
Abstract
Neurofibromatosis type 1 (OMIM 613,113, NF1) is a neurocutaneous disorder caused by pathogenic genetic alteration in NF1 gene, which exhibits nearly full penetrance and affects multiple systems. Previously two association studies of optic pathway glioma and NF1 protein domains, derived from 215 and 381 patients, respectively, obtained contradicting results, reflecting different datasets can lead to different conclusions and there is a need for a larger dataset to reach a solid conclusion. There is another association study based on 832 patients considering protein domains, clinical features, and types of variants. But it only investigated the GTPase-activating protein domain and non-truncating variants. In this study, an extended association analysis involving eight protein domains, two types of variants, namely truncating and non-truncating variants, and 32 clinical features, was performed based on a combined dataset of 1663 NF1 patients consisting of 738 cases recruited in Hong Kong and 925 reported cases from 25 studies. In summary, this study has identified 121 statistically significant associations between clinical features, types of variants, and protein domains, with 120 of them being novel findings. These new insights about the genotype–phenotype association promote better clinical management.
Similar content being viewed by others
Introduction
Neurofibromatosis type 1 (OMIM 613,113) is a relatively common and well known neurocutaneous disorder caused by pathogenic genetic alterations of the NF1 gene, which has a high mutation rate1. NF1 is a multisystem disorder characterized by skin pigmentary features, multiple neurofibromata, optic pathway glioma (OPG), Lisch nodules, sphenoid dysplasia, and pseudoarthrosis of a long bone. The NF1 gene is mapped to chromosome 17q11.2. It consists of 61 exons and spans over 282 kb of genomic DNA. It encodes the protein neurofibromin which is one of the players in the RAS/MAPK, PI3K/mTOR, and cAMP pathways2. It is also regarded as a tumour suppressor, with a role in regulation of cellular growth, proliferation and differentiation3. The neurofibromin consists of 9 functional domains namely the cysteine/serine-rich domain (CSRD), tubulin-binding domain (TBD), GTPase-activating protein-related domain (GRD) Sec14-like domain (Sec14), pleckstrin homology-like domain (PH), HEAT-like repeat regions (HLR), C-terminal domain (CTD), nuclear localization signal region (NLS), and the syndecan-binding region (SBR)4. The diagnosis of NF1 can be made with reference to the revised NIH criteria which include ≥ 6 café au lait macules, axillary or inguinal freckling, ≥ 2 neurofibromas or Lisch nodules, plexiform neurofibromas, optic pathway glioma, distinctive osseous lesion, and the germline NF1 pathogenic variant5. The mutation spectrum of NF1 is wide and includes single nucleotide variants and indels scattered across the entire gene, single/multiple exon deletion/duplication, and whole gene deletion, etc. There was no clear genotype–phenotype correlation except for specific variants6, such as the c.5425C > T p.(Arg1809Cys) missense variant which correlates with facial features of Noonan-syndrome, and the c.3826C > T (p.(Arg1276*) nonsense variant which correlates with cardiovascular disorders7,8. This lack of genotype–phenotype correlation has made it difficult for the medical professionals to predict the clinical outcome of their patients and to tailor follow-up plan based on the molecular findings.
Single nucleotide variants and indels can generally be classified as truncating or non-truncating. Protein function is easily affected by the shortening of the protein by truncating variants9, while non-truncating variants often affect the protein binding site or enzyme active site or alter the protein 3D structure10,11,12. A protein domain is defined as the functional and/or structural unit in a protein. Non-truncating variants of the same type in the same protein domain tend to produce similar phenotypes because protein function is similarly affected. In addition, protein domains and variant types have been included in the ACMG 2015 variant interpretation guideline13. An increasing number of genes have been found to have multiple disease-causing mechanisms associated with domains and types of variants14,15. Domain-specific genotype–phenotype studies are required and should also consider the type of variant in the studies.
Previous domain-specific analyses have demonstrated the ability to guide clinical management based on molecular evidence16. Patients with non-truncating variants in the Ras GTPase-activating protein domain (RAS-GAP, or GRD) were found to have a significantly higher percentage of congenital heart anomalies. A complete analysis involving other domains of this gene, as well as truncating and non-truncating variants, would facilitate variant interpretation and disease mechanism studies.
In this study, we used data from our own NF1 cohort as well as data from 12 publications to conduct an in-depth investigation of the relationship between clinical features, domains, and types of variants, which can be used to guide the variant interpretation to benefit clinical care planning and support the investigations into disease-causing mechanisms.
Methods
Ethics statement
This project was reviewed and approved by the University of Hong Kong Human Research Ethics Committee (HREC reference number: EA240038). This was a secondary data analysis project, and informed consent was not required by the Committee. All procedures were performed in accordance with relevant guidelines and regulations.
Our cohort
Clinical features and genotype data were obtained from 738 individuals with molecularly confirmed NF1 in the Clinical Genetics Service Unit, Hong Kong Children’s Hospital, Hong Kong SAR, China (hereinafter referred to as the CGS cohort. For details see Ho et al., 2022).
Public cohort
Among 121 search results of ‘NF1 variant’ and ‘genotype’/ ‘phenotype’ in PubMed (on 17 December 2022), only 25 publications provided both clinical features and NF1 variants of each case7,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40. The large amount of variants prevents us to re-classified all of them according to ACMG, ACGS guidelines, or ClinGen recommendations, but sequence variants were further filtered and only variants with at least one pathogenic or likely pathogenic ClinVar submission were kept to construct the public cohort with reported pathogenic and likely pathogenic variants. A collection of 925 NF1 sequence variants and clinical features were obtained from these 25 publications.
Variant analysis
The transcript NM_000267.3 has the largest number of variations in Leiden Open Variation Database (LOVD) NF1 database (https://databases.lovd.nl/shared/transcripts/00014502). For comparison with the previous publication of our institute16, transcript NM_000267.3 was used. Based on transcript NM_000267.3, variants were classified as truncating or non-truncating variants by variant consequences41,42. Variant consequences involving splice acceptor variant, splice donor variant, splice region variant, start lost, nonsense, frameshift variants, and microdeletions were considered as truncating variants. While non-truncating variant consequences involve missense variant, intron variant, inframe insertion, and inframe deletion. For variants with RNA study supporting the out-of-frame splicing effect, involving c.1466A > G, c.479G > C, and c.5791 T > C, are classified as truncating variants43.
Clinical features studied in this study are listed in Table 1.
Protein domains were obtained from a recent publication4 instead of from UniProt which contains only 2 of the protein domains and were based on 3 previous publications44,45,46.
The number of truncating and non-truncating variants per domain was obtained by bedtools v2.25.047. The Fisher’s exact test was used to compare distributions of categorical variables48. A p-value < 0.001 was considered statistically significant.
Results
Demographic data
A total of 1663 individuals were included in the NF1 dataset comprising 809 females, 801 males, and 53 individuals of unreported sex. There are 1,210 (72.76%) ≥ 8 years old individuals. Table 1 summarises the frequencies of 32 clinical features. The top 3 most frequent clinical features are CALMs (Café-Au-Lait Spots or Café-Au-Lait Macules) (94.58%), freckling (71.04%), and cutaneous neurofibromas (42.92%). All of them are included in the NIH diagnostic criteria of NF15.
Domain VS Clinical Features
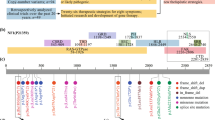
All 9 protein domains of the neurofibromin were studied (Fig. 1).

Distribution of NF1 variants (Reference transcript: NM_000267.3). Stacked bar plot showing the number of unique variants in exons. Splice site mutations are included in the closest exon. Protein domains and tertiles are shown below the stacked bar plot. CSRD: cysteine/serine-rich domain. TBD: tubulin-binding domain. GRD: GTPase-activating protein-related domain. Sec14: Sec14-like domain. PH: pleckstrin homology-like domain. HLR: HEAT-like repeat regions. CTD: C-terminal domain. NLS: nuclear localization signal region. SBR: syndecan-binding region.
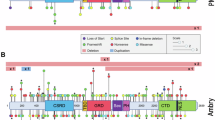
Considering all the truncating and non-truncating variants, a total of 34 pairs out of the 288 investigated pairs of domains and clinical features were identified to have statistically significant associations (p-value < 0.001) and all of them are novel (Table 2, Table S1, Figs. 2, and 3). For clinical features with common age of onset ≥ 8 years old, only samples fulfil the age criteria were included in the comparison.

Categories of clinical features associated with domains. Number in the sub-bar represents the number of clinical features associated with a domain.

Clinical features with statistically significant associations with domains.
Associations between domains and clinical features were further grouped by categories because clinical features of the same categories are usually evaluated together during the diagnostic process (Fig. 2). Variations in frequencies between different domains and clinical features outlined in the NIH criteria for neurofibromatosis type 1 have been noted. In addition, among the domains analysed, it was observed that variants in the CSRD, TBD, GRD and PH domains were positively associated with clinical features overlapping with Noonan syndrome. Clinical features in NIH criteria and features overlapping with Noonan syndrome are further discussed below.
Clinical features in NIH criteria
Eight NIH criteria-related clinical features were included in this study: CALMs, freckling, Lisch nodules, optic pathway glioma, pseudoarthrosis of a long bone, sphenoid bone dysplasia, plexiform neurofibromas, and cutaneous neurofibromas. Half of these clinical features were found to have association with domains, except freckling, optic pathway glioma, pseudoarthrosis of a long bone and sphenoid bone dysplasia.
Patients with variant in the CSRD domain were found to have statistically significant higher frequencies of 3 NIH criteria-related clinical features: Lisch nodules (47.50% vs. 40.39%, OR = 2.06, CI 1.44–2.95, p < 0.001), plexiform neurofibromas (35.52% vs. 16.63%, OR = 2.76, CI 1.91–397, p < 0.001), and cutaneous neurofibromas (70.62% vs. 48.83%, OR = 2.62, CI 1.78–3.59, p < 0.001).
Clinical features overlapping with Noonan-syndrome
Phenotypic overlap between neurofibromatosis type 1 and Noonan-syndrome has been discussed in many studies 19,49. In this study, we included nineteen clinical features that overlapped with Noonan syndrome and investigated the domain-specific genotype–phenotype correlation. Four groups of clinical features were studied, including 5 facial features, 6 neurological and behavioural features, 4 cardiovascular abnormalities, as well as 4 growth and musculoskeletal features.
Noonan-syndrome facial features
Five Noonan-syndrome facial features in this study include broad nasal bridge, hypertelorism, ptosis, down-slanting palpebral fissures, and bulbous nose. Frequencies of certain Noonan-syndrome facial features increased in the middle tertile (TBD, GRD, and PH) of the NF1 gene (Fig. 2).
Ptosis was found to have positive associations with variants in GRD and PH, respectively. Patients with the GRD variants were found to have an increased frequency of ptosis compared to other patients (36.52% vs. 1523%, OR = 3.20, CI 2.17–4.70, p < 0.001). A similar pattern can be seen in comparing patients with the PH variants (63.64% vs. 15.97%, OR = 9.18, CI 5.23–16.44, p < 0.001) to other patients. Increased frequency of hypertelorism was found in patients with TBD (45.83% vs. 19.67%, OR = 3.45, CI 1.81–6.52, p < 0.001) and GRD (32.50% vs. 18.47%, OR = 2.12, CI 1.41–3.17, p < 0.001) variants compared to other patients.
Neurological and behavioural features
The neurological and behavioural features in this study include delayed cognitive development, mental retardation, speech delay, attention deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), and Epilepsy. Similar to the Noonan-syndrome facial features, frequencies of certain neurological and behavioural features increased in the middle tertile (TBD, GRD, and PH) of the NF1 gene (Fig. 2).
When comparing the frequency of clinical features in patients with variants in the domain to other patients, the frequency of delayed cognitive development was found to be increased in patients with variants in the PH domain (52.12% vs. 27.75%, OR = 2.83, CI 2.01–3.99, p < 0.001).
Likewise, higher frequencies of speech delay were observed in patients with variants in TBD (32.56% vs. 12.52%, OR = 3.37, CI 1.58–6.86, p < 0.001), GRD (24.31% vs. 11.25%, OR = 2.53, CI 1.56–4.05, p < 0.001) and PH domain (44.74% vs. 12.04%, OR = 5.89, CI 2.81–12.22, p < 0.001).
In a similar manner, patients with variants in the PH domain (37.84% vs. 14.34%, OR = 3.63, CI 1.67–7.62, p < 0.001) were found to have a higher frequency of ADHD when comparing with other patients without variants in the specific domain.
Cardiovascular abnormalities
Increased frequencies of cardiovascular abnormalities were found in patients with variants in the GRD domain (18.30% vs. 6.92%, OR = 3.01, CI 2.02–4.47, p < 0.001) when compared to other patients without variants in the specific domain. This finding demonstrates the importance of regular cardiac follow-up for patients with variants in the GRD.
Growth and musculoskeletal features
Scoliosis, macrocephaly, short stature, and pectus deformity were included in the growth and musculoskeletal features in this study. An association between the PH domain and pectus deformity was observed. Patients with PH domain variants were 8.16 times (CI 3.80–17.14) more likely to develop pectus deformity (p < 0.001) and 3.15 times (CI 1.60–6.03) more likely to develop short stature (p < 0.001). Similarly, short stature was found to have a higher frequency in patients with the GRD variants (29.31% vs. 12.18%, OR = 2.98, CI 1.78–4.95, p < 0.001) compared to other patients.
Based on the above, the frequency of certain growth and musculoskeletal features was increased in the middle tertile (GRD and PH) region of the NF1 gene.
Types of variants VS clinical features
To investigate whether different types of variants disrupt NF1 function by affecting the protein domain, we compared the frequencies of clinical features between individuals with a specific type of variants in the domain and other individuals with the same type of variants but outside of this domain.
For non-truncating variants, twenty-six statistically significant pairs of clinical features and domains were found (Table S2). Twenty-five pairs of them are new findings, except the patients with variants in the GRD domain have a higher frequency of congenital heart disease mentioned in the previous study16. Pectus abnormalities considered more frequent in patients with non-truncating variants in the GRD domain16 were found to have no such association in this study. Similarly, 2 clinical features (Lisch nodules and cutaneous neurofibromas) considered less frequent in patients with non-truncating variants in the GRD domain16 were found to have no such association in this study.
2 clinical features were found to have statistically significant positive associations with non-truncating variants in domains while no such significant associations were found in when considering all variants in domains. Non-truncating variants in PH domain are positively associated with epilepsy and hypertelorism.
When comparing the prevalence of specific clinical features of patients with truncating variants in the domain to that of patients with truncating variants in other domains, no pairs of domains and clinical features with statistically significant associations (p-value < 0.001) (Table S3) were found.
Comparison of truncating variants and non-truncating variants with or without the clinical features was conducted to further explore the association between the types of variants and clinical features regardless of the domain. Sixteen clinical features were found to have significant positive associations with truncating variants or non-truncating variants (Table S4). Higher frequencies of Lisch nodules and CALMs were found in the patients with truncating variants compared with non-truncating variants.
Variant consequences VS clinical features
To investigate whether certain variant consequences have statistically significant associations with clinical features, we compared the frequencies of clinical features between individuals with a specific type of variant and other individuals with other variant consequences (Table S5). Four variant consequences, frameshift variant, stop-gained, missense variant, and in-frame deletion, were found to have positive associations with a total of twenty clinical features.
The associations between variant consequences, domains, and clinical features were studied for the top 3 variant consequences in this study: missense (48%), stop gained (15%), and frameshift (13%) variants. Twenty-eight clinical features have statistically significant associations with missense variants (Table S6). But only one clinical feature, speech delay, has a statistically significant association with frameshift (Table S7). No significant association for stop-gained variants (Table S8).
Variants not in any domain
Besides variants in domains, there are 446 variants not in any domains included in this study, including 245 truncating variants and 201 non-truncating variants. Distributions of clinical features, types of variants, domains and not in any domains were studied. When considering all variants, eighteen out of thirty-two clinical features were found to have higher frequencies in variants that do not belong to any known domains compared with variants in domains (Table S9). Similarly, the numbers become twenty-nine for truncating variants (Table S10) and two for non-truncating variants (Table S11).
Discussion
Larger sample collections yield more precise associations
To the best of our knowledge, the largest NF1 study16 that studied the associations between domains and clinical features only considered the GRD domain and non-truncating variants. Since this study is twice the size of the previous NF1 study considering all domains and both types of variants, it enables us to discover 99.17% (120/121) novel findings with statistically significantly different frequencies (p-value < 0.001). Consistent with the previous study which only considered the GRD domains, congenital heart disease was found to be associated with the non-truncating variants in the GRD domain. This study has demonstrated the same analysis workflow can be applied to other domains of NF1 gene.
Despite the emergence of numerous new findings, there is still considerable scope for improvement in our understanding. Regarding optic pathway glioma, two previous studies50,51 present conflicting results, whereas the most recent study aligns with our findings that there is no statistically significant correlation between various domains and optic pathway glioma. However, it is worth noting that patients with PH domain variants exhibited a lower risk of developing optic pathway glioma, as supported by this study. Notably, our research encompassed a cohort of 970 patients aged 7 years and older, nearly tripling the participant count of previous optic pathway glioma studies. Surprisingly, none of the patients with PH domain variants in our study were diagnosed with optic pathway glioma. Furthermore, we observed associations between all domains of NF1 and at least one clinical feature, except for NLS and SBR domains, which had a limited number of variants. Similarly, clinical features with low frequencies within our cohort did not exhibit associations with any specific domain. These suggest that there is still room for improvement in the diversity of samples in the cohort.
Overall, this analysis workflow can be extended to other genes as long as clinical details and molecular information were collected properly. Complex population studies, like the 100,000 Genomes Project52, Singapore 10 K Genome Project53, All of Us Research Program54, and on-going Hong Kong Genome Project55, can boost genotype–phenotype association discovery by providing both clinical details and molecular information.
Diversified association patterns between categories of clinical features and domains
Distinct groups of clinical features exhibit varying patterns of associations with different domains. 3 clinical features involved in NIH NF1 diagnostic criteria were found to have significantly increased frequencies in patients with variants in CSRD when compared to patients without variants in the corresponding domain.
When compared to patients without variants in the corresponding domain, patients with the middle tertile domain GRD variants were found to have increased frequency of clinical features across all four categories of clinical features overlapped with Noonan syndrome. While patients with TBD or PH variants only have increased frequency related to two and three categories of clinical features overlapped with Noonan syndrome, respectively. Overall, different categories of clinical features demonstrate distinct patterns of associations with the domains which suggests variants in domains might affect the protein function in different ways leading to various damages of different systems.
The PH domain is the second smallest domain of NF1 (101 amino acids). Variant c.5425C > T p.(Arg1809Cys) in this domain is associated with facial features of Noonan-syndrome including hypertelorism, ptosis, low-set ears, webbed neck, and triangular face7. In this study, variants in the PH domain are positively associated with a Noonan-syndrome facial feature, ptosis. This reaffirmed the previous finding based on a single variant and extended it to a domain level. For cases with variants in the PH domain and ptosis, 31 out of 42 were impacted by the variant c.5425C > T. This suggests the association between the PH domain and Noonan features is mainly due to this variant.
A previous study showed the variant c.3826C > T p. (Arg1276*) associated with cardiovascular abnormalities8. The variant c.3826C > T is in the GRD domain. There are only 2 patients with this variant in our cohort. One of them does not have the congenital heart disease and hypertension. The other one does not have the information of these two clinical features. In this study, variants in the GRD domain are positively associated with 2 cardiovascular features: hypertension and congenital heart disease. The GRD domain is positively associated with congenital heart disease when considering only non-truncating variants. However, no significant association was found when considering only truncating variants. This suggests that the association between the GRD domain and cardiovascular features is mainly due to non-truncating variants.
Truncating variants escaping nonsense-mediated decay
Truncating variants can lead to shortened mRNA or shuffled parts of the mRNA sequence by changing the reading frame. These abnormal mRNAs are believed to be eliminated by Nonsense-mediated mRNA decay (NMD). Observing truncating variants in NF1 patients with various clinical features implies that there should be some abnormal mRNA escaping the NMD and leading to abnormal phenotypes which is consistent with the hypothesis mentioned in a previous study56.
In addition, the significant associations between truncating variants in specific domains and clinical features imply that there are various lengths of the gene products that involve different functional domains and these domains which might be truncated or partially altered will eventually lead to different clinical features. However, further analyses involving RNA and protein expression studies are required to help scientists to articulate the story.
Conclusions
This study is the first study for eight domains of the NF1 gene on domain-specific and types of variants-specific basics. Utilisation of the clinical features of individuals with molecularly confirmed variants of the CGS cohort and publications enables us to associate the phenotypes, domains, and types of variants, the result of which can act as a practical guideline for clinical management. Yet, persistently comprehensive collections of molecular evidence and clinical features are required to promote the NF1 study further.
Data availability
The datasets used and/or analysed during the current study is available from the corresponding author on reasonable request.
Change history
25 February 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41598-026-41593-1
References
Isakov, O., Wallis, D., Evans, D. G. & Ben-Shachar, S. Exhaustive non-synonymous variants functionality prediction enables high resolution characterization of the neurofibromin architecture. EBioMedicine 36, 508–516. https://doi.org/10.1016/j.ebiom.2018.09.039 (2018).
Kiuru, M. & Busam, K. J. The NF1 gene in tumor syndromes and melanoma. Lab. Invest. 97, 146–157. https://doi.org/10.1038/labinvest.2016.142 (2017).
Ratner, N. & Miller, S. J. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 15, 290–301. https://doi.org/10.1038/nrc3911 (2015).
Xu, M. et al. Identification of mutation regions on NF1 responsible for high- and low-risk development of optic pathway glioma in neurofibromatosis type I. Front. Genetics 9, 270. https://doi.org/10.3389/fgene.2018.00270 (2018).
Legius, E. et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet. Med. 23, 1506–1513. https://doi.org/10.1038/s41436-021-01170-5 (2021).
Friedman, J. M. Neurofibromatosis 1. GeneReviews (2022).
Pinna, V. et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Europ. J. Human Genet. 23, 1068–1071. https://doi.org/10.1038/ejhg.2014.243 (2015).
Napolitano, F. et al. Genotype-phenotype correlations in neurofibromatosis type 1: identification of novel and recurrent NF1 gene variants and correlations with neurocognitive phenotype. Genes 13, 1130 (2022).
DeBoever, C. et al. Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study. Nat. Commun. 9, 1612. https://doi.org/10.1038/s41467-018-03910-9 (2018).
Ittisoponpisan, S. et al. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated?. J. Mol. Biol. 431, 2197–2212. https://doi.org/10.1016/j.jmb.2019.04.009 (2019).
Li, M., Petukh, M., Alexov, E. & Panchenko, A. R. Predicting the impact of missense mutations on protein-protein binding affinity. J Chem Theory Comput 10, 1770–1780. https://doi.org/10.1021/ct401022c (2014).
Liu, J. et al. Functional identification of two novel variants and a hypomorphic variant in ASS1 from patients with Citrullinemia type I. Front. Genet. 14, 1172947. https://doi.org/10.3389/fgene.2023.1172947 (2023).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Qiu, W. et al. Structural insights into Noonan/LEOPARD syndrome-related mutants of protein-tyrosine phosphatase SHP2 (PTPN11). BMC Struct. Biol. 14, 10. https://doi.org/10.1186/1472-6807-14-10 (2014).
Motta, M. et al. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum. Mol. Genet. 28, 1007–1022. https://doi.org/10.1093/hmg/ddy412 (2018).
Ho, S. K. L. et al. Old and new perspectives on Neurofibromatosis type 1: Clinical and molecular characterization of 832 patients from a single centre over 16 years. Eur. J. Med. Genetics 65, 104474. https://doi.org/10.1016/j.ejmg.2022.104474 (2022).
Upadhyaya, M. et al. The spectrum of somatic and germline NF1 mutations in NF1 patients with spinal neurofibromas. Neurogenetics 10, 251–263. https://doi.org/10.1007/s10048-009-0178-0 (2009).
Peters, H., Lüder, A., Harder, A., Schuelke, M. & Tinschert, S. Mutation screening of neurofibromatosis type 1 (NF1) exons 28 and 29 with single strand conformation polymorphism (SSCP): Five novel mutations, one recurrent transition and two polymorphisms in a panel of 118 unrelated NF1 patients. Hum. Mutat. 13, 258–258. https://doi.org/10.1002/(SICI)1098-1004(1999)13:3 (1999).
De Luca, A. et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-noonan syndrome. Am. J. Hum. Genet. 77, 1092–1101. https://doi.org/10.1086/498454 (2005).
Bendova, S. et al. Novel mutations in the NF1 gene in Czech patients with neurofibromatosis type 1. J. Mol. Neurosci. 31, 273–279. https://doi.org/10.1385/jmn:31:03:273 (2007).
Koczkowska, M. et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 41, 299–315. https://doi.org/10.1002/humu.23929 (2020).
Koczkowska, M. et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): an update of genotype-phenotype correlation. Genet. Med. 21, 867–876. https://doi.org/10.1038/s41436-018-0269-0 (2019).
Koczkowska, M. et al. Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844–848. Am. J. Hum. Genet. 102, 69–87. https://doi.org/10.1016/j.ajhg.2017.12.001 (2018).
Rojnueangnit, K. et al. High incidence of noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum. Mutat. 36, 1052–1063. https://doi.org/10.1002/humu.22832 (2015).
Ekvall, S. et al. Novel association of neurofibromatosis type 1-causing mutations in families with neurofibromatosis-noonan syndrome. Am. J. Med. Genet. Part A 164, 579–587. https://doi.org/10.1002/ajmg.a.36313 (2014).
Santoro, C. et al. Arg (1809) substitution in neurofibromin: further evidence of a genotype-phenotype correlation in neurofibromatosis type 1. Eur. J. Hum. Genet. 23, 1460–1461. https://doi.org/10.1038/ejhg.2015.93 (2015).
Nyström, A.-M. et al. A severe form of Noonan syndrome and autosomal dominant café-au-lait spots – evidence for different genetic origins. Acta Paediatr. 98, 693–698. https://doi.org/10.1111/j.1651-2227.2008.01170.x (2009).
Bausch, B. et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J. Clin. Endocrinol. Metab. 92, 2784–2792. https://doi.org/10.1210/jc.2006-2833 (2007).
Bottillo, I. et al. Germline mosaicism in neurofibromatosis type 1 due to a paternally derived multi-exon deletion. Am. J. Med. Genet. A. 152, 1467–1473. https://doi.org/10.1002/ajmg.a.33386 (2010).
Douben, H. C. W. et al. High-yield identification of pathogenic NF1 variants by skin fibroblast transcriptome screening after apparently normal diagnostic DNA testing. Hum Mutat 43, 2130–2140. https://doi.org/10.1002/humu.24487 (2022).
Hsiao, M. C. et al. Decoding NF1 intragenic copy-number variations. Am J Hum Genet 97, 238–249. https://doi.org/10.1016/j.ajhg.2015.06.002 (2015).
Kang, E. et al. Phenotype categorization of neurofibromatosis type I and correlation to NF1 mutation types. J. Hum. Genet. 65, 79–89. https://doi.org/10.1038/s10038-019-0695-0 (2020).
Mantripragada, K. K. et al. Identification of novel deletion breakpoints bordered by segmental duplications in the NF1 locus using high resolution array-CGH. J. Med. Genet. 43, 28–38. https://doi.org/10.1136/jmg.2005.033795 (2006).
Pacot, L. et al. Contribution of whole genome sequencing in the molecular diagnosis of mosaic partial deletion of the NF1 gene in neurofibromatosis type 1. Hum. Genet. 142, 1–9. https://doi.org/10.1007/s00439-022-02476-3 (2023).
Ponti, G. et al. Clinico-pathological and biomolecular findings in Italian patients with multiple cutaneous neurofibromas. Hered Cancer Clin. Pract. 9, 6. https://doi.org/10.1186/1897-4287-9-6 (2011).
Tsipi, M. et al. Phenotypic expression of a spectrum of Neurofibromatosis Type 1 (NF1) mutations identified through NGS and MLPA. J. Neurol. Sci. 395, 95–105. https://doi.org/10.1016/j.jns.2018.10.006 (2018).
Wang, X. et al. Breast cancer risk and germline genomic profiling of women with neurofibromatosis type 1 who developed breast cancer. Genes Chromosomes Cancer 57, 19–27. https://doi.org/10.1002/gcc.22503 (2018).
Wu-Chou, Y.-H. et al. Genetic diagnosis of neurofibromatosis type 1: Targeted next- generation sequencing with multiple ligation-dependent probe amplification analysis. J. Biomed. Sci. 25, 72. https://doi.org/10.1186/s12929-018-0474-9 (2018).
Zheng, Y. et al. Two novel mutations of NF1 gene identified in Chinese patients with severe neurofibromatosis type 1. Indian J. Dermatol. Venereol. Leprol. 86, 76–81. https://doi.org/10.4103/ijdvl.IJDVL_1037_18 (2020).
Zhu, L. et al. Clinical and molecular characterization of NF1 patients: single-center experience of 32 patients from china. Medicine (Baltimore) 95, e3043. https://doi.org/10.1097/md.0000000000003043 (2016).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. https://doi.org/10.1038/nature19057 (2016).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122. https://doi.org/10.1186/s13059-016-0974-4 (2016).
Wimmer, K. et al. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5’ splice-site disruption. Hum Mutat 28, 599–612. https://doi.org/10.1002/humu.20493 (2007).
D’Angelo, I., Welti, S., Bonneau, F. & Scheffzek, K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep 7, 174–179. https://doi.org/10.1038/sj.embor.7400602 (2006).
Welti, S., Fraterman, S., D’Angelo, I., Wilm, M. & Scheffzek, K. The Sec14 homology module of neurofibromin binds cellular glycerophospholipids: mass spectrometry and structure of a lipid complex. J. Mol. Biol. 366, 551–562. https://doi.org/10.1016/j.jmb.2006.11.055 (2007).
Welti, S. et al. Structural and biochemical consequences of NF1 associated nontruncating mutations in the Sec14-PH module of neurofibromin. Hum. Mutat. 32, 191–197. https://doi.org/10.1002/humu.21405 (2011).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. https://doi.org/10.1093/bioinformatics/btq033 (2010).
Upton, G. J. G. Fisher’s exact test. J. R. Stat. Soc. Ser. A Stat. Soc. 155, 395–402. https://doi.org/10.2307/2982890 (2018).
Yapijakis, C., Pachis, N., Natsis, S. & Voumvourakis, C. Is Neurofibromatosis type 1–noonan syndrome a phenotypic result of combined genetic and epigenetic factors?. In Vivo 30, 315–320 (2016).
Xu, M. et al. Identification of mutation regions on NF1 responsible for high- and low-risk development of optic pathway glioma in neurofibromatosis type I. Front. Genet. 9, 270. https://doi.org/10.3389/fgene.2018.00270 (2018).
Melloni, G. et al. Risk of optic pathway glioma in neurofibromatosis type 1: No evidence of genotype-phenotype correlations in a large independent cohort. Cancers 11, 1838 (2019).
Turnbull, C. et al. The 100 000 genomes project: Bringing whole genome sequencing to the NHS. BMJ 361, k1687. https://doi.org/10.1136/bmj.k1687 (2018).
Wu, D. et al. Large-scale whole-genome sequencing of three diverse Asian populations in Singapore. Cell 179, 736–749. https://doi.org/10.1016/j.cell.2019.09.019 (2019).
Denny, J. C. et al. The “All of Us” Research Program. N Engl J Med 381, 668–676. https://doi.org/10.1056/NEJMsr1809937 (2019).
Chu, A. et al. Potentials and challenges of launching the pilot phase of Hong Kong Genome Project. J. Transl. Genet. Genomics 6, 290–303. https://doi.org/10.20517/jtgg.2022.02 (2022).
Litchfield, K. et al. Escape from nonsense-mediated decay associates with anti-tumor immunogenicity. Nat. Commun. 11, 3800. https://doi.org/10.1038/s41467-020-17526-5 (2020).
Ou, M. et al. Protein Domain-Specific Genotype-Phenotype Correlation Study of Neurofibromatosis Type 1. SSRN (2024). https://doi.org/10.2139/ssrn.4752924
Acknowledgements
The authors acknowledge the patients with NF1 and their families for contributing the clinical data for this study. The preprint version57 of this manuscript is available on https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4752924.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Min Ou: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Data curation, Writing—original draft, Visualization. Stephanie KL. Ho: Data curation, Resources, Writing—review & editing. Ho-Ming Luk: Conceptualization, Resources, Data curation, Writing—review & editing, Project administration, Supervision. Michael Man-Kit Lee: Writing—review & editing. Shirley SW. Cheng: Writing—review & editing. Tak-Wah Lam: Resources, Writing—review & editing, Supervision. Ivan Fai-Man Lo: Resources, Writing—review & editing, Supervision. Ruibang Luo: Resources, Writing—review & editing, Supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in the name of the author Michael Man-Kit Lee, which was incorrectly given as Micahel Man-Kit Lee.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ou, M., Ho, S.K., Luk, HM. et al. Protein domain-specific genotype–phenotype correlation study of neurofibromatosis type 1. Sci Rep 16, 2709 (2026). https://doi.org/10.1038/s41598-025-32422-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-32422-y
