
Abstract
To explore the regulatory effect of c-Jun N-terminal kinase (JNK) inhibitor (SP600125) on forkhead box protein L2 (FOXL2) gene in human ovarian granulosa cell tumor cells (KGN cells). The main pathogenic gene FOXL2 of ovarian cancer was screened by bioinformatics method. KGN cells were randomly divided into control group and experimental group. Different concentrations of SP600125 (0.1, 1, 5, 10, 50 µM) were added to the experimental group, and an equal volume of dimethyl sulfoxide (DMSO) was added to the control group. The cells were incubated for 48 h. Cell RNA was extracted and reverse transcribed into cDNA. The mRNA expression level of FOXL2 was detected by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Proteins were extracted, and the expression level of FOXL2 protein was detected by Western blot. The proliferation ability of KGN cells treated with SP600125 was detected by MTT assay. Cell scratch assay was used to detect its migration ability. Different concentrations of JNK inhibitor reduced the expression of FOXL2 in ovarian granulosa cells KGN, and 1 µM had the best inhibitory effect. JNK inhibitor reduces the expression of FOXL2 in ovarian granulosa cell tumor KGN.
Similar content being viewed by others

Introduction
Ovarian granulosa cell tumors are related to the increase in ovarian mass, such as abdominal pain and bloating, and often present with endocrine manifestations of estrogen excess, such as increased menstruation or postmenopausal vaginal bleeding. Some patients may also have shown endometrial hyperplasia or malignancy1,2.
Forkhead box protein L2 (FOXL2) is involved in biological processes such as ovarian differentiation, cell apoptosis, stress response, and cell cycle regulation. FOXL2 is associated with sex determination, premature ovarian failure, infertility, and other related conditions3. The human FOXL2 gene is approximately 2.7 Kb long, located in the 3q2.02.3 region of the long arm of chromosome 3, encoding a forkhead transcription factor consisting of 376 amino acids. The amino acids at positions 52–142 form a transmembrane structural domain protein4,5,6. The FOXL2 gene is also a gene carried on human autosomes that has been confirmed to be related to ovarian maintenance and premature ovarian failure. FOXL2 gene mutations can be detected in a considerable number of patients with premature ovarian failure. Previous studies by our research group found that estrogen can regulate FOXL2 through the estrogenG protein-coupled receptor 30(GPR30) pathway, thereby promoting apoptosis of ovarian cancer cells7. As research on FOXL2 continues to deepen, researchers have found that this gene is involved in multiple physiological processes. For example, FOXL2 plays an important role in craniofacial development and is involved in almost all stages of ovarian development. As a transcription factor, it plays an important role in the network regulation of the entire organism.
Previous studies by the authors have suggested that estrogen regulates FOXL2 to promote apoptosis in human ovarian granulosa cell tumors8,9. Mutations in the FOXL2 gene have been found in 95–97% of adult ovarian granulosa cell tumor patients, making FOXL2 gene mutation an important focus of research in ovarian granulosa cell tumors10,11,12.
c-Jun N-terminal kinase (JNK) is a crucial member of the mitogen-activated protein kinase (MAPKs) family and serves as a major signaling pathway that induces cell apoptosis in response to cellular stress13. It plays a significant role in the process of apoptosis when cells are subjected to stressful stimuli14. The JNK signaling pathway is divided into two parts: the classical pathway and the non-classical pathway. Both pathways activate MAP kinases through kinase phosphorylation cascades. Kinases MKK4 (MAP kinase kinase 4, a JNK kinase) and Hep (Hemipterous, MAPKK7, a JNK kinase) lead to the activation of JNK, a key protein kinase that regulates many physiological factors, including cell differentiation, apoptosis, stress response, and the occurrence and development of various human diseases. JNK plays a crucial role in these processes, making the JNK signaling pathway a critical regulatory target in normal and diseased cellular states15,16,17,18.
To date, research has mainly focused on FOXL2 as a transcription factor regulating its downstream target genes, while relatively ignoring its own transcription and translation regulation19,20,21,22, which is a limitation in FOXL2 research. Therefore, this study conducts research on the regulatory mechanisms of transcription and translation of this gene. By studying the inhibitory effect of JNK inhibitor on FOXL2, FOXL2 is incorporated into the overall network of human regulation to improve its connecting function. Thus, it provides new ideas for the prevention and treatment of clinically related diseases.
Materials and methods
Materials
The KGN granulosa cells were obtained from the Stem Cell Bank, Chinese Academy of Sciences; fetal bovine serum (FBS) and 1640 culture medium were purchased from Gibco; MEM and Trizol were obtained from Shanghai Biotech Co., Ltd.; pancreatin from BBI; rabbit anti-human FOXL2 antibody from Beijing Zhongshan Gold Bridge Biotechnology Co., Ltd.; DAB chromogenic reagent kit from Beijing Zhongshan Gold Bridge Biotechnology Co., Ltd.; RT-PCR kit and related experimental reagents from Takara, Japan; DNA Marker from TIANGEN; RIPA lysis buffer from Pulilai Gene Technology Co., Ltd. JNK inhibitor (SP600125) from abcam.
Data analysis
In this study, we manually downloaded the matrix and platform files of dataset GSE34526. Using the GEOquery package in R version 4.3.2, we retrieved the expression matrix and clinical information. The matrix data was then subjected to standardization, and probe IDs were converted to gene names based on the platform file. For genes with the same name in the expression matrix, the average expression level was calculated. Batch effects were removed using the removeBatch function in the limma package. Differential expression genes between ovarian cancer and normal ovarian tissues in the training set were identified using the limma package. Selection criteria of |logFC|>1 and adj.P.Value < 0.05 were applied. The pheatmap package was used to construct a hierarchical clustering heatmap of the top 100 differentially expressed genes, while the ggplot2 package was utilized to create a volcano plot of differentially expressed genes, with annotations for the top 20 differentially expressed gene names. Enrichment analysis of differentially expressed genes was conducted using the clusterProfiler package, including Gene Ontology analysis of biological processes, cellular components, and molecular functions. Visualization of the enrichment results was performed using the ggplot2 package.
Cells culture
KGN cells (Innovatbio, Beijing, China) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% FBS and 1% penicillin-streptomycin solution, maintained at 37 °C in a 5% CO2 humidified cell culture incubator. When the cell confluence reached approximately 80%, cells were passaged using 0.05% Trypsin-EDTA and further utilized for experimental procedures.
The detection of FOXL2 mRNA
The cells were randomly divided into control and experimental groups. KGN cells were seeded in a 6-well cell culture plate and allowed to grow to a density of 70-80%. Different concentrations of JNK inhibitor (0.1, 1, 5, 10, 50µM) were applied to the KGN cells for 48 h, with dimethyl sulfoxide (DMSO) used as the solvent control. RNA extraction was performed using Trizol (Solarbio, Beijing, China) from both total cells and ovarian tissue. RNA concentrations were measured by absorbance at 260 nm using a micro-spectrophotometer (ThermoScientific, USA). RNA quality was assessed by electrophoresis in a 1% agarose gel. The cDNA was synthesized using a Fast Super RT Kit cDNA (with gDNase) (B002004018, Bioteke, Beijing, China). Primer pairs were designed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward primer: 5′-AGCCAAAAGGGTCATCATCTCT-3′, reverse primer: 5′-AGGGGCCATCCACAGTCTT-3′) and FOXL2 (forward primer: 5′-TCACGCTGTCCGGCATCTACCA-3′, reverse primer: 5′-GCGGCACCTTGATGAAGCACTC-3′) using Primer version 5.0 software. Taq DNA Polymerase (9618080604, ABclonal, Wuhan, China) was used for polymerase chain reaction (PCR) amplification. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using the SYBR Green Real-time PCR Master Mix in a real-time PCR system (TOYOBO, Japan) following the manufacturer’s protocol. The qRT-PCR parameters were as follows: 94 °C for 3 min, followed by 40 cycles of 94 °C for 30 s, 60 °C for 30 s, and a final extension at 72 °C for 5 min. Data analysis was conducted using the formula: R = 2^−[ΔCt sample − ΔCt control], where R represents the relative expression level, ΔCt represents the difference between the Ct of the gene and the average GAPDH in the experimental sample, and ΔCt control represents the difference between the Ct of the gene and the average GAPDH in the control sample. Experiments were carried out in triplicate with independent experimental samples.
Protein extraction and Western blotting
The KGN cells and tissue utilized for protein extraction underwent grinding in radio-immunoprecipitation assay (RIPA) buffer (Solarbio, Beijing, China) supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF) and phosphatase inhibitors, symbolizing a meticulous approach to sample preparation. Following centrifugation at 10,000 g at 4 ℃ for 10 min, the protein-enriched supernatant was meticulously collected. The protein concentrations were determined using the Bradford method23, showcasing a commitment to precise quantification. Subsequently, protein samples (30 µg) were meticulously subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and precisely electro-transferred onto a 0.22 µM nitrocellulose membrane, illustrating a meticulous technique of protein separation and transfer. The resulting membranes were diligently blocked with a blocking buffer (2% BSA in TBS buffer: 10 mM Tris-HCl, 150 mM NaCl, pH = 7.5) for 1 h at room temperature, demonstrating a thorough experimental procedure. The membrane was then meticulously incubated with the antiserum anti-FOXL2 against human (Abcam, ab5096, USA) (1:10,000 dilution in blocking buffer) overnight at 4 °C, underscoring the meticulous attention to detail in antibody incubation. After being washed twice with TBST buffer (10 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20, pH = 7.5) for 10 min each time, the membrane underwent further incubation with an alkaline phosphatase-conjugated goat anti-rabbit IgG secondary antibody (ABclonal, AS009, Wuhan, China) (1:1000 dilution in blocking buffer) at room temperature for 3 h, emphasizing a methodical secondary antibody incubation process. Following a thorough washing with TBS buffer three times for 10 min, the target protein signal was meticulously visualized using 45 µL of nitroblue tetrazolium (NBT) (Sigma, USA) and 35 µL of 5-bromo-4-chloro-3-indolyl phosphate (BCIP) (Sigma, USA) incubated in 10 mL of TBS in the dark for 10 min, highlighting a precise approach.
MTT assay for measuring cell proliferation
KGN cells in exponential growth were inoculated into three 96-well plates at 3 × 103 cells/well respectively, and the cells were treated with DMSO and JNK inhibitor (1 µM). After being cultured in a constant temperature incubator for 24 h, 48 h and 72 h respectively. Record the optical Density (OD) value of each well at 490 nm on a microplate reader. Taking the culture time as the abscissa and the OD value as the ordinate, draw the cell proliferation curve and calculate the cell proliferation rate. Cell proliferation rate = OD value of group A and group B in the experimental group/OD value of group C. The experiment was repeated three times.
Statistical methods
SPSS statistical software was used for analysis. Data are expressed as mean ± standard deviation. One-way analysis of variance was used for comparison between groups. Student–Newman–Keuls (SNK) test was selected for pairwise comparison. P < 0.05 was considered statistically significant.
Results
Differential gene expression screening
The data set GSE14407 was selected from the GEO database for analysis, based on the criteria of corrected P-value < 0.05 and |log2 FC(fold change)|≥1, which were utilized to screen differentially expressed genes. According to the screening criteria, differential analysis was performed on the gene expression levels in ovarian cancer and normal ovarian samples, resulting in a total of 5145 differentially expressed genes, including 4,027 upregulated and 1,118 downregulated genes. Hierarchical clustering and volcano plots of differentially expressed genes were generated (Fig. 1). The research shows that FOXL2 is one of the down-regulated genes of ovarian cancer, which is consistent with the author’s previous research.
Upon querying the NCBI database, the gene sequence analysis of FOXL2 revealed that the FOXL2 gene is approximately 2.7 Kb in length, located in the 3q22.3 region of the long arm of chromosome 3 (Fig. 2A). It encodes a forkhead transcription factor consisting of 376 amino acids, with amino acids 52–142 forming a transmembrane structural domain protein (Fig. 2B).

Screening of differentially expressed genes in ovarian cancer. Dataset GSE34526 from gene expression omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). Using the GEO query package in R version 4.3.2.

The gene sequence and protein structural domains of FOXL2.
cDNA PCR and gel electrophoresis results
Total RNA of KGN cells was successfully extracted. The OD value of each group of cells was measured by an ultraviolet spectrophotometer. The OD value of KGN cells was all between 1.8 and 2.0, indicating good RNA purity. The primer design GAPDH fragment length is 230 bp, and the FOXL2 fragment length is 260 bp. By comparing the marker bands, the gel imaging results clearly show a band corresponding to GAPDH mRNA (1–6) and a band corresponding to FOXL2 mRNA (7–12), proving that the expression of GAPDH and FOXL2 genes was detected in both the control group and the five different concentration experimental groups (Fig. 3), fully proving the accuracy of the primers and the feasibility of the experiment.

Results of total mRNA 1% agarose gel electrophoresis in twelve groups of cells. 1–6 and 7–12 represented GAPDH and FOXL2 in DMSO, co-clutured with 100 nM, 1, 5, 10, 50 μM JNK inhibitor group respectively. Number 13 refers to the DNA marker (TIANGEN).
Detection of the FOXL2 gene via Real-time fluorescence quantitative PCR
The fluorescence quantitative PCR amplification curves of the reference gene and target gene were satisfactory, with single-peaked melting curves indicating the absence of primer dimers. The relative mRNA expression levels of each group were calculated using the 2−ΔΔCt method. The expression of FOXL2 mRNA in the JNK inhibitor group was lower than that in the reference group, with the lowest mRNA expression level of the FOXL2 gene observed at a final concentration of 1µM in the experimental group. This difference was statistically significant (P < 0.05, Fig. 4). These findings suggest that, on the one hand, the FOXL2 gene is regulated by JNK and may be inhibited in its expression by a certain mechanism.

After treatment with the JNK inhibitor, the mRNA expression levels of FOLX2 were detected in KGN cells by qRT-PCR. (*p < 0.05; **p < 0.01).
The detection of FOXL2 by Western blot
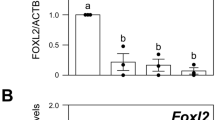
After treating the cells with JNK inhibitor at a concentration of 1 µM, proteins were extracted from the normal control group, experimental group (JNK inhibitor group) after 48 h and subjected to electrophoresis. The primary antibodies were GAPDH antibody and FOXL2 antibody. The expression of FOXL2 protein in the JNK inhibitor group was significantly reduced. The expression of FOXL2 protein in the experimental group cells decreased by 40% compared with the control group, and the differences were statistically significant (P < 0.05, Fig. 5).

Protein expression levels of FOXL2 after treatment with JNK inhibitor.
Cell scratch assay
The two-dimensional migration experiment was conducted by treating cells with DMSO and JNK inhibitor (1 µM) for 48 h. Scratches were made on the culture dish, and the average migration distance of cells was evaluated after 48 h. This assay can reflect the migratory characteristics of cells by observing the ability of cells to migrate into the scratched area after creating scratches on the cell culture surface, and it is widely used in the study of cancer cell invasion and migration.
The migration ability of KGN cells may be related to the expression level of the FOXL2 gene. Our research shows that the JNK inhibitor SP600125 can significantly reduce the expression of FOXL2 (as demonstrated by the qRT-PCR and Western blot experiments in the results section of this article).
The migration ability of the JNK inhibitor group was weaker than that of the control group. JNK partially inhibits cell migration through FOXL2 (Fig. 6).

Cell scratch assay.
The measuring cell proliferation ability by the MTT assay
The proliferation ability of KGN cells was measured at different time points of 24 h, 48 h, and 72 h using optical microscopy and MTT assay. Through microscopic observation, the three groups of cells showed rapid proliferation at 24 h, reaching approximately 80% coverage at 48 h, with no significant differences observed among the groups at 72 h under optical microscopy. The absorbance values measured at 24 h were 0.3 ± 0.05 for the control group and 0.31 ± 0.08 for the experimental group, with P = 0.02; at 48 h, the absorbance values were 0.70 ± 0.01 and 0.65 ± 0.05, with P = 0.03; at 72 h, the absorbance values were 1.01 ± 0.04 and 0.75 ± 0.05, with P = 0.03. The above quantitative data underwent analysis of variance with multiple independent samples, showing a statistically significant difference with P < 0.05. The proliferation curves of KGN cells at different time points indicated that the values of the experimental group were lower than those of the control group, suggesting a lower proliferation capacity compared to the control group. MTT results showed that JNK inhibitor treatment inhibited the proliferation of KGN cells after 72 h (Fig. 7).

The proliferation of KGN at different time periods.
Discussion
This study has found, through bioinformatics analysis and literature review, that FOXL2 may be regulated by the JNK signaling pathway24. The experimental results of this study also indicate that JNK regulates FOXL2, thereby affecting the expression of KGN.
JNK is one of the important members of the MAPKs family and is the main signal transduction pathway for cell apoptosis induced by cellular stress responses. It plays an important role in the process of apoptosis of cells when they are subjected to stressful stimuli14,25. This study found that JNK inhibitor can significantly inhibit the expression of FOXL2 gene in ovarian granulosa cells. We speculate that the FOXL2 gene may mainly regulate ovarian granulosa cells through the JNK pathway. In this study, PCR detection of primers for GAPDH and FOXL2 was first performed, and positive results were found in both, ensuring the feasibility of the experiment. In qRT-PCR, it was found that the inhibitory effect on the FOXL2 gene was the strongest when the concentration of JNK inhibitor was 1 µM, and the inhibitory effect weakened at 5 µM and 10 µM, and increased again at 50 µM. Considering that the concentration at 50 µM is too high, it is caused by a large cytotoxic effect. The inhibitor with a concentration of 1 µM was selected for incubation, and Western blot, cell scratch assay and MTT assay were performed to detect proliferation, which again verified the inhibitory effect on the FOXL2 gene when the concentration of JNK inhibitor was 1 µM.
In summary, the FOXL2 gene is highly expressed in ovarian granulosa cells. FOXL2 can inhibit the proliferation of ovarian granulosa cells through JNK. It may serve as a novel tumor suppressor, holding potential value in the metastasis and recurrence of ovarian cancer. However, further research is required to deepen our understanding of its mechanisms.
Data availability
GSE34526 can be accessed from GEO database. The data supporting the findings of this study are available from the corresponding authors upon reasonable request.
References
Barcellini, A. et al. Granulosa cell tumors (GCTs) of the ovary: what is the role of radiotherapy? Crit. Rev. Oncol. Hematol. 181, 103889 (2023).
Mangili, G. et al. Recurrent granulosa cell tumors (GCTs) of the ovary: a MITO-9 retrospective study. Gynecol. Oncol. 130, 38–42 (2013).
Fryns, J. P., Strømme, P. & van den Berghe, H. Further evidence for the location of the blepharophimosis syndrome (BPES) at 3q22.3-q23. Clin. Genet. 44, 149–151 (1993).
Herman, L. et al. A cellular model provides insights into the pathogenicity of the oncogenic FOXL2 somatic variant p.Cys134Trp. Br. J. Cancer (2024).
Nagy, A., Niu, N., Ratner, E., Hui, P. & Buza, N. Novel FOXL2 mutation in an ovarian adult granulosa cell tumor: report of a case with diagnostic and clinicopathologic implications. Int. J. Gynecol. Pathol. (2024).
Llano, E. et al. The oncogenic FOXL2 C134W mutation is a key driver of granulosa cell tumors. Cancer Res. 83, 239–250 (2023).
Fryns, J. P., Strømme, P., van den Fau -, H. & van den Berghe, H. Further evidence for the location of the blepharophimosis syndrome (BPES) at 3q22.3-q23.
Wu, J. et al. Estrogen regulates forkhead transcription factor 2 to promote apoptosis of human ovarian granulosa-like tumor cells. J. Steroid Biochem. Mol. Biol. 194, 105418 (2019).
Rosario, R., Cohen, P. A. & Shelling, A. N. The role of FOXL2 in the pathogenesis of adult ovarian granulosa cell tumours. Gynecol. Oncol. 133, 382–387 (2014).
Guerrieri, C., Hudacko, R. & Anderson, P. Composite FOXL2 mutation-positive adult granulosa cell tumor and serous borderline tumor of the ovary. Int. J. Gynecol. Pathol. 42, 500–507 (2023).
Singh, N. et al. Adult granulosa cell tumour-like areas occurring in ovarian epithelial neoplasms: report of a case series with investigation of FOXL2 mutation status. Histopathology 64, 626–632 (2014).
Yanagida, S. et al. Clinical and genetic analysis of recurrent adult-type granulosa cell tumor of the ovary: persistent preservation of heterozygous c.402C > G FOXL2 mutation. PLoS One 12, e0178989 (2017).
Bildik, G. et al. Endogenous c-Jun N-terminal kinase (JNK) activity marks the boundary between normal and malignant granulosa cells. Cell. Death Dis. 9, 421 (2018).
Wu, Q. et al. JNK signaling in cancer cell survival. Med. Res. Rev. 39(6), 2082–2104 (2019).
Abdelrahman, K. S. et al. JNK signaling as a target for anticancer therapy. Pharmacol. Rep. 73, 405–434 (2021).
Garg, R. et al. JNK signaling pathway in metabolic disorders: an emerging therapeutic target. Eur. J. Pharmacol. 901, 174079 (2021).
Kumar, A. et al. JNK pathway signaling: a novel and smarter therapeutic targets for various biological diseases. Future Med. Chem. 7, 2065–2086 (2015).
Shah, S. P. et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl. J. Med. 360, 2719–2729 (2009).
Tucker, E. J. The genetics and biology of FOXL2. Sex. Dev. 16, 184–193 (2022).
Fuller, P. J., Nguyen, T., Alexiadis, M. & Chu, S. FOXL2(C134W): much ado about something!(†). J. Pathol. 256, 1–3 (2022).
Caburet, S. et al. The transcription factor FOXL2: at the crossroads of ovarian physiology and pathology. Mol. Cell. Endocrinol. 356, 55–64 (2012).
Köbel, M., Gilks, C. B. & Huntsman, D. G. Adult-type granulosa cell tumors and FOXL2 mutation. Cancer Res. 69, 9160–9162 (2009).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
Tang, G. et al. Inhibition of JNK activation through NF-kappaB target genes. Nature 414(6861), 313–317 (2001).
Lee, M. H. et al. JNK regulates binding of alpha-catenin to adherens junctions and cell-cell adhesion. FASEB J. 25(2), 613–623 (2011).
Funding
This work was supported by Linyi Natural Science Foundation Grant Number 2022YX0053.
Author information
Authors and Affiliations
Contributions
Nan Bai and Jun Wu: conceived and supervised this projectYuzhu Zhang: conducted cell experiments and performed the data extraction Yu Wang: analysis procedures and wrote the original manuscriptYuan Gu: helped with the data analysis and manuscript writing procedures Yang Liu and Guohua Liu: helped the data analysis procedures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Wang, Y., Gu, Y. et al. Regulation of FOXL2 gene in ovarian granulosa cell tumor by JNK inhibitor. Sci Rep 15, 12167 (2025). https://doi.org/10.1038/s41598-025-85439-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85439-8

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}