
Abstract
ABCC6 pathogenic variants affect ischemic and hemorrhagic lesions in multiple organs and represent the third most common cause of hereditary cerebral small vessel diseases (CSVD) in Japan, yet their CSVD phenotypes remains unclear. We conducted a multicenter, cross-sectional study of 158 consecutive Japanese adults with severe CSVD. Genetic analysis was performed, and clinical and brain MRI findings were compared among ABCC6-related CSVD, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), and genetically undetermined CSVD. Univariate and multivariate analyses assessed the association between ABCC6 pathogenic variants and hemorrhagic stroke, adjusting for age and hypertension. Nine patients (5.7%) carried pathogenic heterozygous ABCC6 variants. Hemorrhagic stroke was more frequent in ABCC6-related CSVD than in CADASIL (55.6% vs 13.6%, P = 0.039) and undetermined CSVD (55.6% vs 10.3%, P = 0.010). Multivariate analysis confirmed an independent association (odds ratio 8.4, 95% CI 2.0–38.4, P = 0.004). On MRI, ABCC6-related CSVD showed fewer anterior temporal lesions but more multiple microbleeds than CADASIL. The direction of these findings remained consistent when biallelic cases were included. These findings indicate a distinct, hemorrhage-predominant CSVD phenotype associated with ABCC6 in Japanese patients, support a contributory role of ABCC6, and warrant validation in larger, multi‑ethnic cohorts.
Similar content being viewed by others

Introduction
Cerebral small vessel disease (CSVD) is a leading cause of lacunar infarction, intracerebral hemorrhage, and dementia1. Although most cases are sporadic, several monogenic forms have been identified, with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) being the most prevalent2. While CADASIL has been characterized in detail, facilitating improved diagnostic accuracy, the clinical spectrum of other hereditary CSVDs remains insufficiently defined3,4. Clarifying the phenotype of each hereditary CSVD is essential not only for accurate diagnosis but also for personalized management, prognostication, mechanistic insight, and future therapy development. ABCC6-related CSVD is one such under-characterized entity2,5.
Loss of function in ABCC6, the major gene for pseudoxanthoma elasticum (PXE), leads to calcification and fragmentation of elastic fibers, resulting in vascular fragility6,7. PXE is an autosomal recessive disorder caused by biallelic ABCC6 pathogenic variants; heterozygous carriers are not considered to have PXE, although subclinical or partial phenotypes have been reported8,9. Recently, ABCC6 has also been implicated in CSVD: in a Japanese cohort of severe CSVD, 5.7% carried ABCC6 pathogenic variants, the third most common genetic cause, and in a Korean young-onset stroke cohort, 5.1% harbored pathogenic variants, suggesting relevance to cerebrovascular disease in Asian populations5,10. Nevertheless, the CSVD phenotype attributable to ABCC6 remains unclear, and adequately powered comparative studies are scarce. ABCC6 pathogenic variants have also been reported to be associated with ischemic coronary disease and gastrointestinal hemorrhagic manifestations11,12, raising the possibility that ABCC6‑related CSVD may exhibit a distinct phenotype within CSVD.
We therefore compared the clinical features of ABCC6-related CSVD with those of CADASIL and genetically undetermined CSVD. Regarding imaging characteristics, we conducted detailed comparisons with CADASIL, for which imaging criteria are well established. To directly address carrier effects, we prespecified a heterozygous‑only primary analysis of pathogenic ABCC6 variants, with pooled pathogenic analyses irrespective of zygosity as sensitivity.
Methods
Ethical approval
This study received central ethical approval from the Niigata University Ethics Board (approval numbers 801, 802, and G2020-0032), and all participating centers formally relied on this approval under a central IRB model. Each participating institution entered a formal agreement for reliance on the central IRB. Written informed consent was obtained from all participants or, where necessary, their legally authorized representatives or family members, including consent for publication. All data were anonymized to protect participant privacy. This study was conducted in accordance with the Declaration of Helsinki, and all methods were carried out in accordance with relevant guidelines and regulations.
Study design and participants
We conducted a cross-sectional study across Japan from October 2010 to February 2022. Patients were consecutively referred to our institute for genetic testing when hereditary CSVD was clinically suspected. The referral criteria were standardized across centers and included the presence of symmetrical white matter hyperintensity (Fazekas grade ≥ 2/II) together with at least one additional CSVD-related finding (e.g., lacunar infarcts, dilated perivascular spaces, external capsule lesions, or microbleeds) on brain magnetic resonance imaging (MRI)13,14, and neurological symptoms such as stroke, cognitive impairment, or gait disturbance. The age at onset of neurological symptoms or signs was between the ages of 20 and 60 years (cognitive impairment was defined as a score of ≤ 25 on the Montreal Cognitive Assessment (Japanese edition)15). All referred patients who met these predefined criteria were included in the study, and no cases were excluded after referral.
Genetic analysis
Genetic testing followed a previously reported protocol5. Genomic deoxyribonucleic acid was extracted from blood samples. Initially, conventional genetic tests for NOTCH3 and HTRA1 were performed to identify CADASIL and HTRA1-related CSVD cases. Patients with cysteine-altering NOTCH3 or NOTCH3 p.R75P variants were diagnosed with CADASIL. Using outsourcing services (Macrogen, Korea), whole-exome sequencing was performed on the remaining patients to investigate CSVD causative genes (see Supplementary Fig. S1 online). Copy number variants were analyzed using copy number estimation by the Copy Number estimation by a Mixture Of Poissons (cn.MOPS) package in R (version 4.1.0) and confirmed via droplet digital polymerase chain reaction16.
Variant classification
Allele frequencies for each variant were referenced from two public datasets: the Genome Aggregation Database (gnomAD v4.1.0; https://gnomad.broadinstitute.org/), comprising approximately 807,000 individuals of diverse ancestry, and the Japanese Multi-Omics Reference Panel (14KJPN), which includes whole-genome sequencing data from 14,000 healthy Japanese individuals (28,000 alleles). Allele frequencies from 14KJPN were accessed via the NCBI dbSNP database (https://www.ncbi.nlm.nih.gov/snp/), and values were confirmed as of October 3, 2024.
To assess variant pathogenicity, we used several resources: the ClinVar database for known clinical significance (https://www.ncbi.nlm.nih.gov/clinvar/), in silico analysis tools (PolyPhen2, SIFT, and Provean) on the VaProS website to predict functional impairments (http://p4d-info.nig.ac.jp/vapros/), PHRED scores from combined annotation-dependent depletion to estimate deleteriousness (https://cadd.gs.washington.edu/), and previous literature. Variants were classified according to American College of Medical Genetics and Genomics (ACMG) standards17, ensuring consistent and reliable genetic variant interpretation. Only ABCC6 variants classified as pathogenic according to ACMG were included; this encompassed PXE-reported variants and novel loss-of-function variants meeting ACMG (e.g., pathogenic very strong (PVS) 1 with appropriate supporting evidence). Missense variants of uncertain significance were excluded from the primary analyses but included in a heterozygous sensitivity analysis, considering that such variants, although not classified as pathogenic in PXE due to insufficient evidence, may still have unrecognized pathogenic potential. Participants harboring ABCC6 variants were identified irrespective of zygosity; primary analyses were restricted to heterozygous pathogenic carriers.
Clinical and imaging analysis
Clinical findings were compared among three groups: ABCC6-related CSVD, CADASIL, and genetically undetermined cases. Clinical data, including vascular risk factors, neurological signs and symptoms, and disability levels, were collected using a standardized survey sheet to ensure consistency. Family history was defined as a documented episode of dementia, stroke, or leukoencephalopathy in first- or second-degree relatives. Disability was assessed using the modified Rankin Scale and Barthel Index18,19.
For imaging analysis, ABCC6-related CSVD was compared with CADASIL and referenced against the established CADASIL imaging phenotype. We did not designate the genetically undetermined group as a primary imaging comparator because the absence of known pathogenic variants does not exclude undetected or yet-to-be-identified genetic causes, rendering this group heterogeneous and therefore unsuitable as a primary imaging comparator. Brain MRI scans were acquired using 3.0 Tesla scanners across all centers. T2*-weighted images were primarily used for the evaluation of microbleeds; susceptibility-weighted imaging (SWI) was used when T2* images were not available (n = 1 in the ABCC6-related CSVD group, n = 2 in the CADASIL group). Minor variability in sequence parameters across centers may have existed, but all images were reviewed centrally to ensure consistency of evaluation. All scans were independently assessed by two authors (S.K. and S.A.), blinded to the patients’ genotypes and clinical information; disagreements were adjusted by a third author (M.U.), and final ratings were determined by consensus. Inter-rater reliability was not calculated because consensus ratings after adjudication were used. White matter hyperintensities were graded with the Fazekas scale; anterior temporal lesion severity was rated on a four-point scale (0–3) as described previously20. Lacunar infarcts and microbleeds were defined according to the Standards for Reporting Vascular Changes on Neuroimaging21.
Statistical analysis
Primary analysis focused on heterozygous carriers with pathogenic ABCC6 variants. Sensitivity analyses were performed in two steps: first, including all pathogenic cases irrespective of zygosity (heterozygous and biallelic), and second, adding heterozygous carriers with variants of uncertain significance to assess robustness to variant classification. We did not perform formal kinship filtering due to data limitations, but confirmed from clinical and pedigree information that no known relatives were included in the cohort. For microbleed (MB)–related outcomes, we repeated comparisons after excluding outliers defined by the Tukey fence, applied independently within each diagnostic group. In the heterozygous-pathogenic ABCC6 set, Q1 and Q3 were 12 and 35 (IQR = 23), giving an upper fence of 69.5; one case (Patient 7; total MBs = 524) exceeded this threshold and was excluded in a sensitivity analysis (Supplementary Table S3). In the CADASIL group, Q1 and Q3 were 1.0 and 16.5 (IQR = 15.5), yielding an upper fence of 39.75; four cases exceeded this threshold and were excluded in a parallel sensitivity analysis (two cases originally lacked microbleed data, yielding n = 38 for analysis).
The statistical analysis systematically addressed our primary research questions. First, we investigated whether p.M848fs variant frequency in ABCC6 (relatively common in the Japanese population) is higher in patients with CSVD than in a Japanese public database or an amyotrophic lateral sclerosis cohort from our institute (see Supplementary Table S1 online). We used Fisher’s exact test for this comparison, with statistical significance at P < 0.05 (one-tailed test).
To compare clinical features among the three groups (ABCC6-related CSVD, CADASIL, and undetermined cases), we used the Kruskal–Wallis test for continuous variables and the chi-square test for categorical variables. Post-hoc analyses were conducted using the Dunn–Bonferroni test for continuous variables and the Bonferroni correction for categorical variables to account for multiple comparisons.
To assess the effects of ABCC6 pathogenic variants on hemorrhagic stroke, we developed two logistic regression models: Model 1 included hypertension and age of onset of neurological signs or symptoms, the two most consistently reported risk factors for CSVD and hemorrhagic stroke in Asian populations22. Given the limited sample size and number of outcome events in our cohort, additional covariates were not entered to avoid model overfitting. Model 2 included only hypertension. Adjusted odds ratios (ORs) with 95% confidence intervals (CIs) were calculated for each model.
To compare imaging findings between ABCC6-related CSVD and CADASIL, we used the Wilcoxon rank-sum test for continuous variables and Fisher’s exact test for categorical variables. Receiver operating characteristic curve analysis identified a threshold of 11.5 microbleeds (sensitivity: 77.8%, specificity: 71.4%) associated with ABCC6-related CSVD, using Youden’s J statistic (see Supplementary Fig. S2 online). Multiple microbleeds were defined as > 11 (i.e., ≥ 12), and Fisher’s exact test based on this cutoff was used for comparisons between the two groups.
Results
ABCC6 variant frequency and characteristics in patients with CSVD
The study cohort comprised 158 consecutive index patients recruited from 91 neurological centers across Japan, ensuring broad nationwide representation (see Supplementary Fig. S3 and Table S2 online). ABCC6 variants were identified in 14 cases (8.9%), including 11 heterozygous carriers, one homozygous mutant, and two compound heterozygous mutants (see Supplementary Fig. S1 online). Notably, one patient (Patient 14) was clinically and pathologically diagnosed with PXE (see Supplementary Table S3 online). The comparison cohort for patients with ABCC6 variants included 44 patients with CADASIL and 78 with genetically undetermined CSVD; patients with other genetic variants were excluded.
The frequency and pathogenicity classification of each ABCC6 variants identified in this study are summarized in Table 1 and Supplementary Table S5. Frameshift variants were the most common, accounting for 58.8% (10/17) of mutant alleles, with p.M848fs being particularly prevalent (52.9%, 9/17). All frameshift variants (p.M848fs and p.L1313fs) were classified as pathogenic based on ACMG guidelines and have been previously reported in PXE23. Nonsense variants were observed in two alleles: p.Q378X (n = 1), previously reported in PXE23, and p.W14X (n = 1), a novel variant occurring early in the coding sequence. Although not previously reported, p.W14X was considered pathogenic due to ACMG (predicted loss-of-function; PVS1). Large deletions involving exons 7–10, 23–25, and 30–31 were identified in one case accounting for 5.9% (1/17) of ABCC6 mutant alleles (see Supplementary Table S4 online). CNVs in ABCC6 have been previously reported in PXE, and such large deletions are expected to lead to truncated or absent protein products, disrupting the ATP-binding cassette transporter function of ABCC6, which may contribute to vascular calcification and small vessel fragility7. Missense variants comprised 23.5% (4/17), including p.N428S (n = 1), p.R419Q (n = 2), and p.S587C (n = 1). Among these, p.R419Q was classified as pathogenic and has been previously reported in PXE23, whereas p.N428S and p.S587C were categorized as variants of unknown significance and excluded from the primary analysis. Although they have not been reported in PXE and lack sufficient evidence of pathogenicity, these two variants were associated with PXE-related ocular manifestations and stroke9,24. Considering that such variants, while not classified as pathogenic due to limited evidence, may still have unrecognized pathogenic potential, they were included only in an exploratory heterozygous analysis. As a result, nine cases (5.7%) carrying pathogenic heterozygous variants in ABCC6 were included in the primary analysis. When three additional patients (1.9%) with biallelic pathogenic variants (two compound heterozygous and one homozygous) were included, the total number of pathogenic cases was 12 (7.6%), which were used in sensitivity analyses. Furthermore, when two carriers (1.3%) with variants of uncertain significance (p.N428S and p.S587C) were added, the total number of heterozygous cases became 11 (7.0%), and this set was analyzed in a supplementary sensitivity analysis. NOTCH3 pathogenic variants diagnosed as CADASIL are listed in Table S5.
The allele frequency of p.M848fs in our CSVD cohort (0.052) was significantly higher than that in the general Japanese population (0.014 in the 14KJPN dataset of 14,000 individuals, 28,000 alleles; P = 0.001) and in the amyotrophic lateral sclerosis cohort from our institute (0.010 in our dataset of 145 individuals, 290 alleles; P = 0.011), supporting its potential relevance.
Clinical characteristics of ABCC6-related CSVD
The median onset and diagnosis ages in the ABCC6-related CSVD group were 44 [interquartile range (IQR): 39–54.0] and 46 [IQR: 45.0–54.0] years, respectively, similar to those in the CADASIL and genetically undetermined groups (Table 2). Hemorrhagic stroke was significantly more common in the ABCC6-related CSVD group compared with the CADASIL (55.6% vs. 13.6%, P = 0.039) and genetically undetermined (55.6% vs. 10.3%, P = 0.010) groups. Hypertension prevalence did not differ significantly between the ABCC6‑related CSVD and CADASIL groups (66.7% vs 31.8%, P = 0.200) and was similar to that in the genetically undetermined group (66.7% vs 62.3%, P = 1.000). Multivariate logistic regression confirmed a significant association between ABCC6 pathogenic variants and hemorrhagic stroke, even after adjusting for age and hypertension (unadjusted: OR: 9.6, 95% CI: 2.3–42.8, P = 0.002; Model 1: OR: 8.4, 95% CI: 2.0–38.4, P = 0.004; Model 2: OR: 9.0, 95% CI: 2.1–41.2, P = 0.003; Table 3). When three additional biallelic cases (two compound heterozygous and one homozygous) were included in sensitivity analyses, the association with hemorrhagic stroke remained significant (Model 1: OR 6.7, 95% CI 1.8–25.0, P = 0.004; Model 2: OR 7.2, 95% CI 2.0–26.5, P = 0.003), and hypertension was significantly more prevalent in the ABCC6 group than in CADASIL (66.7% vs 31.8%; P = 0.045) (see Supplementary Tables S6 and S7 online). In an additional sensitivity analysis that included two heterozygous carriers with variants of uncertain significance (p.N428S and p.S587C), overall patterns were similar to the heterozygous‑only analysis, and the ABCC6–CADASIL difference in hypertension did not reach significance, consistent with the heterozygous‑only pattern (see Supplementary Tables S8 and S9 online). For detailed clinical histories, see the Supplemental Note.
Imaging features of ABCC6-related CSVD and CADASIL
The imaging features of ABCC6-related CSVD were distinct from those of CADASIL (Table 4). The ABCC6-related CSVD group had lower anterior temporal lesion scores (median [IQR]: 0 [0–0] vs. 2 [0–3], P = 0.004). The number of lacunar infarcts tended to be lower in the ABCC6 group but did not differ significantly between the groups (median [IQR]: 1 [0–8] vs 7.5 [2.8–13.3], P = 0.106). By contrast, a significantly higher cerebral hemorrhage rate was observed in the ABCC6-related CSVD group than in the CADASIL group (55.6% vs. 11.9%, P = 0.010), particularly in the infratentorial (22.2% vs. 0%, P = 0.028) and lobar (33.3% vs. 2.4%, P = 0.014) regions. The proportion of patients with multiple microbleeds was also higher in the ABCC6-related CSVD group (77.8% vs. 28.6%, P = 0.010). In the ABCC6-related CSVD group, microbleeds were frequently observed in the lobar region (median [IQR]: 8.5 [0.8–20] vs. 0.5 [0–4.8], P = 0.031), similarly to intracerebral hemorrhages. A trend toward increased microbleeds in the infratentorial regions was observed in the ABCC6-related CSVD group, although the difference was not statistically significant (median [IQR]: 2 [0.75–8.5] vs. 0 [0–1.8], P = 0.050) (Figs. 1 and 2).

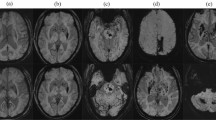
Brain magnetic resonance imaging (MRI) findings of the most severely affected patient in the cohort (Patient 7). T2*‑weighted images show multiple cerebral microbleeds in the infratentorial (a) and lobar (b) regions.

Imaging characteristics of hemorrhagic lesions in ABCC6-related cerebral small vessel diseases (CSVD) and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Violin plots showing the number of microbleeds (MBs) between two groups: (a) total, (b) lobar, (c) infratentorial, and (d) deep MBs. (e) A bar chart showing multiple MBs prevalence between two groups. Statistical analyses used the Wilcoxon rank-sum test (a–d) and Fisher’s exact test (e). P < 0.05 was considered significant.
When three biallelic pathogenic cases (two compound heterozygous and one homozygous) were included in the sensitivity analysis, the overall imaging pattern was consistent with the results presented in Table 4, but the difference in lacunar infarcts (median [IQR]: 1 [0–7.3] vs 7.5 [2.8–13.3], P = 0.021) and infratentorial microbleeds (median [IQR]: 2 [0.8–8.5] vs 0 [0–1.8], P = 0.031) became significant, while the difference in lobar microbleeds observed in the heterozygous analysis was no longer significant (median [IQR]: 8.5 [0.75–20] vs 0.5 [0–4.8], P = 0.082) (see Supplementary Table S10 online). The biallelic cases themselves showed a low lacune burden (0–3 in total). When two heterozygous carriers with variants of uncertain significance (p.N428S and p.S587C) were added in a supplementary sensitivity analysis, the results were similar to those of the heterozygous-only analysis: no significant difference in lacunes, whereas lobar microbleeds remained significantly higher and infratentorial microbleeds also reached significance (P = 0.019) in the ABCC6 group (see Supplementary Table S11 online).
After excluding outliers defined by the Tukey fence (Patient 7 in the ABCC6 group and four cases in the CADASIL group), the difference in multiple microbleeds (≥ 12) remained significant (6/8 vs 8/38, P = 0.006). The ABCC6-related CSVD group continued to show a higher total microbleed count (median [IQR]: 17 [9–32.8] vs 3.5 [1–11], P = 0.080) and significantly greater involvement in the infratentorial (median [IQR]: 2 [0–4.3] vs 0 [0–1], P = 0.043) and lobar regions (median [IQR]: 8.5 [1.5–15.5] vs 0 [0–2], P = 0.018) (Supplementary Table S12). These findings confirm that the hemorrhage-predominant pattern in ABCC6-related CSVD remained robust even after symmetrical outlier exclusion in both groups.
Discussion
We identified a hemorrhage-prone CSVD phenotype associated with ABCC6 pathogenic variants in a nationwide, multicenter Japanese cohort, with higher odds of hemorrhagic stroke after adjustment for age and hypertension. In the primary analysis of nine heterozygous carriers, this association was significant, and sensitivity analyses including biallelic pathogenic cases and heterozygous carriers with variants of uncertain significance confirmed the robustness of the findings. Imaging differences—fewer anterior temporal lesions and a greater burden of hemorrhagic markers—were evident in comparison with CADASIL, a monogenic CSVD with well-established imaging criteria that allowed detailed head-to-head assessment. The difference in lacunes was not significant in the heterozygous analysis but became significant when biallelic cases were included. Compared with prior case series, our analysis leveraged a nationwide, multicenter cohort drawn from 91 neurology centers across Japan, enhancing generalizability within this population.
The increased bleeding tendency observed in ABCC6-related CSVD likely mirrors PXE pathology; elastic fiber calcification and fragmentation can compromise blood vessel integrity, potentially increasing hemorrhagic event risk25,26. Similar pathological changes in PXE occur in gastric arteries27, suggesting analogous processes in cerebral small vessels. Furthermore, altered capillary morphology and extracellular matrix composition may further predispose patients with ABCC6-related CSVD to hemorrhage28,29.
Our imaging analysis identified characteristic ABCC6-related CSVD features, particularly hemorrhagic patterns. Patients with ABCC6 pathogenic variants showed significantly more lobar microbleeds and a borderline increase in infratentorial microbleeds (P = 0.050), alongside significantly more lobar and infratentorial ICH and minimal anterior temporal involvement compared with CADASIL. Importantly, hemorrhagic events are not uncommon in East Asian—including Japanese—CADASIL; a meta‑analysis reported higher ICH occurrence in Asian cohorts (17.7%) than in European cohorts (2.0%)30, and a nationwide Japanese survey found cerebral microbleeds in 47.7% of CADASIL patients31, supporting CADASIL as an appropriate comparator in this population. Moreover, our cohort included six patients with NOTCH3 p.R75P, a variant associated with hemorrhage‑prone CADASIL (Supplementary Table S5)32.
In terms of distribution, the characteristic lobar and infratentorial hemorrhagic pattern may be useful for differential diagnosis. Notably, since lobar hemorrhages are rare in COL4A1/2-associated CSVD, which also features hemorrhagic lesions33, the lobar and infratentorial hemorrhage pattern may suggest an ABCC6-related pathology.
Across sensitivity analyses, results were broadly consistent: the hemorrhage‑predominant imaging signature was preserved; only the inclusion of biallelic cases shifted the significance profile (lacunes significant; lobar microbleeds not significant), whereas adding carriers of variants of uncertain significance (p.N428S and p.S587C) reproduced the heterozygous‑only findings. After outlier handling in both groups, the difference in multiple microbleeds (≥ 12) remained significant (6/8 vs 8/38, P = 0.006); lobar microbleeds were also significantly higher in the ABCC6 group (P = 0.018), supporting robustness (Supplementary Table S12). In our cohort, p.M848fs predominated, consistent with Japanese PXE data22, whereas p.R1141X, the common European allele34, was not observed. This population‑specific spectrum raises the possibility of variant‑dependent effects (e.g., p.R1141X linked to ischemic coronary disease11) but our sample size precluded formal subgroup analyses; it may also help explain discrepancies with predominantly non‑Japanese reports that emphasized ischemic stroke35, whereas our Japanese cohort showed a hemorrhage‑prone phenotype. Regarding zygosity, most ABCC6-related CSVD cases in this study were heterozygous (11/14), with one homozygote and two compound heterozygotes. Given reports of subclinical/partial phenotypes in heterozygous carriers8,9, we cannot exclude heterozygote effects on the observed CSVD features; however, the clinical significance of monoallelic pathogenic variant remains uncertain and warrants further study in larger, multi-ethnic cohorts.
Our findings should be interpreted as risk associations rather than causal effects. Nonetheless, the association between ABCC6 pathogenic variants and hemorrhagic stroke remained significant after adjustment for age and hypertension, and was accompanied by convergent imaging differences relative to CADASIL, fewer anterior temporal lesions, with a greater burden of hemorrhagic markers, but not by differences in overall stroke occurrence or ischemic stroke. The ABCC6 group tended to have a higher prevalence of hypertension than the CADASIL group (66.7% vs 31.8%), and we did not adjust for dyslipidaemia or other metabolic mediators; experimental data suggest ABCC6 deficiency can promote dyslipidemia and atherosclerosis36, which could contribute to the hemorrhagic burden. In our cohort, however, dyslipidemia prevalence was comparable across groups (ABCC6 37.5%, CADASIL 40.9%, undetermined 40.3%), with no significant differences. Clinically, ABCC6 pathogenic variants may act as risk modifiers of hemorrhagic CSVD phenotypes, underscoring the need for comprehensive vascular risk assessment and validation in independent, multi-ethnic cohorts.
This study has limitations. First, the modest sample size and cross-sectional design limit robustness and preclude causal inference. Second, MRI acquisition used both T2* and SWI with minor inter-center protocol variability; despite central, standardized ratings, such differences may have influenced microbleed detection. Third, the ROC-derived threshold for “multiple microbleeds” (> 11) is exploratory and lacks external validation. Fourth, imaging comparisons were conducted only against CADASIL; head-to-head data versus other monogenic CSVDs were not available. Fifth, the NOTCH3 p.R75P variant, relatively common in East Asia and associated with hemorrhagic lesions32, was identified in only six patients (see Supplementary Table S5 online), precluding meaningful subgroup analyses by R75P status. Sixth, we did not assess APOE ε2/ε4, which limits formal exclusion of coexisting cerebral amyloid angiopathy (CAA); however, given the cohort’s relatively young age at onset and an infratentorial-predominant microbleed pattern without a significant excess of cortical superficial siderosis, major confounding by CAA appears unlikely. Seventh, lipid profiles and antithrombotic exposure were not incorporated into multivariable models; future studies should evaluate these potential mediators and confounders explicitly. Eighth, because exome sequencing can miss deep intronic or structural variants, some apparent heterozygotes could harbor undetected second hits in ABCC6 (as suggested by PXE‑related ocular findings in individual cases), which may bias zygosity‑specific estimates. Finally, because patient recruitment was based on referral for suspected hereditary CSVD, the study cohort may have been enriched for relatively severe or typical cases showing extensive and symmetrical white matter hyperintensity. Mild or atypical cases might have been underrepresented if they were not referred. We did not have systematic data on potentially eligible but non‑referred patients. However, as the referral criteria were standardized across participating centers and all referred patients who met these predefined criteria were included, the impact of referral processes on within‑cohort comparisons is unlikely to be large, although generalizability beyond severe CSVD may be limited. In addition, clinical judgment at referring sites may have introduced some selection effects that we could not quantify.
ABCC6 pathogenic variants are associated with a distinct CSVD phenotype in Japanese patients, potentially characterized by an increased hemorrhagic burden and hemorrhagic-dominant imaging features. These findings support a contributory role of ABCC6 in CSVD and underscore the need for validation across diverse populations.
Data availability
The corresponding author will provide the anonymized data related to this study upon reasonable request.
References
Wardlaw, J. M., Smith, C. & Dichgans, M. Small vessel disease: Mechanisms and clinical implications. Lancet Neurol. 18, 684–696 (2019).
Manini, A. & Pantoni, L. Genetic causes of cerebral small vessel diseases: A practical guide for neurologists. Neurology 100, 766–783 (2023).
Pescini, F. et al. The Cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) scale: A screening tool to select patients for NOTCH3 gene analysis. Stroke 43, 2871–2876 (2012).
Koizumi, T. et al. The CADASIL scale-J, A modified scale to prioritize access to genetic testing for Japanese CADASIL-suspected patients. J. Stroke Cerebrovasc. Dis. 28, 1431–1439 (2019).
Uemura, M. et al. High frequency of HTRA1 AND ABCC6 mutations in Japanese patients with adult-onset cerebral small vessel disease. J. Neurol. Neurosurg. Psychiatry. 94, 74–81 (2023).
Germain, D. P. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 12, 85 (2017).
Cutan, J., Hubbard, E. & Lebwohl, M. Vascular effects of pseudoxanthoma elasticum. Skin: J. Cutan Med. 5(453), 461 (2021).
Nollet, L. et al. Clinical and subclinical findings in heterozygous ABCC6 carriers: results from a Belgian cohort and clinical practice guidelines. J Med Genet. 59, 496–504 (2022).
Katagiri, S. et al. ABCC6 gene analysis in 20 Japanese patients with angioid streaks revealing four frequent and two novel variants and pseudodominant inheritance. J. Ophthalmol. 2017, 1079687 (2017).
Park, H. K. et al. Prevalence of mutations in Mendelian stroke genes in early-onset stroke patients. Ann. Neurol. 93, 768–782 (2023).
Trip, M. D. et al. Frequent mutation in the ABCC6 gene (R1141X) is associated with a strong increase in the prevalence of coronary artery disease. Circulation 106, 773–775 (2002).
Lebwohl, L. & Phelps, R. Association between pseudoxanthoma elasticum and bleeding. Skin J. Cutan. Med. 6, 8–13 (2022).
Wardlaw, J. M. et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 12, 822–838 (2013).
O’Sullivan, M. et al. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology 56, 628–634 (2001).
Ihara, M., Okamoto, Y. & Takahashi, R. Suitability of the Montreal cognitive assessment versus the mini-mental state examination in detecting vascular cognitive impairment. J. Stroke Cerebrovasc. Dis. 22, 737–741 (2013).
Klambauer, G. et al. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 40, e69 (2012).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17, 405–424 (2015).
van Swieten, J. C., Koudstaal, P. J., Visser, M. C., Schouten, H. J. & van Gijn, J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke 19, 604–607 (1988).
Mahoney, F. I. & Barthel, D. W. Functional evaluation: The Barthel index. Md. State Med. J. 14, 61–65 (1965).
Tomimoto, H. et al. Small artery dementia in Japan: radiological differences between CADASIL, leukoaraiosis and Binswanger’s disease. Dement Geriatr. Cogn. Disord. 21, 162–169 (2006).
Duering, M. et al. Neuroimaging standards for research into small vessel disease advances since 2013. Lancet Neurol. 22, 602–618 (2023).
Hilal, S. et al. Prevalence, risk factors and consequences of cerebral small vessel diseases: Data from three Asian countries. J. Neurol. Neurosurg. Psychiatry. 88, 669–674 (2017).
Iwanaga, A. et al. Analysis of clinical symptoms and ABCC6 mutations in 76 Japanese patients with pseudoxanthoma elasticum. J Dermatol. 44, 644–650 (2017).
Nomura, E. et al. A case of a heterozygous ABCC6 mutation showing recurrent ischemic strokes and intracranial hemorrhages. Neurol. Clin. Neurosci. 10, 98–101 (2022).
Kranenburg, G. et al. Prevalence and severity of arterial calcifications in pseudoxanthoma elasticum (PXE) compared to hospital controls. Novel insights into the vascular phenotype of PXE. Atherosclerosis 256, 7–14 (2017).
Favre, G. et al. The ABCC6 transporter: A new player in biomineralization. Int. J. Mol. Sci. 18, 1941 (2017).
Kundrotas, L., Novak, J., Kremzier, J., Meenaghan, M. & Hassett, J. Gastric bleeding in pseudoxanthoma elasticum. Am. J. Gastroenterol. 83, 868–872 (1988).
Stumpf, M. J. et al. Pseudoxanthoma elasticum – also a microvascular disease. Vasa. 49, 57–62 (2020).
Plümers, R., Lindenkamp, C., Osterhage, M. R., Knabbe, C. & Hendig, D. Matrix metalloproteinases contribute to the calcification phenotype in pseudoxanthoma elasticum. Biomolecules 13, 672 (2023).
Lai, Q.-L. et al. Occurrence of intracranial hemorrhage and associated risk factors in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: A systematic review and meta-analysis. J. Clin. Neurol. 18, 499–506 (2022).
Shindo, A. et al. A nationwide survey and multicenter registry-based database of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy in Japan. Front Aging Neurosci. 12, 216 (2020).
Ishiyama, H. et al. Pro-hemorrhagic cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy associated with NOTCH3 p. R75P mutation with low vascular NOTCH3 aggregation property. Ann. Neurol. 95, 1040–1054 (2024).
Lanfranconi, S. & Markus, H. S. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: A systematic review. Stroke 41, e513–e518 (2010).
Saeidian, A. H. et al. Genetic heterogeneity of heritable ectopic mineralization disorders in a large international cohort. Genet. Med. 24, 75–86 (2022).
De Vilder, E. Y. G. et al. Pathogenic variants in the ABCC6 gene are associated with an increased risk for ischemic stroke: Pathogenic ABCC6 Variants Increase Risk for Stroke. Brain Pathol. 28, 822–831 (2018).
Brampton, C. et al. ABCC6 deficiency promotes dyslipidemia and atherosclerosis. Sci. Rep. 11, 3881 (2021).
Acknowledgements
The authors appreciate the cooperation of Dr. Tomoko Sakai from Komatsu Municipal Hospital and her contribution to this study.
Funding
The study was funded by a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control; 26117006) from MEXT, Grant-in-Aid for a Practical Research Project for Rare/Intractable Diseases (19ek0109236h0003) from AMED, Grant-in-Aid for Scientific Research (A) (19H01043), Grant-in-Aid for Medical Research from the Takeda Science Foundation, and Grant-in-Aid for Research on Intractable Disease (21FC0201), (24FC1011) from the Japanese Ministry of Health, Labour and Welfare, Japan.
Author information
Authors and Affiliations
Contributions
Conceptualization: Sho Kitahara, Masahiro Uemura, and Osamu Onodera. Data curation: Sho Kitahara, Shoichiro Ando, Masahiro Uemura, and Osamu Onodera. Patient data acquisition: Hiroaki Nozaki, Daisuke Hirozawa, Emi Nomura, Kohei Okubo, Asami Munekane, Takumi Tashiro, Kanako Asai, Minako Yamaoka, Tomone Taneda, Yutaka Honma, Hajime Kondo, Ryo Itabashi, and Hitoshi Aizawa. Formal analysis: Sho Kitahara, Shoichiro Ando, Masahiro Uemura, and Osamu Onodera. Genetic testing: Hiroaki Nozaki, Akira Iwanaga, Hiroyuki Murota, and Tomohiko Ishihara. Funding acquisition: Osamu Onodera. Investigation: Sho Kitahara, Shoichiro Ando, and Masahiro Uemura. Methodology: Sho Kitahara, Shoichiro Ando, and Masahiro Uemura. Resources: Osamu Onodera. Software: Sho Kitahara, Masahiro Uemura, and Yutaka Honma. Supervision: Osamu Onodera. Writing—original draft preparation: Sho Kitahara. Writing—review & editing: Sho Kitahara, Shoichiro Ando, Masahiro Uemura, Hiroaki Nozaki, Akira Iwanaga, Hiroyuki Murota, and Osamu Onodera.
Corresponding author
Ethics declarations
Competing interests
OO received speaker honorarium from Kyowa Hakko Kirin, Bristol-Myers Squibb, Ono Pharmaceutical, Mitsubishi Tanabe Pharma, Takeda, Daiichi-Sankyo, FUJIFILM, SANOFI, and FP-pharm. All other authors have no conflicts of interests to declare.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kitahara, S., Ando, S., Uemura, M. et al. ABCC6 pathogenic variants are associated with hemorrhagic phenotypes in Japanese patients with severe cerebral small vessel disease. Sci Rep 16, 3858 (2026). https://doi.org/10.1038/s41598-025-33934-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-33934-3
