
Abstract
Due to the diverse climate in Iran, there has been an opportunity for the emergence of different domestic sheep breeds. On the other hand, two of the seven wild sheep species have settled in the broad areas of Iran. This study investigated the introgression between wild and domestic Iranian sheep using the whole genome sequencing data for 55 domestic and 19 wild sheep. The study reveals introgression signals from Asiatic mouflon and urial in Iranian domestic breeds. The results of Treemix unveiled the introgression from the common ancestor of Iranian domestic sheep to Asiatic mouflons, which sheds light on the dynamics of gene flow between domestic and wild sheep in Iran. There were 36 genomic regions (containing 26 protein coding genes) introgressed from both Asiatic mouflon and urial into the genome of their domestic relatives. In the genomic windows containing 19 of the mentioned genes, we observed a noticeable drop in the dxy value of the urial vs. domestic and Asiatic mouflon vs. domestic sheep. Also, a decrease in nucleotide diversity was observed in these regions for wild and domestic sheep, suggesting the occurrence of a selective sweep in the mentioned genomic regions. The study found important genes in these regions that are involved in fertility (TTC29, STPG2), spatial memory and learning (CAMK1G), taste perception (OTOP1), visual system (DYRK2) and olfactory (LOC101110674). The results demonstrate that most genes affected by introgression between wild and domestic sheep are related to survival, reproduction and adaptation. This information can enhance our understanding of the impact of wild species on domesticated livestocks, providing us with clearer insights for management, breeding, and conservation of these species.
Similar content being viewed by others

Introduction
Iran has a highly diverse climate, including cold areas in the northwest, hot and desert areas in the center and east (Fig. 1a), Hyrcanian forests with high precipitation in the north (Fig. 1b), coastal areas in the south, and mountainous areas in the west (Zagros mountains) and north (Alborz mountains). The aforementioned climatic conditions and the different goals and production purpose of sheep breeders have led to the emergence of many domestic sheep breeds in Iran. There are at least 27 indigenous breeds in Iran1. Moghani (MOG), Ghezel (GEZ), Makui (MAK), Afshari (AFS), Shal (SHA), Grey-Shiraz (GRE), Karakul (KAR), Baluchi (BAL), Kermani (KER) and Zel (ZEL) are among the most important native sheep breeds of Iran. Also, the western and eastern half of Iran (with a geographically overlapping area) are part of the distribution areas of two species of wild sheep, namely Asiatic mouflon (A mouflon, Ovis orientalis) and Urial (Ovis vignei). The geographical overlap of the habitat of wild sheep and their domestic relatives is one factor that provides the chance of introgression between them. The study of introgression as a source of providing effective alleles to improve the adaptation abilities of domestic sheep has been one of the areas of attention for researchers in recent years2,3,4. The availability of a suitable number of whole genome sequencing (WGS) data for Iranian domestic sheep gave us this chance to study the genomic introgression between two wild sheep species and different Iranian domestic sheep breeds with more focus. In this study, the population structure of Iranian domestic and wild sheep was investigated usisng whole genome sequencing data. We also identified gene flow between the two mentioned groups and pinpointed specific genomic regions that have been introgressed from wild sheep into their domestic relatives. Furthermore, we discuss the genes located within these introgressed regions and their potential impacts on the adaptation of Iranian domestic sheep.

Temperature (a) and precipitation (b) map of Iran (2000–2023).
Materials and methods
Data collection and quality filtration
For this study, genomic data for Asiatic mouflon (N = 17), Urial (N = 11), 10 Iranian domestic sheep breeds, including Afshari (N = 5), Moghani (N = 3), Makui (N = 3), Ghezel (N = 3), Shal (N = 3), Grey-Shiraz (N = 3), Karakul (N = 7), Baluchi (N = 3), Kermani (N = 3) and Zel (N = 4), along with 20 other domestic Iranian sheep (samples were taken from the northwest of Iran without registering their breed) and one goat sample (accession number: SRR17775436; breed: Iraq; region: Iraq) as outgroup were obtained from NCBI database.
The quality of downloaded data was assessed using FastQC v.0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The sliding windows (5:20) approach from the Trimmomatic v.0.39 program5 was used for quality filtration. Other options used for the filtration of the genomic reads were LEADING:5, TRAILING:5 and MINLEN:40. Also, the HEADCROP and ILLUMINACLIP options were applied to some samples that needed to remove base content noises and adapter contamination, respectively.
Genomic variant calling
Clean reads were mapped to sheep (ovis aries) reference genome (GCF_016772045.1) using the mem approach of BWA v.0.7.176. Sorting in coordinate order and removing duplicated reads was performed by Picard (http://broadinstitute.github.io/picard). The RealignerTargetCreator and IndelRealigner approaches from Genome Analysis Toolkit (GATK) v.3.77 program was performed for local realignment around INDELs. We used GATK for SNPs calling along with BCFtools and VCFtools8 for quality filtration of identified SNPs as described by Khalkhali-Evrigh et al.9, except that in the current study -max-missing and –hwe were 0.95 and 0.001, respectively. It should be noted that to prevent the presence of related individuals in the analyses, before any other step, pairwise kinship between all samples was estimated using –kinship flag in KING v.2.2.710. After identifying related individuals and removing one of the paired individuals with high kinship, SNP detection and filtration were repeated using unrelated samples.
Population structure
Before principal component analysis (PCA), detected SNPs were pruned by -indep-pairwise 50 10 0.2 option (SNP windows size:50, SNP step size:10 and r2 threshold: 0.2) in the Plink1.911. In fact, pairs of SNPs within 50-SNP sliding windows with a 10-SNP step size that show r2 greater than 0.2, are identified as linked SNPs and subsequently pruned. The r2 is one of the most commonly used statistics to measure the degree of linkage disequilibrium (LD) between two SNPs. The neighbor-joining (NJ) tree was constructed by Plink1.9 and visualized using FigTree v.1.4.4. Analysis of coancestry relationship among samples was done using ADMIXTURE v.1.312 based on pruned SNPs. For this, the number of ancestral populations (K) ranged from 2 to 12, and finally, the plot of obtained results was made in PONG13. The PopLDdecay14 and Plot_MultiPop.pl were performed to measure and plot the decay of LD in each Ovis group, respectively.
Introgression and gene flow
Dsuite15 and Treemix16 programs were used to investigate the status of introgression and gene flow between native domestic and wild populations of the genus Ovis. We performed the ABBA-BABA test (D statistic) on genome-wide autosomal bi-allice SNPs with the Dtrios tool from Dsuite to test for introgression between domestic and wild populations. To implement the ABBA-BABA test, four populations are needed with the phylogeny pattern (((P1, P2), P3), O), where P1 and P2 are sister populations/species and potential targets of introgression, P3 is potential source of gene flow to P1 (negative D statistic) or P2 (positive D statistic), and O is an outgroup population/specie that was a goat sample in the present study. Trios with a Z-score greater than + 3 were considered as combinations with introgression between P2 and P3 populations. Next, the fdm statistics17 with sliding windows of 100 SNPs and steps of 25 SNPs were used to localize introgressed genomic regions between P3 to P2 in the selected trios. Windows with values greater than the 99th percentile of fdm were defined as putative introgressed regions in each trio.
The populations command of the Stacks v.2.65 program was used to convert a multi-sample VCF file to an input file for Treemix. Treemix was performed to construct maximum-likelihood phylogenetic trees with different numbers of migration events ranging from 0 to 12 and 10 replicates for each event. Then, a web-based optM program18 was used to identify the optimal number of migration events. Finally, we used the BITE package19 to construct trees with different migration events with 100 bootstrap replicates and block sizes containing 1000 SNPs.
Window-based mean pairwise sequence divergence (dxy) and nucleotide diversity (pi)
Two scripts available at https://github.com/simonhmartin/genomics_general were used to calculate window-based Dxy and Pi. The multi-sample VCF file was converted to geno format in the first step using parseVCF.py script. Then, the popgenWindows.py script was performed to calculate Dxy and pi in windows size equal to 50 kb with 20 kb overlap (step size). The results obtained for the desired introgressed regions were visualized using the ggplot2 and tidyverse R packages.
Annotation and gene ontology (GO) analysis
Annotation of introgressed regions was performed using BEDtools20 and GTF files related to the reference genome. The web-based g: Profiler server was performed to characterize gene lists and assign them to the biological process (BP), molecular function (MF), and cellular component (CC) categories21. The calculated p-values were corrected using the Benjamini-Hochberg FDR, and enriched terms were considered statistically significant at adjusted p-value ≤ 0.05.
Maps
Precipitation and temperature maps were created using the Google Earth Engine platform, utilizing MODIS satellite data for the long-term average from 2000 to 2023.
Results
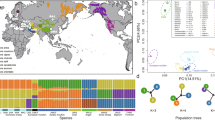
The individuals with high kinship calculated by the KING program were removed from the analyses. Accordingly, one domestic sample due to geographical differences from other domestic sheep with the unknown breed was removed from the study. Finally, 74 individuals, including 55 Iranian domestic sheep, 10 Asiatic mouflons and 9 urial sheep, were used for subsequent analyses. The average sequence coverage was 14.19 × (5.92–22.11x), 11.83 × (4.09–14.6x) and 15.38 × (12.34–18.9x) for domestic sheep, Asiatic mouflons and urials, respectively (Supplementary Table S1). A high genetic similarity between Moghani and Makui sheep breeds was revealed by domestic-specific PCA plot. Based on ADMIXTURE analysis and especially the constructed phylogenetic tree (Supplementary Fig. S1), these two breeds were placed in the same group as Ma-Mo. The first principle component (PC1) of the PCA plot for all domestic and wild sheep individuals clearly separated domestic and wild sheep, and the PC2 revealed the separation between Asiatic mouflons and urials (Fig. 2a). The constructed phylogenetic tree for all studied samples confirmed the PCA results (Fig. 2d). The LD level for all Iranian domestic sheep breeds (except NW-Iran) was higher than the two wild sheep species studied (Fig. 2b). Experiencing a population bottleneck by a species can lead to an increase in LD level in that population22, similar to the conditions experienced by domestic sheep during domestication. Based on ADMIXTURE results, two groups of domestic and wild sheep were separated at K = 2, and at K = 6. It should be mentioned, in ADMIXTURE analysis, K represents the hypothesized number of ancestral populations contributing to the genetic structure of the samples. Specifying K is crucial, as it determines how the software partitions genetic data into distinct populations, influencing the interpretation of genetic diversity and relationships among the groups studied. Five genetic groups were formed among the studied sheep, which included Asiatic mouflons, urials, Karakul, Ma-Mo and other samples (Fig. 2c). Also, the results of ADMIXTURE (K = 5 and K = 6) clarified that based on the available genomic data, there are at least two genetic subgroups for A. mouflons in Iran.

Population structure analysis results, including PCA (a), LD decay (b), admixture (c) and phylogenetic tree (d) for Iranian wild and domestic sheep.
The ABBA-BABA test was performed to identify possible introgressions between two species of wild sheep and different Iranian domestic sheep breeds. Since the Dtrios procedure of the Dsuite program examines all possible combinations of trios between populations, we selected five trios, where population P3 (as donor) was a wild species, and populations P1 and P2 (as possible acceptors) were domestic sheep breeds. The mentioned trios included ((Baluchi, MA_Mo), A.mouflon), ((Baluchi, NW-Iran), A.mouflon), ((Baluchi, Zel), A.mouflon), ((Baluchi, MA_Mo), Urial) and ((NW-Iran, Zel), Urial) (Table 1).
Based on the criteria defined for placing a genomic window in the list of introgressed regions, 1863 (containing 792 protein-coding genes), 1924 (containing 798 protein-coding genes), 2124 (containing 824 protein-coding genes), 2459 (containing 946 protein-coding genes) and 1990 (containing 828 protein-coding genes) genomic windows were identified as introgressed segments between P3 and P2 in trios 1 to 5, respectively (Table 2). Results of GO analysis for obtained introgressed genes from trios 1 to 5 are presented in Supplementary Tables S2 to S6.
Since optM designated one migration event (Δm = 13.64) as the optimal number, the tree with one migration event revealed a gene flow between the common ancestor of the Iranian domestic sheep and the Asiatic mouflon (Fig. 3a). This model placed Iranian domestic sheep breeds into three categories: the first category only included Ma-Mo, the second category included Shal, Ghezel, Afshari and NW-Iran, and the third category included Kermani, Baluchi, Karakul, Grey-Shiraz and Zel. Except for the Zel breed, which was expected to be placed in a separate branch due to distinct phenotypic characteristics and geographic region, the rest of the breeds were divided based on distribution area (Fig. 3b).

Treemix analysis results with m = 1 (a) and phylogenetic relationship among different domestic sheep breeds and two wild sheep species with goat as outgroup species (b).
The second largest Δm (11.68) belonged to the tree with four migration events (Supplementary Fig. S2). This tree revealed gene flow from the lineage leading to Ma-Mo into Ghezel, NW-Iran, Afshari branch and Zel. This model showed a gene flow event from NW-Iran to Zel. The tree with four migration events and the results of the ABBA-BABA test revealed a geographical-wise introgression among Iranian domestic sheep breeds (Supplementary Table S7). The tree with six migration events also revealed evidence of gene flow from A. mouflon to Iranian domestic sheep (Supplementary Fig. S3).
First, the common genomic widows introgressed from A. mouflon and urial to each domestic breeds were picked to identify the common introgressed genomic windows from wild sheep to domestic sheep. The results revealed that there are 277 (containing 234 protein coding genes) common genomic regions in all domestic sheep breeds that were transferred from A. mouflon, and this number was 139 (containing 110 protein coding genes) for urial. Finally, we found 36 (containing 26 protein coding genes) common genomic regions that were introgressed from A. mouflon and urial into the genome of Iranian domestic sheep. The results of GO analysis for the two mentioned gene sets (introgressed genes from urial to domestic sheep had no significant GO terms) are presented in Supplementary Tables S8 and S9.
Discussion
Designing new studies and re-analyzing accumulated genomic data in public databases has been one of the interest to researchers in recent years. This study used the genomic data of Iranian wild and domestic sheep for a more comprehensive search on introgression between them. Analyzing WGS enables the identification of a larger number of variants, as it encompasses the complete sequence of an organism’s genome. The detection of more variants at the genomic level enhances the potential to uncover introgressed genomic segments between the studied species. Several studies examining introgression in sheep using whole genomic data have observed that this data can reveal regions (especially shorter segments) that are undetectable with BeadChips due to the limited number of variants and the considerable distances between each SNP4,23. One of the reasons for the importance of introgression is its role in improving adaptation abilities in domestic animals. There is considerable evidence that domestic sheep have acquired alleles associated with adaptation from their wild relatives2,24.
We identified a new clade in Iranian domestic sheep that included two breeds, MOG and MAK. The results of ADMIXTURE, phylogenetic tree and domestic-specific PCA plot confirmed this clade. According to ADMIXTURE (K = 6) results, this clade has a genetic effect on all Iranian sheep breeds (except KER; probably due to a small number of samples) and even the A. mouflon. According to the treemix tree (m = 1), a gene flow event from this clade to A. mouflons was observed, although the ABBA-BABA test did not confirm this. On the other hand, based on the treemix tree with 4 migration events, we observed gene flow from Ma_Mo to ZEL and northwestern sheep (GEZ, AFS and NW-Iran), also from NW-Iran to ZEL. The ABBA-BABA test confirmed this result.
In the list of genes that were identified as introgressed genes from urial to Iranian domestic sheep (set1), two genes from the cytochrome P450 family, including CYP2C19 and CYP3A24 (also introgressed from A. mouflon to Iranian domestic sheep) were observed. Some genes belonging to the cytochrome P450 family (CYP2C19) are involved in the transformation of arachidonic acid to 19 S-HETE as a vasodilator factor of the renal preglomerular vessels that stimulate water reabsorption25. This process is one of the success factors of some species, including Bactrian camel26, sheep27, chicken28 and rodents29 to survive in dry and hot conditions i.e., desert conditions.
The UMOD gene is a kidney-specific protein that plays an important role in kidney homeostasis, and its defects are related to different kidney diseases. UMOD modulates salt reabsorption and blood pressure control at the tubular cell level by regulating apical transport systems operating in the thick ascending limb and the distal convoluted tubule30. Also, the excretion of this protein in the urine may provide defense against urinary tract infections caused by uropathogenic bacteria. The LOC101104728 gene encodes an antimicrobial peptide (NK-lysin) that has an inhibitory effect against Gram-negative and Gram-positive microorganisms31. The protein product of the LAMB3 gene is one of three subunits that form laminin-5. The laminin-5 is a component of the anchoring complex that is involved in the connection of the dermis and epidermis and is considered a key factor in the stability and homeostasis of the skin. This glycoprotein leads to an increase in the recovery of skin injuries32. Evidence has revealed that some mutations in the LAMB3 are associated with herlitz junctional epidermolysis bullosa33.
Also, we found one synaptonemal complex (SC) coding gene, including SYCE1. The SC mediates the synapse of homologous chromosomes and plays an essential role in genome haploidisation and the formation of normal gametes. A study on SYCE1 knockout mice revealed that this gene correctly forms synapses between homologous chromosomes during meiosis, and mice with SYCE1−/− were infertile34. Also, a rare frameshift mutation in SYCP1 is associated with infertility in men35. We identified two olfactory-related genes (LOC101113790, LOC101120028) as introgressed genes from urial to domestic sheep.
Investigation of the introgressed genes from A. mouflons to Iranian domestic sheep (set2) based on the literature review provided us with exciting results. The obtained evidence showed that the PLA2G4E gene is involved in vascular smooth muscle contraction in Australian Boer goats36, and it is probably one of the reproduction-related genes in pigs37. We found 11 olfactory receptor genes (LOC101106361, LOC101106108, LOC101110938, LOC101105851, LOC101121810, LOC101107892, LOC101110674, LOC114109605, LOC101102436, LOC101117708 and LOC101104643) that introgressed from A. mouflon to Iranian domestic sheep. It seems that this genetic transfer was probably influential in increasing the success of domestic sheep in processes such as the perception of reproductive pheromones2. The olfactory system can also play a crucial role in helping animals identify various odors, including the odor of predators38.
The TSHR gene is another vital gene identified as one of the introgressed genomic regions from mouflons to domestic sheep in the present study. The encoded protein by this gene is a receptor for thyrothropin and thyrostimulin and is associated with metabolic regulation and photoperiod control of reproduction in chickens and sheep39,40. Also, some studies41,42 have revealed the link between the TSHR gene and the litter size in sheep. The CIDEA gene controls lipid droplets and storage in brown and white adipose tissue43 and regulates lipid hemostasis44. In a study, this gene was introduced as the best candidate for the single gene estimator of intramuscular fat percentage in cattle and sheep45. We also found DGKH as a lipid metabolism and growth-related gene belonging to the DGK family. This gene plays a role in the growth process in cattle by affecting the secretion of growth-related hormones in the pituitary gland46. The LEPR, AACS, SCAP, and OSBPL11 genes were among the other lipid-related genes in the present study. The OC90 gene was associated with adaptation to climate in chickens28 and great tit47.
Among the introgressed genes from A. mouflons to Iranian domestic sheep, there were two genes belonging to the spermadhesin family gene (LOC101111242 and LOC101111505). Spermadhesins constitute an important part of the seminal proteome. They are the second most abundant protein in the seminal plasma of cattle48. These proteins have ligand-binding abilities to carbohydrates, phospholipids and protease inhibitors, reflecting their possible role in the evolution of sperm function in the male reproductive tract and subsequent sperm-egg binding in the female reproductive tract49. The ERCC6 gene (a DNA repair gene) is one of the set of genes that are involved in response to ultraviolet (UV) radiation50 and adaptation to high altitude51. The RNF168 gene is another DNA repair gene introgressed from A. mouflons to their domestic relatives. This finding suggests that these genes probably reduce DNA damage caused by stresses such as high altitude in mountainous areas and high UV radiation in desert areas in domestic Iranian sheep.
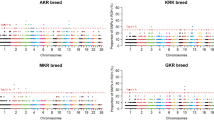
We found 36 genomic windows containing 26 protein coding genes introgressed from A. mouflons and urials into Iranian domestic sheep (set3). A close focus on the introgressed windows associated with 19 of the mentioned genes revealed a remarkable decrease in the dxy value of the urial vs. domestic and A.mouflon vs. domestic sheep (Supplementary Fig. S4 to S17). Also, a reduction in nucleotide diversity was observed in all three species in these regions, suggesting the occurrence of a selective sweep in the mentioned genomic regions. The Dxy and Pi distribution of the remaining 7 genes did not show a pattern that could confirm our results (Supplementary Fig. S18 to S23).
One of the important genes in the set3 was TTC29 (Fig. 4a-b) that was considered a male fertility-related gene. TTC29 has testis-specific expression and encoded protein in the sperm flagellum and plays a vital role in sperm motility and male fertility52. Studies report that some mutations in this gene are related to asthenozoospermia and infertility in males52,53. Also, we found another fertility-related gene (STPG2) that showed a similar pattern to TTC29 (Fig. 4c-d). Based on evidence, the STPG2 gene plays a role in testicular development and spermatogenesis54, and the presence of some mutations in it leads to male infertility55. Also, STPG2 is potentially linked with pigs’ litter size56 and prolificacy in sheep57.

Dxy and Pi of the introgressed genomic region on chromosome 17 containing TTC29 gene (a and b) and on chromosome 6 containing STPG2 gene (c and d) in wild sheep compared to domestic sheep.
DYRK2 belongs to DYRKs as a conserved family of protein kinases associated with neuron system development and disorders. The study of the DYRK2 gene in Drosophila has revealed that this gene plays a role in developing the olfactory and visual systems. DYRK2 null flies showed impaired olfactory sense, impaired visual integrity, and a subtle but measurable defect in the eye58. Also, there is evidence that this gene is involved in mammary gland development and health59. The LOC101110674 was another olfactory-relates genes in s3. A study on rats found that the CAMK1G gene is one of the learning and memory related genes, especially spatial memory. Down-regulation of the CAMK1G gene, along with other memory-related genes in aged rats, is probably one of the important causes of age-associated spatial learning impairment60. Identifying this gene as one of the introgressed genes from wild sheep to domestic sheep is important because evidence shows spatial memory is crucial for sheep to find the distribution of sites with a preferred food at pasture61.
Another possible important role of CAMK1G is its impact on emotion processing and stress-related conditioned and unconditioned fear. CAMK1G has a high expression in the central nucleus of the amygdala, ventromedial hypothalamic nucleus and bed nucleus of the stria terminalis. The mentioned parts of the brain are involved in conditioned fear responses, stress hormone release, olfactory system function, innate defensive reactions to dangers (e.g. predators) and sexual behaviours62. Thus, the genomic region carrying the mentioned gene can be considered as one of the most important genomic regions introgressed from wild to domestic sheep due to its connection with the ability to survive in challenging conditions. The introgression of the OTOP1 gene as a taste-related gene63 can be attributed to its possible role in the grazing behaviour of wild and domestic sheep, which can help in the selection of non-toxic and preferred plants.
Conclusions
The results showed an interesting information about the introgression from A. mouflon and urial on the genomic architecture of Iranian domestic sheep breeds. Identification of the introgressed genomic regions for both wild species and Iranian domestic sheep breeds (from A. mouflon to domestic and urial to domestic) revealed that these regions carry genes related to fertility, olfactory, lipid metabolism, nervous system function (e.g. memory) as well as genes involved in adaptation. The results revealed how introgression between the wild and domestic species can lead to transferring biological capabilities and genetic-based survival skills (as mentioned about the CAMK1G gene) between them. In addition to the findings of the current study, utilizing genomic data to identify distinct genomic regions between domestic and wild sheep, as well as examining their effects on various adaptive traits of each species, could enhance our understanding of how genomic architecture influences the interaction between these species and their environment.
Data availability
All genomic data used in this study were downloaded from NCBI. The information of the mentioned data is presented in the Table S1.
References
Tavakolian, J. An Introduction to Genetic Resources of Native farm Animals in Iran (Animal Science Genetic Research Institute, 2000).
Cao, Y. H. et al. Historical introgression from wild relatives enhanced climatic adaptation and resistance to pneumonia in sheep. Mol. Biol. Evol. 38, 838–855 (2021).
Chen, Z. H. et al. Whole-genome sequence analysis unveils different origins of European and Asiatic Mouflon and domestication-related genes in sheep. Commu Biol. 4, 1307 (2021).
Cheng, H. et al. Long divergent haplotypes introgressed from wild sheep are associated with distinct morphological and adaptive characteristics in domestic sheep. PLoS Genet. 19, e1010615 (2023).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Khalkhali-Evrigh, R., Hedayat, N., Ming, L. & Jirimutu Identification of selection signatures in Iranian dromedary and bactrian camels using whole genome sequencing data. Sci. Rep. 12, 9653 (2022).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. AJHG 81, 559–575 (2007).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Behr, A. A., Liu, K. Z., Liu-Fang, G., Nakka, P. & Ramachandran, S. Pong: fast analysis and visualization of latent clusters in population genetic data. Bioinformatics 32, 2817–2823 (2016).
Zhang, C., Dong, S. S., Xu, J. Y., He, W. M. & Yang, T. L. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019).
Malinsky, M., Matschiner, M. & Svardal, H. Dsuite-fast D‐statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 21, 584–595 (2021).
Pickrell, J. & Pritchard, J. Inference of population splits and mixtures from genome-wide allele frequency data. Plos Genet. 8, e1002967 (2012).
Malinsky, M. et al. Genomic islands of speciation separate cichlid ecomorphs in an east African crater lake. Science 350, 1493–1498 (2015).
Fitak, R. R. OptM: estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 6, bpab017 (2021).
Milanesi, M. et al. BITE: an R package for biodiversity analyses. BioRxiv 181610. https://doi.org/10.1101/181610 (2017).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Kolberg, L. et al. G: profiler—interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 51, W207–W212 (2023).
Slatkin, M. Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 9, 477–485 (2008).
Takahashi, L. K. Olfactory systems and neural circuits that modulate predator odor fear. Front. Behav. Neurosci. 8, 72 (2014).
Hu, X. J. et al. The genome landscape of tibetan sheep reveals adaptive introgression from argali and the history of early human settlements on the Qinghai–Tibetan Plateau. Mol. Biol. Evol. 36, 283–303 (2019).
Carroll, M. A. et al. Cytochrome P-450-dependent HETEs: profile of biological activity and stimulation by vasoactive peptides. Am. J. Physiol. Regul. Integr. Comp. Physiol. 271, R863–R869 (1996).
Jirimutu. Sequencing, T. B. C. G. & Analysis Consortium. Genome sequences of wild and domestic bactrian camels. Nat. Commun. 3, 1202 (2012).
Yang, J. I. et al. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 33, 2576–2592 (2016).
Tian, S. et al. Genomic analyses reveal genetic adaptations to tropical climates in chickens. Iscience 23, 101644 (2020).
Cheng, J. et al. Similar adaptative mechanism but divergent demographic history of four sympatric desert rodents in eurasian inland. Commun. Biol. 6, 33 (2023).
Joseph, C. B. et al. Meta-GWAS reveals novel genetic variants associated with urinary excretion of uromodulin. J. Am. Soc. Nephrol. 33, 511–529 (2022).
Yacoub, H. A. et al. Effect of Nk-lysin peptides on bacterial growth, MIC, antimicrobial resistance, and viral activities. Anim. Biotechnol. 35, 2290520 (2024).
Nishiyama, T. et al. The importance of laminin 5 in the dermal–epidermal basement membrane. J. Dermatol. Sci. 24, 51–59 (2000).
Nakano, A., Pfendner, E., Pulkkinen, L., Uitto, J. & Hashimoto, I. Herlitz junctional epidermolysis bullosa: novel and recurrent mutations in the LAMB3 gene and the population carrier frequency. JID 115, 493–498 (2000).
de Vries, F. A. et al. Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Gene Dev. 19, 1376–1389 (2005).
Nabi, S. et al. A rare frameshift mutation in SYCP1 is associated with human male infertility. Mol. Hum. Reprod. 28, gaac009 (2022).
Yang, B. G. et al. Genome-wide selection signal analysis of Australian Boer goat reveals artificial selection imprinting on candidate genes related to muscle development. Anim. Genet. 52, 550–555 (2021).
Zhao, Y. et al. Identification and analysis of genes related to testicular size in 14-Day-old piglets. Animals 14, 172 (2024).
Lv, F. H. et al. Whole-genome resequencing of worldwide wild and domestic sheep elucidates genetic diversity, introgression, and agronomically important loci. MBE 39, msab353 (2022).
Rubin, C. J. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591 (2010).
Liu, Z. et al. Genome-wide analysis reveals signatures of selection for important traits in domestic sheep from different ecoregions. BMC Genom. 17, 1–4 (2016).
Tao, L. et al. Combined approaches identify known and novel genes associated with sheep litter size and non-seasonal breeding. Anim. Genet. 52, 857–867 (2021).
Li, M. et al. Screen of small fragment mutations within the sheep thyroid stimulating hormone receptor gene associated with litter size. Anim. Biotechnol. 34, 658–663 (2023).
Wu, L. et al. Cidea controls lipid droplet fusion and lipid storage in brown and white adipose tissue. Sci. China Life Sci. 57, 107–116 (2014).
Chen, F. J., Yin, Y., Chua, B. T. & Li, P. CIDE family proteins control lipid homeostasis and the development of metabolic diseases. Traffic 21, 94–105 (2020).
Guo, B. et al. P. A gene expression estimator of intramuscular fat percentage for use in both cattle and sheep. J. Anim. Sci. Biotechno. 5, 1–2 (2014).
Lu, X. et al. Comparative transcriptomic analysis of the pituitary gland between cattle breeds differing in growth: yunling cattle and leiqiong cattle. Animals 10, 1271 (2020).
Stonehouse, J. C. et al. The genomics of adaptation to climate in European great tit (Parus major) populations. Evol. Lett. 8, 18–28 (2024).
Gomes, F. P. et al. Protein signatures of seminal plasma from bulls with contrasting frozen-thawed sperm viability. Sci. Rep. 10, 14661 (2020).
Haase, B. et al. Evolution of the spermadhesin gene family. Gene 352, 20–29 (2005).
Li, M. et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and tibetan wild boars. Nat. Genet. 45, 1431–1438 (2013).
Wu, H. et al. Identifying molecular signatures of hypoxia adaptation from sex chromosomes: a case for tibetan Mastiff based on analyses of X chromosome. Sci. Rep. 6, 35004 (2016).
Liu, C. et al. Bi-allelic mutations in TTC29 cause male subfertility with asthenoteratospermia in humans and mice. AJHG 105, 1168–1181 (2019).
Lorès, P. et al. Mutations in TTC29, encoding an evolutionarily conserved axonemal protein, result in asthenozoospermia and male infertility. AJHG 105, 1148–1167 (2019).
Kubo, S. et al. Native doublet microtubules from Tetrahymena Thermophila reveal the importance of outer junction proteins. Nat. Commun. 14, 2168 (2023).
Chan, C. C. et al. A comprehensive genetic study of microtubule-associated gene clusters for male infertility in a Taiwanese cohort. Int. J. Mol. Sci. 24, 15363 (2023).
Liu, C. et al. Whole-genome analysis of structural variations between Xiang pigs with larger litter sizes and those with smaller litter sizes. Genomics 111, 310–319 (2019).
Zhang, C. L., Zhang, J., Tuersuntuoheti, M., Chang, Q. & Liu, S. Population structure, genetic diversity and prolificacy in pishan red sheep under an extreme desert environment. Front. Genet. 14, 1092066 (2023).
Luebbering, N. et al. Drosophila Dyrk2 plays a role in the development of the visual system. PLoS One. 11, 8:e76775 (2013).
Tolleson, M. W. et al. Association of udder traits with single nucleotide polymorphisms in crossbred Bos indicus–Bos taurus cows. J. Anim. Sci. 95, 2399–2407 (2017).
Uddin, R. & Singh, S. M. Gene network construction from microarray data identifies a key network module and several candidate hub genes in age-associated spatial learning impairment. Front. Syst. Neurosci. 11, 75 (2017).
Dumont, B. & Petit, M. Spatial memory of sheep at pasture. Appl. Anim. Behav. Sci. 60, 43–53 (1998).
Piechota, M. et al. Glucocorticoid-regulated kinase CAMKIγ in the central amygdala controls anxiety-like behavior in mice. Int. J. Mol. Sci. 23, 12328 (2022).
Liang, Z., Wilson, C. E., Teng, B., Kinnamon, S. C. & Liman, E. R. The proton channel OTOP1 is a sensor for the taste of ammonium chloride. Nat. Commun. 14, 6194 (2023).
Acknowledgements
We would like to thank the university of Mohaghegh Ardabili for supporting this study as well as we are very grateful to Dr. Ali Nasiri Khiavi for his assistance in preparing Fig. 1.
Author information
Authors and Affiliations
Contributions
R.K-E. and N.H. designed the project. R.K-E. performed analysis on WGS data and interpreted the results. N.H., R.S., M.S. and E.N.P. supervised the analysis and writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khalkhali-Evrigh, R., Hedayat, N., Seyedsharifi, R. et al. Genomic evidence of improved fertility and adaptation in Iranian domestic sheep attributed to introgression from Asiatic Mouflon and urial. Sci Rep 15, 1185 (2025). https://doi.org/10.1038/s41598-025-85756-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85756-y
This article is cited by
-
Genomic analyses of Asiatic Mouflon in Iran provide insights into the domestication and evolution of sheep
Genetics Selection Evolution (2025)
