communities into their respective PM groups, indicating the presence of normal eukaryotic microbiota. An inaccurate classification may indicate the presence of abnormal microbiota and potential risks.

Spearman correlation of differentially dominant OTUs (A, B) and linear discriminant analysis (C). (A) Bubble diagram of the Spearman pairwise correlations between all significantly different dominant OTUs. (B) Bubble diagram of the Spearman pairwise correlations between the dominant OTUs filtering out the extremely collinear dominant OTUs (|Spearman correlation coefficient| > 0.9). (C) Scatter plot of the results of the constructed linear discriminant model. M3GUL, the pit mud sample was collected at the upper layer (0.5 m below the cellar surface) of the three-grain Baijiu brewing pit. M3GML, the pit mud sample was collected at the middle layer (1.3 m below the cellar surface) of the three-grain Baijiu brewing pit. M3GBL, the pit mud sample was collected at the bottom of the three-grain Baijiu brewing pit. M5GUL, the pit mud sample was collected at the upper layer of the five-grain Baijiu brewing pit. M5GML, the pit mud sample was collected at the middle layer of the five-grain Baijiu brewing pit. M5GBL, the pit mud sample was collected at the bottom of the five-grain Baijiu brewing pit. * P < 0.05; ** P < 0.01; *** P < 0.001.
Discussion
The quality of PM, the main carrier of microorganisms, largely depends on the composition and species diversity of the microbiota. During Baijiu fermentation, a complex organic system is formed by the combination of fermented grains, PM, and PM microorganisms. This results in a series of complex biochemical reactions that produce liquor and contribute to the flavor of the final product. Although the abundance of eukaryotic microorganisms in PM is relatively low compared with that of prokaryotic microorganisms, they still play a crucial role in Baijiu quality29. Fungi in PM primarily originate from fermented grains and Daqu, with yeast and mold being the main sources29. These fungi produce alcohol and their high saccharifying and esterifying enzyme activities enable the effective decomposition of macromolecular organics in the raw materials. Their metabolites directly contribute to the flavor of Baijiu, playing a key role in generating flavor substances30,31,32. Yeast are the main fermentation fungi, often referred to as the “power of liquor fermentation”. They utilize small-molecule sugars and other nutrients to metabolize and produce ethanol, organic acids, and esters, which are essential for alcohol and flavor development in LFL33. Common yeasts used in the production of Baijiu include S. cerevisiae, Hansenula, Candida, P. pastoris, and ester-producing yeast34. These strains play key roles in the brewing and flavor development in LFL. S. cerevisiae is known for its strong ethanol production capacity, which is essential for achieving high liquor yields. However, its activity may decrease or even cease as fermentation temperature increases. Ester-producing yeast and P. pastoris synthesize esters through the action of esterase, which can greatly impact the aroma profile of LFL35. Molds are the primary saccharifying microorganisms used in the brewing process, as they secrete glucoamylase to hydrolyze starch and other nutrients into single molecular sugars. Aspergillus and Rhizopus are particularly effective in this process because of their strong enzymatic activities. In addition to glucoamylase, they secrete glucose oxidase and protease, which provide various organic acids and promote yeast growth and reproduction. This results in higher alcohol production and improves the overall quality and flavor of LFL36,37. Some fungi, such as Penicillium, can also impact Baijiu taste by consuming alcohol substrates and producing metabolites that increase the pH of distillers’ grains38.
Wang et al. 39 used 454 high-throughput sequencing to analyze the eukaryotic microbiota of the PM in LFL. They found that the majority of the microbiota belonged to five phyla: Opisthokonta, Archaeplastida, SAR, Amoebozoa, and Excavata. Our study also detected these dominant phyla, along with five other eukaryotic phyla. However, compared to prokaryotic microorganisms, the abundance of eukaryotic microbiota in the PM was relatively low. Some of these eukaryotes directly originate from fermented grains, whereas others are indirectly introduced through Daqu29. Previous studies have shown that eukaryotic microbial groups in PM include Candida, Pichia, Aspergillus, Rhizopus, Trichosporon, Mortierella, Penicillium, and Saccharomyces40. Further identification of fungi in Huangshui of LFL revealed that S. cerevisiae was the dominant group, accounting for 41.0% of the total fungi in the community41. Genus-level analysis of eukaryotic microorganisms in Daqu identified 11 different genera, including Aspergillus, Pichia, Saccharomyces, and Hyphophia. Among them, the dominant genera were Pichia and Saccharomyces, accounting for 65.95% and 10.97%, respectively42. Our study also detected eight yeast species, such as P. fementans, Pichia sp., S. selenospora, S. cerevisiae, and S. fibuligera, as well as three mold species, including A. fumigatus, Aspergillus sp., and P. chrysogenum (Fig. 2A). These eukaryotic microorganisms play crucial roles in LFL fermentation.
Pichia and Saccharomyces are important functional fungi in the brewing process and have a significant correlation with ethyl caproate and free amino acid content in PM and fermented grains43. Pichia belongs to the non-S. cerevisiae group and can produce β-glucosidase. Additionally, Pichia, an important lipid-producing yeast, can promote esterification reactions and generate many flavor substances, such as terpenes44. However, Saccharomyces is even more critical, as it can produce many alcohols and flavor substances by utilizing various carbon sources, including pentose and hexose, under both aerobic and anaerobic conditions45. In this study, the dominant eukaryotic microorganisms in the upper layer of PM from M5G were S. fibuligera, Saccharomycopsis, S. sidaceae, and P. fermentans. Pichiaceae, Pichia, and Saccharomyces were also detected in the upper PM of M3G. These results indicate that microorganisms in the upper PM layer in the LFL play a crucial role in esterification, alcohol production, and formation of flavor substances.
Aspergillus, the most prevalent mold in Daqu, thrives in harsh environments and maintains its stability34. This adaptability is demonstrated by the production of glucose oxidase, protease, amylase, xylanase, and alcoholic flavor substances, making it a crucial fungus for Baijiu fermentation34,38. Sorghum, the main raw material used in the production of LFL, contains high concentrations of tannins that can negatively impact the quality of the liquor, resulting in a bitter and astringent taste46. However, during fermentation, Aspergillus helps reduce the tannin content in sorghum, resulting in a more balanced and pleasant taste47. Additionally, Penicillium, another type of mold, plays an important role in LFL fermentation by producing a complete glycanase system for degrading natural lignocellulosic materials, as well as a large amount of β-glucosidase and esterase48. This study revealed that Aspergillus and P. chrysogenum were mainly detected in the upper layer of the M3G PM. This indicates that the upper layer of the M3G PM has stronger fermentation ability, which greatly contributes to the overall aroma and taste of the wine. A. fumigatus is commonly found in moldy grains, contaminated food, soil, and moldy materials and can cause aspergillosis in humans and animals, as well as infections in the lungs and other organs41. Interestingly, this study also detected A. fumigatus in both M3G and M5G PMs. This finding highlights the importance of thoroughly understanding PM microbial composition and conducting safety assessments in LFL brewing processes and research.
Eukaryotic microorganisms in PM are mostly aerobic or facultative aerobic microorganisms (such as various yeasts), which directly originate from fermented grains and indirectly from Daqu27. Species diversity and richness of eukaryotic microorganisms in the PM of LFL are generally lower than those of prokaryotic microorganisms. This difference may be attributed to the anaerobic environment of the PM, which is not conducive to the growth of aerobic or facultative aerobic fungi49. The bottom layer of PM, soaked in Huangshui formed by year-round fermentation, tends to have a more anaerobic environment, whereas the PM in the upper layer or pit wall may not be anaerobic until later stages of fermentation, with higher oxygen content. Therefore, different spatial positions significantly influence the composition of eukaryotic microorganisms in the PM49. A study on the microbiota diversity of PM at different spatial positions of the same pit age showed that the eukaryotic microbial diversity in the upper and middle layers of the same pit age was higher than that of the bottom PM50. Long-term production practices and scientific research results have also shown that there are differences in the quality of LFL produced at different pit ages, and liquor quality is significantly correlated with pit age51.
Other studies have shown that both the strains and raw materials affect the total amino acid content in the medium after fermentation. The amino acid contents of different raw materials fermented by the same strain were also different, which was caused by differences in the physical structure and nutritional components of different grain particles52. This study found that the observed OTU number and the Shannon and Chao 1 indices of eukaryotic microorganisms in the middle PM layer of the LFL, regardless of M3G or M5G, were significantly lower than those in the upper and bottom layers. Therefore, pit age, pit space, and brewing materials are key factors that affect the diversity of eukaryotic microorganisms in the PM of LFL.
It is worth noting that only one cellar of each brewing material was analyzed in this study, and the primers used in the PCR amplification amplified many plant and higher animal sequences, resulting in a relatively low proportion of fungi. In future studies, the number of pit samples should be increased. The sequencing process should be further optimized to perform a more comprehensive analysis of eukaryotic microorganisms in the PM of different brewing materials.
Conclusions
The spatial distribution of the eukaryotic microbiota structure in the PM of different parts of the LFL was significantly changed by the combination of different fermentation materials, and the coexistence pattern of eukaryotic microorganisms in the PM was also affected. In addition, OTUs based on 14 types of dominant eukaryotes with significant differences can be used to construct a linear discriminant model that effectively distinguishes all PM samples. This model holds potential for quality detection of PM microbiota.
Data availability
The raw data was delivered into the Sequence Read Archive under the accession number PRJNA1212826 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1212826).
Abbreviations
- LFL:
-
Luzhou-flavored liquor
- M3G:
-
Three-grain liquor
- M5G:
-
Five-grain liquor
- OUT:
-
Operational taxonomic unit
- PM:
-
Pit mud
- rRNA:
-
Ribosomal RNA
- SSU:
-
Small subunit
References
Zheng, X. W. & Han, B. Z. B. Chinese liquor: History, classification and manufacture. J. Ethn. Foods. 3, 19–25 (2016).
Fan, W. & Xu, Y. History and technology of Chinese liquor. In Science and Engineering of Chinese Liquor (Baijiu) (Xu, Y. eds) . 3–41. https://doi.org/10.1007/978-981-19-2195-7_1 (Springer, 2023).
Fu, G. et al. Analysis of microbial community, physiochemical indices, and volatile compounds of Chinese Te -flavor Baijiu Daqu produced in different seasons. J. Sci. Food Agric. 101, 6525–6532 (2021).
Zou, W., Ye, G. & Zhang, K. Diversity, function, and application of Clostridium in Chinese strong flavor Baijiu ecosystem: A review. J. Food Sci. 83, 1193–1199 (2018).
Tao, Y. et al. Prokaryotic communities in pit mud from different-aged cellars used for the production of Chinese strong-flavored liquor. Appl. Environ. Microbiol. 80, 2254–2260 (2014).
Wang, H. Y., Zhang, X. J., Zhao, L. P. & Xu, Y. Analysis and comparison of the bacterial community in fermented grains during the fermentation for two different styles of Chinese liquor. J. Ind. Microbiol. Biotechnol. 35, 603–609 (2008).
Lin, Z. et al. Isolation and identification of cellulose-degrading strains from yellow water of Baijiu (Chinese liquor) and its cellulase activity. China Brew. 35, 59–63 (2016).
Zou, W., Zhao, C. & Luo, H. Diversity and function of microbial community in Chinese strong-flavor Baijiu ecosystem: A review. Front. Microbiol. 9, 671 (2018).
You, L. et al. Distribution and function of dominant yeast species in the fermentation of strong-flavor Baijiu. World J. Microbiol. Biotechnol. 37, 26 (2021).
Bangchao, H. et al. Research and application of functional yeast in Chinese liquor production. Liquor Mak. 44, 13–18 (2017).
Zhang, G. et al. Isolation and characterization of yeast with benzenemethanethiol synthesis ability isolated from Baijiu Daqu. Foods 12, 2464 (2023).
Ma, J. et al. Screening of yeasts isolated from Baijiu environments for producing 3-methylthio-1-propanol and optimizing production conditions. Foods 11, 3616 (2022).
Fan, G. et al. Screening of yeasts isolated from Baijiu environments for 2-phenylethanol production and optimization of production conditions. 3 Biotech. 10, 275 (2020).
Qu, C. et al. Screening ester-producing yeasts to fortify the brewing of rice-flavor Baijiu for enhanced aromas. Bioengineered 14 (2023).
Dong, W. et al. Characteristics and functions of dominant yeasts together with their applications during strong-flavor Baijiu brewing. Foods 13, 2409 (2024).
Wang, J. W. et al. Genetic diversity and population structure of the amylolytic yeast Saccharomycopsis fibuligera associated with Baijiu fermentation in China. J. Microbiol. 59, 753–762 (2021).
Duan, Z. et al. Evolution of fermented grain yeast communities in strong-flavored Baijiu and functional validation of yeasts that produce superior‐flavored substances. J. Sci. Food Agric. 104, 5973–5981 (2024).
Wang, C., Tang, J. & Qiu, S. Profiling of fungal diversity and fermentative yeasts in traditional Chinese Xiaoqu. Front. Microbiol. 11 (2020).
Cheng, W. et al. Comparison of the correlations of microbial community and volatile compounds between pit-mud and fermented grains of compound-flavor Baijiu. Foods 13, 203 (2024).
Kang, J. et al. Deciphering the shifts in microbial community diversity from material pretreatment to saccharification process of Fuyu-flavor Baijiu. Front. Microbiol. 12 (2021).
Cai, W. et al. The depth-depended fungal diversity and non-depth-depended aroma profiles of pit mud for strong-flavor Baijiu. Front. Microbiol. 12 (2022).
Zhao, J. & Gao, Z. Dynamic changes in microbial communities and flavor during different fermentation stages of proso millet Baijiu, a new product from Shanxi light-flavored Baijiu. Front. Microbiol. 15 (2024).
Zhang, J. et al. The improvement of Hovenia acerba-sorghum co-fermentation in terms of microbial diversity, functional ingredients, and volatile flavor components during Baijiu fermentation. Front. Microbiol. 14 (2024).
Wang, Y., Quan, S., Xia, Y., Wu, Z. & Zhang, W. Exploring the regulated effects of solid-state fortified Jiuqu and liquid-state fortified agent on Chinese Baijiu brewing. Food Res. Int. 179, 114024 (2024).
Yu, X. et al. Effects of six commercially available Koji (Chinese Xiaoqu) on the production of Ethyl acetate, Ethyl lactate, and higher alcohols in Chinese Baijiu (distilled spirit) brewing. Heliyon 9, e17739 (2023).
Li, J. et al. Microbial community Spatial structures in Luzhou-flavored liquor pit muds with different brewing materials. PeerJ 10, e12987 (2022).
Lejzerowicz, F. et al. High-throughput sequencing and morphology perform equally well for benthic monitoring of marine ecosystems. Sci. Rep. 5, 13932 (2015).
Schön, M. E. et al. Single cell genomics reveals plastid-lacking Picozoa are close relatives of red algae. Nat. Commun. 12, 6651 (2021).
Hu, X., Du, H., Ren, C. & Xu, Y. Illuminating anaerobic microbial community and cooccurrence patterns across a quality gradient in Chinese liquor fermentation pit muds. Appl. Environ. Microbiol. 82, 2506–2515 (2016).
Maoke, L. et al. Recent advances in research on the community, isolation, and application of microbes in the pit mud used in manufactrue of Chinese strong-flavor Baijiu. Microbiol. China. 44, 1222–1229 (2017).
Xiaolong, H. et al. Microbial community succession pattern and spatial heterogeneity in fermented grains of strong-flavor Baijiu. Food Ferment. Ind. 46, 66–73 (2020).
Lv, X. C. et al. Characterization of fungal community and dynamics during the traditional brewing of Wuyi Hong qu glutinous rice wine by means of multiple culture-independent methods. Food Control. 54, 231–239 (2015).
Sen, Z., Cheng, W., Shaobin, Z., Wenjun, Z. & Jingguo, L. Fungal community diversity of strong-flavor Baijiu Daqu based on high-throughput sequencing technology. China Brew. 40, 55–59 (2021).
Wang, C., Shi, D. & Gong, G. Microorganisms in daqu: a starter culture of Chinese Maotai-flavor liquor. World J. Microbiol. Biotechnol. 24, 2183–2190 (2008).
Wu, Q., Chen, L. & Xu, Y. Yeast community associated with the solid state fermentation of traditional Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 166, 323–330 (2013).
Shikuan, W., Ming, P., Yanli, X., Chengjin, Y. & Haiguang, Y. Changes of fungi during strong flvour Daqu preparation. China Brew. 29, 42–45 (2010).
Wang, H. Y., Gao, Y. B., Fan, Q. W. & Xu, Y. Characterization and comparison of microbial community of different typical Chinese liquor Daqus by PCR-DGGE. Lett. Appl. Microbiol. 53, 134–140 (2011).
Bing, L. et al. Research progress on funcitional microbes and enzymes in Daqu of Baijiu. China Brew. 38, 7–12 (2019).
Wang, F. Analysis of Microbiol Diversity’s Distribution Pattern in Fermentation of Liquor (Inner Mongolia University, 2014).
Liu, M. et al. Determination of the fungal community of pit mud in fermentation cellars for Chinese strong-flavor liquor, using DGGE and illumina miseq sequencing. Food Res. Int. 91, 80–87 (2017).
Li, K. Microbial Community and Diversity of Fermentated Yellow Water in Chinese Intense Flavor Liquor Cellar (Xihua University, 2014).
Li, K. Analysis of Microflora and Falvor Compounds in the Production of Special-Flavor Liquor (Nanchang University, 2017).
Zhang, M. Study on Wenwanggong Baijiu Microbial Community Structure and Its Association with Main Flavoring (Hefei University of Technology, 2021).
Mou, H. Screening Yeast Strains for the Winemaking of Aroma Enhancement from the Brewing Ecosystem of Luzhou Flavor Liquor in South Sichuan (Northwest Agriculture and Forestry University, 2015).
Zheng, X. W. et al. Microbiota dynamics related to environmental conditions during the fermentative production of Fen-Daqu, a Chinese industrial fermentation starter. Int. J. Food Microbiol. 182–183, 57–62 (2014).
Lesschaeve, I. & Noble, A. C. Polyphenols: factors influencing their sensory properties and their effects on food and beverage preferences. Am. J. Clin. Nutr. 81, 330S–335S (2005).
Shi, B., He, Q., Yao, K., Huang, W. & Li, Q. Production of ellagic acid from degradation of valonea tannins by Aspergillus niger and Candida utilis. J. Chem. Technol. Biotechnol. 80, 1154–1159 (2005).
Tao, W. et al. Diversity of molds producing esterifying enzyme during fermentation of strong-flavor liquor. Food Ferment. Ind. 38, 37–41 (2012).
Zheng, J. et al. Characterization of microbial communities in strong aromatic liquor fermentation pit muds of different ages assessed by combined DGGE and PLFA analyses. Food Res. Int. 54, 660–666 (2013).
Qin, X. et al. Comparative analysis of microbial community diversity and physicochemical factors of Nongxiangxing Baijiu pit mud at different ages and cellar locations. Food Sci. 44, 165–174 (2023).
Liu, H. & Sun, B. Effect of fermentation processing on the flavor of Baijiu. J. Agric. Food Chem. 66, 5425–5432 (2018).
Dlamini, B. C., Buys, E. M. & Taylor, J. R. Effect of sorghum type and malting on production of free amino nitrogen in conjunction with exogenous protease enzymes. J. Sci. Food Agric. 95, 417–422 (2015).
Acknowledgements
The authors would like to thank Jiajia Ni at Guangdong Meilikang Bio-Science Ltd. (Foshan, China) for his assistance with data analysis and visualization. We also thank Lei Zheng and Zixuan Ma from Shandong Baimaiquan Wine Co., Ltd. (Jinan, China) for their assistance in sampling.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.L.; methodology, M.Z. and J.L.; software, J.L.; validation, C.W.; formal analysis, M.Z. and J.L.; investigation, M.Z., J.L., Y.Z., K.W., W.H., and C.W.; resources, J.L.; data curation, W.H.; writing—original draft preparation, M.Z.; writing—review and editing, J.L.; visualization, M.Z., J.L. and Y.Z.; supervision, J.L.; project administration, J.L.; funding acquisition, J.L. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, M., Li, J., Zhang, Y. et al. Different brewing materials change the vertical distribution pattern of eukaryotic communities in Luzhou-flavor liquor pit muds. Sci Rep 16, 2258 (2026). https://doi.org/10.1038/s41598-025-32011-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-32011-z
