
Abstract
Individuals with Turner syndrome (TS) phenotypes may exhibit short stature, ovarian dysfunction, and neurocognitive disorders. Their genomes can include ring chromosomes formed from the X chromosome (RCX). Here, we present cytogenomic and clinical findings from seventeen individuals with TS who bore mosaic forms of RCX and frequently presented with short stature and concern for TS. The subjects were retrospectively included and tested at a single-site cytogenetics laboratory for over 42 years. Here, we illustrate each subject’s comprehensive cytogenetic workup and phenotypes. The cohort shows comorbidities and sexual characteristics associated with mosaic RCX. These cytogenetic findings and clinical features are distinct from those of individuals with non-mosaic TS. Studying the pattern of X-activation across tissues in this cohort could provide additional data on a postulated source of phenotypic variability. Current guidelines recommend karyotype as the first-line test rather than SNP microarray analysis when aneuploidy is suspected. Conventional cytogenetics is still necessary to understand structural abnormalities, provide genomic context, and detect low-level mosaicism. These cases add to the knowledge of mosaic RCXs and offer new clinical laboratory information that is important for diagnosis and useful for comprehensively caring for and managing TS patients.
Similar content being viewed by others

Introduction
Turner syndrome (TS) was first described as a clinical syndrome before the availability of karyotype analysis1. The causes include complete or partial deletion or structural abnormality involving the X chromosome, including ring chromosome X (RCX). Further cytogenetic workup should occur when a small ring/marker chromosome is diagnosed2. Complete rings can occur via telomere-telomere fusions without deletion of distal chromosomal material. Alternatively, rings can result in breakage and rejoining, with subsequent loss of genomic material at their distal ends. Numerous factors can influence the severity of the RCX-associated phenotype.
Laboratory test utilization has changed in the number of metaphases assessed by karyotype and the types of probes used for fluorescence in situ hybridization (FISH). Peripheral blood is the most common specimen for genetic testing for TS, as it is more accessible.
Current TS clinical guidelines address typical and variant karyotypes, which include RCX1. Commonly, secondary loss of RCX due to mitotic instability can result in the presence of a subset of cells mosaic for monosomy X (45,X). Females with RCX and mosaicism for the subsequent loss of the X might also have a typical 46,XX chromosomal complement that survives into the postnatal period and, when present in the reproductive tissues, supports a finding of fertility in females with RCX. It is postulated that TS patients survive to term with “pure monosomy X” owning to a cryptic 46,XX line3.
In 2010, chromosomal microarray (CMA) or SNP array analysis was recommended as a first-tier test for individuals affected with developmental disabilities or multiple congenital anomalies. CMA provides a whole genome-wide copy number analysis that includes sex chromosomes4. Laboratory guidelines indicate that working up recognizable chromosomal disorders, such as TS, should include conventional cytogenetic studies as an initial screening before CMA which might miss low-level mosaicism4.
The TS clinical practice guidelines for females recognize the need for multidisciplinary care1. Unlike TS arising from pure monosomy X, RCX can be associated with variable intellectual impairment5. SNP array can determine the extent of functional disomy of the genes on RCX, confirming the XIST locus’s status and identifying genome dosage changes. SNP array testing with a conventional karyotype has largely replaced metaphase FISH in clarifying the nature and extent of genomic imbalance. While there is not always a correlation between the presence of XISTin the ring and the cognitive phenotype, the active ring size appears strongly correlated with the phenotypic outcome2.
TS is diagnosed in 1 in 5,000 live female births, with estimates that 6–15% of TS patients are mosaic for an RCX6,7. First described in 19628, over 109 cases of mosaic RCX have been described9, but only a subset has received CMA testing10,11,12,13,14. We report data from 16 novel mosaic RCX cases and one previously described15. Seven subjects have informative findings from SNP array.
Results
Clinical evaluation
The demographic data included n = 13 white and n = 4 black subjects. The average age of the first ascertainment was 10.83 years old, with five neonatal at first cytogenetic analysis. The upper limit of the first ascertainment was 60 years old in a patient who had been aware of her diagnosis since her teens. The average cohort age was about 24 years old, ranging from neonatal to 66 years. The total years with data available was 34 years in one. To date, two (RCX-13 and RCX-16) passed away at ten months and 66 years of age, respectively.
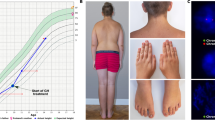
The features identified in our subjects with RCX are listed in Table 1 and Supplementary Table 1 (S1) and included the presence of short stature, developmental delay, craniofacial features, hearing loss, cardiovascular, endocrine, skeletal, genitourinary, skin, autoimmune, and neurologic findings.
Cardiac features included bicuspid aortic valve, coarctation of the aorta, hypoplastic left heart, aortic aneurysm, and patent ductus arteriosus (5/17 subjects; 29%). Renal anomalies included renal malrotation, a duplicated collecting system, and a pelvic kidney (4/17 subjects; 24%). Hearing impairment was present (5/17 subjects; 29%). Abnormalities of the thyroid gland were diagnosed in a subset (5/17 subjects; 29%). Ophthalmologic findings included papilledema, strabismus, and esotropia. Growth hormone deficiency was observed (7/17 subjects; 41%) with at least two receiving growth hormone, and cognitive impairment and/or developmental delay (8/17 subjects; 47%) were also noted.
Varying degrees of secondary sexual development were noted. Of those subjects whose last age documented was 18 years or older (n = 10), four had Tanner Stage V development, one each had Tanner Stage III, II, or precocious puberty, and three had no comment on their sexual development. Of the subjects aged < 18 (n = 7), one each had Tanner Stage V, IV, II, or some secondary sexual development, and the remaining (n = 3) had either Tanner Stage I or no documented growth.
Of the reproductive-aged subjects greater than 18 years of age (n = 9), menarche was achieved among at least five subjects; however, no documented pregnancies existed. One subject (RCX-15) had a variable anti-müllerian hormone (AMH) level that fluctuated between < 0.03–0.21 that was last reported at 0.11 at age 16. Levels of this hormone can be evaluated clinically to assess ovarian reserve. She chose to undergo serial monitoring of AMH levels every six months. Another adolescent subject had an AMH of 0.01 at age 11. She chose to proceed with unilateral ovarian cryopreservation at age 14 (RCX-12). Hormone replacement therapies were prescribed in at least four.
Non-sexual characteristics of TS observed at a higher frequency in this cohort included short stature (15/17 subjects; 88%) and a high-arched palate (7/17 subjects; 41%). Widely spaced nipples were also observed in 5/17 subjects (29%). A low posterior hairline was seen in 5/17 subjects (29%). A webbed neck was observed in a subset (4/17 subjects; 24%). Lymphedema was not commonly observed (2/17 subjects; 12%).
Other features included hypotonia in two, one of which required mobility assistance. Seizures were also reported in four out of seventeen subjects (24%).
The TS-associated comorbidities confirmed here included increased BMI, diabetes mellitus (DM), autoimmunity, hypothyroidism, hypertension, bone conditions including reduced bone mineral density, and cardiovascular disease: strokes, cardiac arrest, cardiomyopathy, and congestive heart failure. Complications secondary to diabetes included neuropathy, retinopathy, and glaucoma in one. Documented liver disease and cirrhosis were seen in one. Chronic kidney disease was diagnosed in at least two.
Four out of seventeen subjects (24%) had autoimmune features (Celiac disease, Graves disease, Hashimoto thyroiditis, and/or type I diabetes mellitus). Specifically, Celiac disease was noted in two.
Notably, one had a second clinical diagnosis with Crouzon syndrome (RCX-2) based on the presence of craniosynostosis and a genetics assessment. However, to our knowledge, this was not molecularly confirmed.
One subject (RCX-8) had an isochromosome Xq with a secondary RCX observed in a subset of cells. She was described as having a severe r(Xp) phenotype, was non-verbal, and had mobility issues. Her severe presentation was attributed to the presence of the small secondary RCX comprised of Xp material, which lacked XIST. Two of these subjects had a small RCX, though none had a Kabuki-like syndrome diagnosis. Rare individuals having a small and active RCX have been reported in association with Kabuki syndrome14,16.
Conventional cytogenetic analyses
The ages at initial laboratory testing ranged from neonatal to 60 years old. Peripheral blood was sampled at least once during the subject’s lifetime. In one, skin fibroblasts were also collected. Mosaicism for monosomy X and a simple RCX in peripheral blood was observed in 16/17 subjects (94%).
We performed G-banded karyotyping on lymphocytes, revealing mosaicism in seventeen, one of whom had follow-up studies on skin fibroblast cultures (Table 1, Fig. 1, Supplementary Tables 1 and 2 and 3 (S1, S2, and S3), and Fig. S1 S2, and S3). Sixteen had a mosaic karyotype with 46 chromosomes (45,X/46,X,r(X)). In one, we diagnosed an isochromosome Xq with a subset of cells showing a secondary RCX (RCX-8).

The schematic shows the breakpoints and deleted Xp and Xq arms in the seven subjects whose RCX was further clarified by the SNP array. The top panel represents the full-length chromosome X (UCSC Genome Browser). The track below represents the SNP array results for each of the seven subjects. The portions of X contained in the RCX and the normal homolog are depicted as pink bars (1 ~ 2 (CN) copy number). Distal losses of the terminal ends of X are noted in red (CN = 1). A segment of the X with a typical copy number of 2 is depicted in black, likely representing a duplication of the normal X homolog (RCX-14). The sizes of the RCXs ranged from 13.5 Mb (RCX-12) to 105.4 Mb (RCX-17). Levels of mosaicism were observed for all subjects based on the B-allele frequency. Below, the regions of interest track demonstrate that regions PAR1 (SHOX (teal)) and PAR2 are deleted in all seven subjects (purple). The XIST locus was intact in the RCX for all seven subjects (teal). The involvement of the Xp-arm and Xq-arm inclusive of the centromere was confirmed by the SNP array in six subjects; centromeric involvement of the RCX was confirmed via metaphase FISH for RCX-12 (I). The smallest region of RCX overlap is indicated in yellow and is 11.9 Mb in size (chrX:61,719,290–73,569,476). This shared segment is defined by cytobands Xq11.1 to Xq13.2. Recurrent events on the X chromosome are shown (orange) and their corresponding flanking low-copy repeats (black). Below are the G-banded partial karyotypes for the seven subjects whose RCX was resolved by SNP array (RCX-4 (A), RCX-9 (B), RCX-12 (C), RCX-13 (D), RCX-14 (E), RCX-15 (F), and RCX-17 (G)). The RCX (on the right) and the normal homolog (on the left) are shown for comparison. The bottom panel shows the available metaphase FISH images (H-J) (RCX-9 (H), RCX-12 (I), and RCX-17 (J)), which also confirmed that the rings were X chromosomal in origin. RCX-9 (H) was confirmed using whole chromosome paint FISH probes for the X chromosome, while RCX-12 (I) and RCX-17 (J) were confirmed using CEPX/SRY FISH probes.
The RCX was not always appreciated during the initial karyotype, so the total number of cells studied was combined, with mosaicism levels ranging from 1 to 60%. The percentage of cells with RCX could be grouped as mild (< 10%) for three, moderate (10–30%) for five, severe (30–50%) for eight, and very severe (> 50%) for one17. In one (RCX-2), a rare subset of cells having a normal line was confirmed upon repeat karyotyping. This finding indicates a normal XX line present during early zygote formation, likely persisted below the detection limit of the karyotype, and was thought to explain her typical sexual development.
Fragments, markers, or secondary rings likely arising from the mitotic instability of the RCX were observed in four subjects, potentially representing dynamic mosaicism, which prompted repeat cytogenetic studies in a subset.
Molecular methods and cytogenomics
A subset of subjects (14 out of 17) received molecular cytogenetics or molecular methods, highlighting technological changes over the last 42 years. In two predating FISH, extracted genomic DNA was probed for human centromeric regions using alpha-satellite, or alphoid, repetitive DNA sequences specific to the centromeres of human chromosomes X (XC) and Y (YC-2), followed by pulsed-field gel electrophoresis, revealing that the ring was X chromosomal in origin for RCX-3 while ruling out the ring being Y chromosomal in origin in RCX-5 (Table 1)18.
Fluorescence in situ hybridization (FISH) confirmed the ring or secondary events as X chromosomal in origin in eight subjects. FISH confirmed XIST status in at least two subjects with RCX (RCX-8 (subject with isochromosome) and RCX-10, who was also studied with SNP array) (Table 1, Fig. 1, Supplementary Fig. S2). As TS laboratory guidelines recommend ruling out the ring/marker is Y-chromosomal in origin due to concern for gonadoblastoma, a subset was tested by FISH and confirmed the absence of such material. Not all subjects received this recommended testing to exclude the possibility of Y-chromosomal material based on when their study occurred.
We achieved SNP array-aided ring sizing and confirmed the XIST locus status in 7 out of 11 subjects, all with 45,X and 46,X,r(X) (Table 1, Fig. 1). Mosaic RCXs were classified as small (13.5 megabases (Mb) for RCX-12 from Xq11.1 to Xq13.3, 21.6 Mb for RCX-13 from Xp11.22 to Xq13.3, and 19.9 Mb for RCX-14 from Xp11.22 to Xq13.3), intermediate (44.6 Mb for RCX-4 from Xp21.2 to Xq13.3 and 52 Mb for RCX-9 from Xp22.11 to Xq13.3), or large (104 Mb for RCX-15 from Xp22.2 to Xq23 and 105.4 Mb for RCX-17 from Xp11.4 to Xq27.2). The number of genes within these mosaic RCXs ranged from 104 (RCX-12) to 726 (RCX-17) (GeneScout; last accessed 12/8/2023).
We observed the inclusion of the short and long arms and the pericentromeric region of X in six. One subject whose RCX lacked coverage for informative SNP probes in the centromere was ultimately confirmed to include X pericentromeric material using FISH (RCX-12).
These rings, which varied in sizes and breakpoints, all included XIST. The role of XIST has been associated with X-chromosome inactivation in females (OMIM *314,670). The presence of the XIST locus in the ring implies that the ring can be inactivated. The B-allele frequency indicated the presence of mosaicism in the interstitial regions. The smallest region of RCX overlap by the SNP array is defined by cytobands Xq11.1 to Xq13.2 for this cohort and was 11.9 Mb and contained 97 genes, including XIST (GeneScout; last accessed 12/8/2023).
SNP array can miss low-level mosaicism. Indeed, three subjects whose known RCX was at a low frequency showed monosomy X as the sole array aberration (RCX-1, RCX-5, and RCX-10); in another, the resulting data was unsuitable for analysis (RCX-16). Six others did not receive research SNP array testing due to a lack of archival material.
Eight had mosaic simple RCXs that contained XIST by SNP array and/or FISH. In those, the RCX would be preferentially inactivated. However, a lack of expression of XIST from small RCX chromosomes containing the XIST locus is reported to result in more severe phenotypes. Thus, XIST transcription levels and/or cell selection are likely essential for phenotype expression. Six of the eight subjects whose RCXs were confirmed to contain XIST had features of psychotic episodes, altered mental status, depression, ADHD, anxiety, or a learning disorder, among others. Of the three whose RCX was confirmed to contain XIST by the SNP array, only one displayed cognitive impairment (RCX-4). The others only displayed developmental delay (RCX-14 and RCX-17). An isochromosome Xq with a secondary small supernumerary RCX that lacked XIST according to FISH (RCX-8) was also observed and deemed explanatory for her severe phenotype.
One subject was known prenatally to be at increased risk for TS by non-invasive prenatal screening (RCX-13). Invasive prenatal cytogenetic testing was declined, and postnatal cytogenetic testing revealed a mosaic RCX.
Discussion
TS can be caused by partial or complete absence of the X chromosome. There are multiple explanations for the phenotypic variability found between TS patients, including the presence of an RCX. This case series provides additional details regarding the genotype–phenotype correlation for RCX. This cohort included subjects with RCX and CMA data to resolve their breakpoints and XIST status simultaneously.
Accordingly, conventional and molecular cytogenetics can evaluate factors such as the karyotype, level of mosaicism of the RCX, size of the genomic imbalance, genes affected, and mitotic instability. Other factors include X-inactivation and somatic distribution within the affected individual. Associated TS features in individuals mosaic for RCX can range from a mild presentation to a more severe phenotype: short stature, primary and secondary amenorrhea, gonadal dysgenesis, development of secondary sexual characteristics, infertility, and intellectual disability. Individuals presenting with a severe phenotype may display intellectual disability, hypotonia, and syndactyly and may be seen in association with an active RCX containing an XIST deletion.
Given that both losses of the RCX and the RCX itself can confer differences in genomic imbalances for the X chromosome, these variables can influence the differences observed in the TS phenotypic spectrum. Here, we focused on subjects mosaic for simple RCXs, which can exhibit typical TS features consistent with those of monosomy X and unique phenotypic features that might be attributed to the RCX phenotype.
RCX can form due to various mechanisms. 1) Breaks at the distal ends result in losses of the terminal regions of the short arm and long arm, which can fuse and form a ring19,20,21; 2) subtelomeric fusion in the absence of distal copy number changes can result in a complete ring19,20,21,22,23; and 3) an inverted duplication deletion (inv del dup) results in inverted duplications contiguous to terminal deletions24,25. Understanding the underlying mechanism might inform familial follow-up in the diagnosed cases.
Here, we present sixteen cases of a simple and mosaic RCX with variable levels of mosaicism for 45,X, and 46,X,r(X) (Fig. 1). Importantly, one neonate (RCX-2) was found to bear thirteen female cells with a normal chromosomal complement upon a second cytogenetic follow-up fifteen years later in the background of monosomy X and an absence of the RCX. This finding indicates that the typical cells were present during early development. A presentation of normal secondary sexual development in a patient with pure 45,X raises the possibility of being mosaic for a cryptic co-occurring karyotype. A seventeenth subject (RCX-8) had an isochromosome Xq with a secondary RCX observed in a subset of cells. In all, a postnatal G-banded karyotype was essential to identify the RCX. Other ancillary methods identified the size and ring structure. Taken together, most of this cohort demonstrated a Turner-like phenotype with differences in phenotypic. Two displayed hypotonia and developmental delay; one had an isochromosome Xq with a secondary RCX. Importantly, neither displayed syndactyly and cognitive impairment, findings seen in association with a more severe TS phenotype.
In this cohort, the RCX was observed in only one affected germ layer tissue in all subjects studied. Regarding the developmental mechanism, functional X-chromosome mosaicism was observed, and differential expression of XIST in these females might be involved. The etiology of the RCX could indicate that the phenotype was due to sizeable genomic imbalance changes. However, the possibility of epigenetic and/or positional effects cannot be excluded.
We found that the RCXs followed a generalized small, intermediate, and large grouping. The smallest in this cohort were under 22 Mb in size (RCX-12, RCX-13, and RCX-14). None was seen in association with a Kabuki-like phenotype, a finding attributed to a small subset with RCX. Medium-sized rings were also observed as 44.6 Mb (RCX-4) and 52 Mb (RCX-9). The largest were greater than 100 Mb (RCX-16 and RCX-17). The smallest region of RCX overlap for the seven individuals was observed at cytobands Xq11.1 to Xq13.2. The length was 11.9 Mb and included XIST (GeneScout; last accessed 12/8/2023). As XIST was confirmed in the RCX in eight subjects (FISH and/or SNP array), XIST inactivation could preferentially silence the abnormal X. Examples of small RCXs lacking XIST have been described in association with a severe phenotype owing to an inability for the genome to silence the structurally abnormal X preferentially. Additionally, a large RCX lacking XISTmight also be associated with a severe phenotype owing to its active status and functional disomy of the genes on the RCX26.
Six subjects tested by SNP array whose RCX included the short and long arms, and the pericentromeric region of X shared the smallest region of overlap that was 18.8 Mb in size (chrX:54,737,366–73,569,476; hg19) and contained XIST. One subject’s short arm breakpoint was not mapped to Xp by the SNP array but indicated XIST inclusion in the RCX. Centromeric region involvement was confirmed via FISH, indicating probe limitations on the SNP array. Therefore, our subjects with a simple RCX were intact for this critical cis-acting X chromosome inactivation regulator. The only subject whose (secondary) RCX lacked XIST was thought to explain her severe presentation (isochromosome X).
Mosaicism is a factor that influences severity. Thus, we classified the fraction of the affected tissue (cells) with RCX into mild, moderate, severe, and very severe using the recommendation provided by Martinez-Glez et al.17. These subjects had RCX levels ranging from 1 to 60%. Three had mild levels (< 10%), five had moderate levels (10–30%), eight had severe levels (30–50%), and one had very severe levels (> 50%)17. These cumulative levels are based on total metaphases examined from combined studies (ranging from 1–3) due to the rarity of the RCX in the individual, which was not definitively diagnosed in each study.
We observed three main groupings: (1) predominantly monosomy X over RCX, (2) equal numbers of cells with monosomy X or RCX, and (3) predominantly RCX over monosomy X. In one, a minority of cells had a normal female chromosomal complement only appreciated upon follow-up, indicating the line must have been present in the early zygote.
Only one subject was tested in skin and peripheral blood; ultimately, only one rare clone with the RCX was seen in skin fibroblasts, which was undetected in blood (RCX-1); this subject was previously described15. This impact of mosaicism on the severity of the clinical phenotype is challenging to determine, as the levels of mosaicism can change in response to aging due to RCX instability. Karyotype studies performed as follow-ups to testing at an outside laboratory (in two subjects) confirmed the previous finding of a mosaic RCX in additional cells based on a larger number of metaphases examined (RCX-6 and RCX-17). We also observed evidence of RCX instability as fragments, markers, or secondary rings in four subjects, potentially representing dynamic mosaicism. Repeated testing confirmed the RCX (RCX-5 and RCX-16) in subjects whose initial karyotypes revealed possible instability.
Another factor in the severity spectrum may be attributed to the length of the observed deletions on the distal ends of both arms of the X chromosome. For the 11 subjects with available material for cytogenomic characterization, all had a simple mosaic 46,X,r(X) co-occurring with monosomy X. When the SNP array was informative (n = 7), we observed recurrent loss of the terminal end of the short arm at Xp22.33, corresponding to genomic position 60,814 (hg19), with variations in the interstitial breakpoint (Xp22.2, Xp22.11, Xp21.2, Xp11.4, Xp11.2, Xp11.22, and Xp11.1). Among the seven subjects, the distal Xp region included the pseudoautosomal region 1 (PAR1), which contains the SHOX gene and some non-coding regulatory elements. Pathogenic variants in SHOX are associated with idiopathic growth retardation, short stature, and Leri-Weill dyschondrosteosis (OMIM *312,865). Fifteen of the seventeen demonstrated short stature; the two subjects without this qualifier were young children. According to the SNP array (n = 7 subjects), the largest loss of Xp was 58.4 Mb (RCX-12), while the smallest was 11.4 Mb (RCX-15). The smallest region of distal Xp loss overlap (Xp22.33 to Xp22.2) was 11.4 Mb and impacted 63 genes, including SHOX (GeneScout; last accessed 12/8/2023). Loss of genes in this interval may indicate an increased risk of unmasking X-linked recessive disorders for these effectively hemizygous females. Only three genes associated with intellectual disability were shared within the smallest overlap region: NLGN4X, ANOS1, and CLCN4 (GeneScout; last accessed 11/8/2024). Typically, loss-of-function variants in ANOS1 are associated with Kallman syndrome in males (MIM:308,700), while such variants in NLGN4X are associated with X-linked complex neurodevelopmental disorder (ClinGen; last curated 8/1/2018; last accessed 11/8/2024). CLCN4has limited evidence for dosage sensitivity (ClinGen; last curated 10–20–2017; last accessed 11/8/2024). Additionally, terminal Xp deletions may be associated with premature ovarian insufficiency (POI). However, even when more than two-thirds of Xp is deleted, fertility may be retained based on historical cytogenetic studies of Xp alterations27.
Xq deletions might support the causes of ovarian dysgenesis. Xq13- > Xq28 is necessary for normal ovarian function. Deletions involving Xq13- > q27 may result in ovarian insufficiency and can be further stratified by Xq13-Xq21 and Xq23-Xq2728 into POI critical regions I and 2, respectively. We also observed recurrent loss of the terminal end of the long arm at Xq28, corresponding to different genomic positions, with variation in the interstitial breakpoint on the long arm (Xq13.3 in five subjects, with one subject each for Xq23 and Xq27.2). The PAR2 region was included in the loss involving the long arm for all seven subjects. According to the SNP array, the size of the largest loss of Xq was 81.2 (RCX-9), and the smallest was 12.2 Mb (RCX-17). A shared loss of 12.2 Mb was observed for the loss of distal Xq material at cytobands Xq27.3 to Xq28, including PAR2, and impacted 182 genes (GeneScout; last accessed 12/8/2023). 29 genes within the smallest overlap region on Xq are associated with intellectual disability (GeneScout; last accessed 11/8/2024). Of these, nine are curated to display haploinsufficiency (ClinGen; last accessed 11/8/2024): FMR1, AFF2, IDS, MTM1, SLC6A8, BCAP31, ABCD1, L1CAM, and MECP2.
Again, the gene deletion in this interval might confer an increased risk of unmasking X-linked recessive disorders for these effectively hemizygous females. In one, we observed a segment representing a duplication of the normal homolog on Xq28 (RCX-14).
Those with proximal losses may experience normal menstruation and fertility, while the more terminal losses are expected to be associated with POI29. Rare instances of fertility exist in the RCX literature30,31,32,33. To our knowledge, none of the subjects in this cohort has successfully achieved a pregnancy. However, at least 10 exhibited some level of secondary sexual development. Menarche was achieved in at least five subjects. Two subjects were evaluated during adolescence: RCX-12, with a secondary RCX with loss of Xq11.1- > Xq13.3, which is 80 Mb according to the SNP array, notably had follicles present and underwent cryopreservation via a unilateral oophorectomy.
Moreover, RCX-15 is noted to have a stable but diminished oocyte supply based on AMH level. The SNP array showed a loss of Xq23- > Xq28, which was 39.8 Mb in the latter subject and included the FMR1gene. POI has been associated with terminal Xq28 chromosome deletions34 and can include Fragile X-associated POI (MIM# 311,360).
Both RCX-12 and RCX-15 may benefit from these early fertility interventions and monitoring to aid their childbearing in the future.
While typically de novo, maternal transmission is rare30,31,32,33. Only one was confirmed de novo (RCX-8), with most parents not routinely studied here.
Phenotypic variation may exist within different karyotypes and could be confounded by somatic mosaicism. The dataset illustrates phenotypic variation within a TS karyotype variant. Larger datasets of TS individuals with different karyotypes, such as RCX, could be assessed to improve their care. Others recommend that systematic comparisons of larger TS cohorts focus on clarifying morbidity and mortality, risk stratification, and phenotypes between karyotype groups35. The cohort provides data encompassing previously associated comorbidities and sexual characteristics in an aging TS population with mosaic RCX.
Karyotypes can detect mosaicism and suspected aneuploidy. However, the sizes of events must exceed 5–10 Mb to be reliably detected. The number of metaphases examined can rule out cytogenetic mosaicism at specified confidence levels36. As mosaicism in TS can be common, laboratory guidelines recommend that at least 30 cells be counted, except if mosaicism is documented within the first 20 cells2. As some studied here were diagnosed before these guidelines, 20 to 50 cells are observed per subject study, with a subset studied multiple times (n = 6). With the ability to detect these low-level events, karyotypes proved informative when the SNP array was not.
FISH is a complementary tool in determining the status of specific loci at the cellular level. Various FISH probes have been utilized in this laboratory to confirm whether the ring contained X-chromosome centromeric material, lacked Y-chromosomal material, and whether XIST was present. A limitation is that these selected probes cannot provide sizing or genomic coordinates of the RCX detected. Indeed, severe phenotypes have been associated with RCX and the absence of XIST. However, small active RCXs intact for XISThave been described, implicating a more complex mechanism37,38. Even large RCXs lacking XIST may also present with a severe phenotype, highlighting the need for further clarification of the composition and transcriptional activity of the RCX. Over time, this single-site institution began implementing cytogenomic-based methods for simultaneous sizing and XIST status determination in 2011.
CMA or SNP array can help provide an X-chromosome level assessment, detecting net copy number imbalances such as gains or losses. Notably, the SNP array may miss balanced rearrangements or low-level mosaicism (< 20%). Indeed, monosomy X was the sole aberration in three whose RCX was present at a low frequency on karyotype. Reports of SNP array data focusing on the RCX approximation are growing10,11,12,13,14. While the SNP array can confirm the status of the XIST locus, the assay cannot determine the expression levels of genes predicted to escape X inactivation or of XIST itself.
There is an opportunity to determine the parental origin and confirm the functional disomy of genes on the RCX and X-inactivation status of the derivative chromosomes in different tissues for improved genotype–phenotype correlation. Additionally, epigenetic modifiers could influence the epigenetic signatures and subsequent phenotypic expression. Pursuing single-cell differences in RCX patients may enhance understanding of the association between RCX karyotypes and TS phenotypes.
Future applications like optical genome mapping or long-read sequencing must consider the presence of structural rearrangements, such as rings, that might be appreciated only with the identification of terminal copy number changes. A limitation herein is that cytogenetically identified events cannot confirm the presence of additional genomic complexity.
Methods
Study population
A retrospective analysis was performed for cases with constitutional RCX by routine cytogenetic testing. We evaluated patients with RCXs detected in postnatal specimens referred to the Hopkins Cytogenomics Laboratory beginning in 1981 when the first RCX was definitively diagnosed in this laboratory, through July 1, 2023. This case series highlights seventeen instances of mosaic constitutional RCX in individuals affected with TS from this single institutional cytogenetics laboratory. The subjects described here were diagnosed with RCX, and the cases were numbered chronologically. The institutional review board of the hospital approved this study. All methods were performed in accordance with the relevant guidelines and regulations.
Cytogenetic analyses
Peripheral blood specimens and, where available, skin fibroblasts were obtained from the proband and available family members as part of their routine clinical care. Chromosome analysis was performed using available methods at the time: solid staining, Q-banding, R-banding, or G-banding according to standard cytogenetic protocols, and the cytogenetic nomenclature was defined using the International System for Human Cytogenetic Nomenclature Version used at that time.
FISH analysis
We performed fluorescence in situ hybridization (FISH) analysis following a standard protocol on cultured cells. The Vysis probes included CEPX (DXZ1) and HIRA/TUPLE1 (22q11.2), and the control probe ARSA, AneuVysion (13/X/Y), (DXZ1, DYZ1/DYZ3) and SRY. The Oncor probes used included LSI DMD (Xp21) and LSI XIST (Xq13.2). A whole chromosome paint (cosmid) probe was applied on a research basis.
DNA analysis
Genomic DNA was extracted and probed for human centromeric regions using alpha-satellite, or alphoid, repetitive DNA sequences specific to the centromeres of human chromosomes 6 (D6Z1), X (XC), and Y (YC-2) and pulsed-field gel electrophoresis18 was applied to samples from probands and, if available, their family members.
Single nucleotide polymorphism (SNP) array analysis
We genotyped DNA extracted from peripheral blood per the manufacturer’s recommendations (QIAamp DNA Blood Midi Kit) (QIAGEN). A subset of subjects with remnant cell pellets or stored cell lines underwent DNA extraction (PureGene). We performed genotyping using various Illumina-based Human Genotyping BeadChip versions (HumanQuad610, CytoSNP-850 K v1.0, CytoSNP-850 K v1.1, CytoSNP-850 K v1.2 or CytoSNP-850 K v1.3) (Illumina Inc.) per the manufacturer’s instructions. The copy number and B-allele frequency data were analyzed using KaryoStudio and GenomeStudio software (Illumina Inc.). Genome coordinates are provided according to the hg19/GRCh37 human reference genome.
Data availability
De-identified data are available from the corresponding author upon reasonable request.
References
Gravholt, C. H. et al. Clinical practice guidelines for the care of girls and women with turner syndrome: Proceedings from the 2016 cincinnati international Turner syndrome meeting. Eur. J. Endocrinol. 177, G1–G70 (2017).
Wolff, D. J., Van Dyke, D. L. & Powell, C. M. Laboratory guideline for Turner syndrome. Genet. Med. 12, 52–55 (2010).
Hassold, T., Pettay, D., Robinson, A. & Uchida, I. Molecular studies of parental origin and mosaicism in 45. X concept. Hum. Genet. 89, 647–652 (1992).
Miller, D. T. et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86, 749–764 (2010).
Leppig, K. A. et al. Phenotype and X inactivation in 45, X/46, X, r(X) cases. Am. J. Med. Genet. A. 128A, 276–284 (2004).
Sybert, V. The adult with Turners syndrome: May 18–21; Gothenburg, Sweden. Edited by: Albertsson-Wikland K, Ranke MB. Amsterdam, Lausanne, New York, Oxford, Shannon, Tokyo: Elsevier (1995).
Jacobs, P. et al. Turner syndrome: A cytogenetic and molecular study. Ann. Hum. Genet. 61, 471–483 (1997).
Lindsten, J. & Tillinger, K.-G. Self-perpetuating ring chromosome in a patient with gonadal dysgenesis. Lancet 279, 593–594 (1962).
Li, P. et al. The past, present, and future for constitutional ring chromosomes: A report of the international consortium for human ring chromosomes. HGG Adv. 3, 100139 (2022).
Prakash, S. et al. Single-nucleotide polymorphism array genotyping is equivalent to metaphase cytogenetics for diagnosis of Turner syndrome. Genet. Med. 16, 53–59 (2014).
Suntharalingham, J. P. et al. Analysis of genetic variability in Turner syndrome linked to long-term clinical features. Front. Endocrinol. (Lausanne) 14, 1227164 (2023).
Luo, H. et al. Characterization of a rare mosaic X-ring chromosome in a patient with Turner syndrome. Mol. Cytogenet. 15, 15 (2022).
Shchelochkov, O. A. et al. Mosaicism for r(X) and der(X)del(X)(p11.23)dup(X)(p11.21p11.22) provides insight into the possible mechanism of rearrangement. Mol. Cytogenet. https://doi.org/10.1186/1755-8166-1-16 (2008).
Rodríguez, L. et al. A small and active ring X chromosome in a female with features of Kabuki syndrome. Am. J. Med. Genet. A. 146A, 2816–2821 (2008).
Berkovitz, G., Stamberg, J., Plotnick, L. P. & Lanes, R. Turner syndrome patients with a ring X chromosome. Clin. Genet. 23, 447–453 (1983).
McGinniss, M. J., Brown, D. H., Burke, L. W., Mascarello, J. T. & Jones, M. C. Ring chromosome X in a child with manifestations of Kabuki syndrome. Am. J. Med. Genet. 70, 37–42 (1997).
Martínez-Glez, V. et al. A six-attribute classification of genetic mosaicism. Genet. Med. 22, 1743–1757 (2020).
Jabs, E. W., Goble, C. A. & Cutting, G. R. Macromolecular organization of human centromeric regions reveals high-frequency, polymorphic macro DNA repeats. Proc. Natl. Acad. Sci. U S A 86, 202–206 (1989).
Guilherme, R. S. et al. Mechanisms of ring chromosome formation, ring instability and clinical consequences. BMC Med. Genet. 12, 171 (2011).
Sigurdardottir, S. et al. Clinical, cytogenetic, and fluorescence in situ hybridization findings in two cases of ‘complete ring’ syndrome. Am. J. Med. Genet 87, 384–390 (1999) ((1999 Wiley-Liss, Inc., United States, 1999)).
Conlin, L. K. et al. Molecular analysis of ring chromosome 20 syndrome reveals two distinct groups of patients. J. Med. Genet. 48, 1–9 (2011).
Vermeesch, J. R., Baten, E., Fryns, J.-P. & Devriendt, K. Ring syndrome caused by ring chromosome 7 without loss of subtelomeric sequences. Clin. Genet. 62, 415–417 (2002).
Le Caignec, C. et al. Inherited ring chromosome 8 without loss of subtelomeric sequences. Ann. Genet. 47, 289–296 (2004).
Rossi, E. et al. Duplications in addition to terminal deletions are present in a proportion of ring chromosomes: Clues to the mechanisms of formation. J. Med. Genet. 45, 147–154 (2008).
Knijnenburg, J. et al. Ring chromosome formation as a novel escape mechanism in patients with inverted duplication and terminal deletion. Eur. J. Hum. Genet. 15, 548–555 (2007).
Dennis, N., Coppin, B., Turner, C., Skuse, D. & Jacobs, P. A clinical, cytogenetic and molecular study of 47 females with r(X) chromosomes. Ann. Hum. Genet. 64, 277–293 (2000).
Lachlan, K. L., Youings, S., Costa, T., Jacobs, P. A. & Thomas, N. S. A clinical and molecular study of 26 females with Xp deletions with special emphasis on inherited deletions. Hum. Genet. 118, 640–651 (2006).
Therman, E., Laxova, R. & Susman, B. The critical region on the human Xq. Hum. Genet. 85, 455–461 (1990).
Mercer, C. L. et al. Detailed clinical and molecular study of 20 females with Xq deletions with special reference to menstruation and fertility. Eur. J. Med. Genet. 56, 1–6 (2013).
Uehara, S. et al. A Turner syndrome woman with a ring X chromosome [45, X/46, X, r(X)(p22.3q27)] whose child also had a ring X chromosome. Fertil. Steril. 67, 576–579 (1997).
Blumenthal, A. L. & Allanson, J. E. Turner syndrome in a mother and daughter: R(X) and fertility. Clin. Genet. 52, 187–191 (1997).
Matsuo, M. et al. Mother and daughter with 45, X/46, X, r(X)(p22.3q28) and mental retardation: Analysis of the x-inactivation patterns. Am. J. Med. Genet. 91, 267–272 (2000).
Sybert, V. P. Turner syndrome. In Management of Genetic Syndromes https://doi.org/10.1002/0471695998.mgs049 (2005).
Toniolo, D. X-linked premature ovarian failure: A complex disease. Curr. Opin. Genet. Dev. 16, 293–300 (2006).
Gravholt, C. H. et al. The changing face of Turner syndrome. Endocr. Rev. 44, 33–69 (2023).
Hook, E. B. Exclusion of chromosomal mosaicism: Tables of 90%, 95% and 99% confidence limits and comments on use. Am. J. Hum. Genet. 29, 94–97 (1977).
Migeon, B. R. et al. Severe phenotypes associated with inactive ring X chromosomes. Am. J. Med. Genet. 93, 52–57 (2000).
Tomkins, D. J., McDonald, H. L., Farrell, S. A. & Brown, C. J. Lack of expression of XIST from a small ring X chromosome containing the XIST locus in a girl with short stature, facial dysmorphism and developmental delay. Eur. J. Hum. Genet. 10, 44–51 (2002).
Prandstraller, D. et al. Turner’s syndrome: Cardiologic profile according to the different chromosomal patterns and long-term clinical follow-Up of 136 nonpreselected patients. Pediatr. Cardiol. 20, 108–112 (1999).
Van Dyke, D. L. et al. Ullrich-Turner syndrome with a small ring X chromosome and presence of mental retardation. Am. J. Med. Genet. 43, 996–1005 (1992).
Kuntsi, J., Skuse, D., Elgar, K., Morris, E. & Turner, C. Ring-X chromosomes: Their cognitive and behavioural phenotype. Ann. Hum. Genet. 64, 295–305 (2000).
Acknowledgements
The authors would like to acknowledge the efforts of the cytogenetics technologists and laboratory technicians of the Johns Hopkins Cytogenomics Laboratory. We want to thank Path Photo for their assistance in generating the figures. We also thank the IDDRC Tissue Culture Repository for their help. We thank the Hopkins Editorial Assistance Services Initiative.
Funding
The Johns Hopkins Cytogenomics Laboratory is an academic laboratory supported by the Johns Hopkins School of Medicine Department of Pathology, and KCV has research support from the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number K12-HD-000849.
Author information
Authors and Affiliations
Contributions
JBM developed the topic and wrote the manuscript. YSZ, VS, DMM, CA, AM, CC, and KCV read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
The Johns Hopkins Medicine Institutional Review Board (IRB) approved this HIPAA-compliant study with a waiver of consent. The secondary research study protocol was approved by the Johns Hopkins School of Medicine Institutional Review Board (IRB00345136).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Madison, A., Applegate, C., Stinnett, V. et al. Cytogenomic characterization of mosaic X-ring chromosomes in seventeen patients with Turner syndrome (TS)-42 years of experience at a single-site institution. Sci Rep 15, 12836 (2025). https://doi.org/10.1038/s41598-025-89843-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-89843-y
