
Abstract
The global incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) continues to rise, primarily driven by the escalating obesity epidemic worldwide. MASLD, a spectrum of liver disorders, can progress to more severe conditions, metabolic dysfunction-associated steatohepatitis (MASH), ultimately culminating in hepatocellular carcinoma (HCC). Given the complex nature of MASLD, there is an urgent need to develop robust risk prediction models and design specialized cancer screening initiatives tailored specifically for individuals with MASLD. This study aimed to identify genes exhibiting trending expression patterns that could serve as potential biomarkers or therapeutic targets. Our approach involved analyzing expression patterns across the five stages of MASLD development and progression. Notably, we introduced an innovative two-phase classification—MASLD occurrence and MASLD progression—instead of categorizing differentially expressed genes (DEGs) into multiple types. Leveraging LASSO regression models, we demonstrated their relatively strong capability to predict and distinguish both MASLD occurrence and progression. Furthermore, our analysis identified CYP7A1 and TNFRSF12A as significantly associated with the prognosis of MASLD progressing to HCC. These findings contribute to the understanding of gene expression dynamics in MASLD and may pave the way for the development of effective prognostic tools and targeted therapies in the realm of liver disease.
Similar content being viewed by others
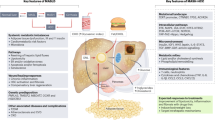


Introduction
The global prevalence of metabolic dysfunction-associated steatotic liver disease (MASLD) is on the rise, affecting nearly 1 billion individuals worldwide1. Surveys indicate varying MASLD prevalence rates from 23 to 32% across different geographical region, with the highest rates observed in South America and the Middle East, followed by Asia, the Americas, and Europe2,3. Moreover, modelling studies expect that the incidence of MASLD and consequent advanced liver disease will continue to increase internationally in the upcoming years4.
MASLD is notably prevalent among individuals burdened with obesity, with prevalence rates ranging from approximately 10–80%, depending on geographical location. Even higher frequencies are observed in severely obese patients5. Studies have indicated that obese individuals face an elevated risk of MASLD and subsequent HCC development6. Studies in mice have suggested that the accumulation of liver triacylglycerols and inflammation associated with obesity may contribute to the development of MASLD7,8. The substantial number of individuals with MASLD sets it apart from other liver diseases, highlighting the importance of identifying those at the greatest risk of developing progressive liver disease as the primary focus of clinical care9,10. As the rates of obesity continue to rise, MASLD has become the most common cause of liver dysfunction worldwide2. The clinical and economic burden brought by MASLD will become enormous. Therefore, a deeper understanding of the molecular mechanisms underpinning MASLD development resulting from obesity may offer a promising avenue for future therapeutic research11,12.
MASLD encompasses a spectrum ranging from simple steatosis (SS) to metabolic dysfunction-associated steatohepatitis (MASH). MASLD and MASH are novel designations. Previously, they were also referred to as non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH)13. MASH is characterized by hepatic steatosis, inflammation, hepatocellular injury, and varying degrees of fibrosis. Notably, MASLD, especially its more advanced form, MASH, constitutes a significant risk factor for hepatocellular carcinoma (HCC), alongside other factors such as chronic hepatitis B and C14. In contrast to SS, MASH extends beyond hepatic steatosis, involving hepatocyte ballooning and inflammatory infiltration, representing a more severe disease spectrum capable of progressing to advanced fibrosis or cirrhosis. Additionally, a significant proportion of MASH patients develop complications such as liver cirrhosis or hepatocellular carcinoma (HCC)15,16. The pathogenesis of HCC related to MASLD involves a complex interplay of mechanisms, including immune and inflammatory responses, DNA damage, oxidative stress, and autophagy9,17. While the incidence of HCC associated with MASLD is lower compared to other causes like hepatitis B14, the substantial prevalence of MASLD calls for immediate action to raise global awareness and address metabolic risk factors, thereby mitigating the looming burden of MASLD-related HCC. Given the escalating global rates of obesity, there is an anticipated increase in the future incidence of MASLD, particularly MASH-related HCC. Hence, the exploration of the exact molecular mechanisms underlying HCC associated with MASLD, along with the development of efficient cancer screening methods for individuals with MASLD, constitute vital endeavors in ongoing research.
MASLD is widely recognized as a complex pathological condition3. Personalized treatment strategies can be tailored by targeting specific stages of disease progression in MASLD patients. Extensive genome-wide association studies and bioinformatics approaches have unveiled genetic factors that play pivotal roles in the occurrence and progression of MASLD, along with potential therapeutic targets of clinical significance. Variations in PNPLA3, TM6SF2, MBOAT7, and GCKR have emerged as crucial genetic and epigenetic modifiers implicated in the pathogenesis of MASLD18. Zeng and colleagues have developed three classifiers based on the expression levels of nine genes (MT1G, MT1X, MT1F, MT1H, MT1M, FABP4, SPP1, MMP7, and CCL2) to distinguish different states of MASLD19. Through Weighted Gene Co-expression Network Analysis (WGCNA) and Protein-Protein Interaction (PPI) network approaches, Wu et al. have identified CYP7A1, GINS2, and PDLIM3 as potential candidate genes influencing MASLD prognosis and serving as therapeutic targets20. Importantly, these genes were also associated with the prognosis of HCC, indicating their underlying involvement in the progression from MASLD to HCC. Despite significant research efforts focused on understanding the molecular changes that occur during the progression of MASLD to HCC, uncertainties remain regarding the influence of distinct stages of obesity on the advancement of MASLD in patients. Consequently, further exploration in this area is warranted.
In this study, we acquired sample data spanning five stages of MASLD development and progression by merging various databases. These stages encompass Healthy Control (HC), Healthy Obesity (HO), Simple Steatosis (SS), Metabolic Dysfunction-associated Steatohepatitis (MASH), and Hepatocellular Carcinoma (HCC) resulting from MASH. Employing the limma differential analysis tool in conjunction with weighted gene co-expression network analysis, we identified genes that potentially play important roles in these processes. Through expression pattern cluster analysis based on temporal trends, we discerned diverse patterns of continuous gene expression from the onset of MASLD to its progression into HCC. Consequently, we emphasize the crucial importance of distinguishing between MASLD occurrence and MASLD progression when constructing predictive models, a distinction confirmed by our LASSO logistic analysis. In our LASSO logistic analysis, we established disease risk assessment models featuring 27 and 14 genes as gene signatures, respectively, for MASLD occurrence and progression. Furthermore, our findings indicate a significant association between CYP7A1 and TNFRSF12A with HCC prognosis in the MASLD progression model.
Results
Merging databases and initial analysis of differential gene expression
The study design is illustrated in Fig. 1, outlining the essential stages of the analysis. Additionally, Table 1 presents the data extracted from publicly available databases. Initially, we searched for datasets associated with the keywords “MASLD”, “NAFLD”, “MASH” and “NASH” (MASLD was called “NAFLD” and MASH was called “NASH” before), and identified three datasets (GSE48452, GSE89632, and GSE164760) from the GEO database based on their substantial sample size and clear distinction for each stage of MASLD. Additionally, these datasets were chosen primarily for their consistent microarray data format, facilitating the process of amalgamation. To counter potential batch effects arising from the integration of these datasets, we employed the “sva” package as a corrective tool. The effective mitigation of batch effects through this method, as illustrated in Fig. 2A and B, enabled us to successfully obtain a harmonized dataset.
The merged dataset incorporates a substantial mRNA expression matrix featuring samples from diverse liver conditions. It comprised 44 samples of healthy liver, 27 samples of obese liver, 34 samples of simple steatosis liver, 111 samples of MASH liver, and 53 samples of MASH-associated hepatocellular carcinoma liver. This dataset includes mRNA expression data for a total of 16,652 genes. To understand he distribution of samples within this dataset, a Uniform Manifold Approximation and Projection (UMAP) was conducted, as shown in Fig. 2C and D.
The “limma” package is a pivotal tool for conducting differential gene analysis, playing a crucial role in comparing mRNA levels among samples representing various liver disease states and healthy liver samples. This empowers the identification of differentially expressed genes (DEGs). By applying stringent statistical criteria (p < 0.05 and |log2FC| > 0.5), we identified a total of 260 genes that exhibited differential expression in the comparison between HO and HC (Fig. 2E). Additionally, 442 genes were identified in the comparison between SS and HC (Fig. 2F), 381 genes in the comparison between MASH and HC (Fig. 2G), and a substantial 866 genes in the comparison between HCC and HC (Fig. 2H). Under consistent screening criteria, the count of differentially expressed genes emerged as a reliable indicator of the variations between samples with distinct liver disease states and healthy samples. Our results illustrate a progressive increase in distinctions when comparing HO, SS, MASH, and HCC liver samples to HC liver samples.
Moreover, we identified 69 genes that consistently exhibit dysregulation across all four disease groups, demonstrating substantial distinctions when compared to healthy liver samples (Fig. 2I). These genes with consistent dysregulation could serve as potential biomarkers or therapeutic targets shared among the diverse liver conditions under investigation.

Flow chart of the whole procedures.

The identification of DEGs (Differentially Expressed Genes) across four distinct disease types using a consolidated matrix (comprising data from GSE48452, GSE89632 and GSE164760). (A, B) The “sva” tool was employed to merge the gene expression matrices from GSE48452, GSE89632, and GSE164760, effectively removing batch effects from the merged matrix. (A) Before removing batch effect, (B) after removing batch effect. (C, D) UMAP representation of (C) different disease stages and (D) different GEO cohorts. (E–H) A Volcano plot displays the DEGs between for each pairwise comparison, including HO vs. HC (E), SS vs. HC (F), MASH vs. HC (G), and HCC vs. HC samples (H). (I) The Venn diagram represents the overlap of DEGs among the four liver disease states, revealing a total of 69 genes that were consistently identified as differentially expressed across all conditions.
Detection of modules associated with the occurrence and progression of MASLD using WGCNA
We employed the WGCNA approach to establish co-expression networks and identify relevant modules associated with the occurrence and progression of metabolic dysfunction-associated steatotic liver disease (MASLD). To enhance the accuracy of WGCNA analysis, we initially eliminated an outlier sample through hierarchical clustering (Suppl. Figure 1 A). Subsequently, a scale-free network was developed by selecting a soft threshold value, leading to the identification of 14 distinct modules (Suppl. Figure 1B and Fig. 3A, B). The number of genes within each module can be found in Table 2, with detailed gene information provided in Suppl. Table 1.
Our study focuses on investigating the impact of obesity induced MASLD on disease progression, therefore the clinical changes in obese patients are our primary focus. It should be noted that the consistency of fibrosis grade and inflammation grade statistical criteria in the clinical information of different databases cannot be guaranteed due to their subjective nature, often relying on expert judgment. However, BMI data, as an objective measure, is consistent across different databases, making it more suitable as a clinically relevant indicator for obesity induced MASLD. BMI reflects patient obesity levels, with higher values indicating greater obesity. Consequently, we utilized BMI data from patients in GSE48452 and GSE89632 to identify co-expression modules associated with patient BMI. As depicted in Fig. 3C, among the 14 co-expression modules analyzed, the red module, the pink module, and the cyan module all have correlation coefficients with BMI above 0.4, demonstrating a relatively strong correlation with BMI. Subsequent reference to diagnosis information from patients in GSE164760 revealed that these BMI-related modules (red module and cyan module) displayed a significant association with MASH and HCC, suggesting their potential involvement in disease progression for MASLD patients towards MASH or HCC (Fig. 3C). However, a strong correlation between the pink module and the progression of MASH and HCC was not observed. Furthermore, gene set enrichment analysis indicated that the red and cyan co-expressed modules, compared to the pink module, showed highly significant p-values in gene sets associated with different liver disease states, especially MASH and HCC (Fig. 3D).
To validate the association between genes within the identified red and cyan modules and the development and progression of MASLD, we conducted functional enrichment analysis. The genes within the red module exhibited associations with critical biological processes, including inflammatory response, apoptosis, and the regulation of RNA polymerase II promoter transcription in GO-BP analysis (Suppl. Figure 2 A). According to KEGG analysis, these genes were associated with pathways such as the TNF signaling pathway, IL-17 signaling pathway, NF-kappa B signaling pathway, and various cancer-related pathways (Suppl. Figure 2B).
The genes identified within the cyan module were found to be associated with key functions such as fatty acid metabolism, amino acid transmembrane transport, and amino acid transport, as revealed in GO-BP analysis (Suppl. Figure 2 C). Additionally, in KEGG analysis, these genes were linked to crucial pathways including metabolic pathways, biosynthesis of cofactors, and glycolysis/gluconeogenesis (Suppl. Figure 2D). Notably, these biological processes and pathways have close links to the development and progression of MASLD21,22.
By cross-referencing the 69 differentially expressed genes with those identified within the red and cyan modules of WGCNA, we successfully identified 62 genes displaying distinctive expression patterns across various stages of liver disease, exhibiting a co-expression characteristic (Fig. 3E). In subsequent investigations, we classified these 62 genes as Differentially Expressed Genes (DEGs) potentially associated with the evolution and advancement of MASLD.

WGCNA applied for the integrated matrix. (A) Displays the clustering dendrogram and the identification of gene co-expression modules, each designated by a distinct color. (B) Features a hierarchical clustering dendrogram and a heatmap presenting co-expression module eigengenes. (C) Exhibits a heatmap representing the correlation between co-expression module eigengenes and sample characteristics. (D) Overall transcriptional regulation of gene modules upon HO, SS, MASH and HCC compared to baseline (HC). P-values are calculated by mean-rank gene set test using geneSetTest function as described in detail in methods. (E) Displays a Venn diagram revealing the shared genes among co-DEGs and the genes in the red and cyan modules, resulting in the identification of 62 DEGs.
The DEGs exhibit a distinctive “V-shaped” expression pattern as MASLD develops and progresses
To fully comprehend the functional significance of the 62 DEGs in relation to the development and progression of MASLD. Moreover, to elucidate their roles across various MASLD stages, we performed GO and KEGG analyses. Our results unveiled that the top 20 enriched GO terms were predominantly associated with inflammatory responses and cellular apoptosis, as illustrated in Suppl. Figure 3. Furthermore, the top 20 enriched KEGG pathways prominently featured the IL-17 signaling pathway, TNF signaling pathway, MAPK signaling pathway, and pathways in cancer, as displayed in Suppl. Figure 4.
To gain deeper insights into the gene expression dynamics among these 62 DEGs during the development and progression of MASLD, we conducted clustering analysis using the “mfuzz” R package (Fig. 4A). Intriguingly, we observed that the distinctive gene expression patterns spanning the entire occurrence and progression process of MASLD were not consistently continuous. Specifically, the initiation of MASLD (from HC to HO to SS) exhibited a relatively continuous pattern, whereas the progression phase (from SS to MASH to HCC) exhibited a distinct continuous pattern. The mRNA expression profiles of these 62 DEGs also exhibited a consistent trend across various liver samples, as showed in the heatmap (Suppl. Figure 5 A).
To validate this particular molecular expression pattern, we utilized the “STEM” software and applied the STEM algorithm to assess the significance of the expression patterns of these 62 DEGs. Our findings revealed that only the “V-shaped” molecular expression pattern held statistical significance (Fig. 4B), with 28 genes displaying this distinctive pattern, resulting in a p-value of 3.3*10^−22 (Fig. 4C).
Alterations in gene expression can significantly impact on cellular functions. In this study, we acquired 50 Hallmark gene sets and 186 KEGG gene sets from the GSEA website and utilized gene set variation analysis (GSVA) to calculate enrichment scores. This analytical approach enabled us to investigate the functional changes associated with the onset and progression of MASLD. Subsequently, we performed Mfuzz clustering analysis on the calculated gene set scores. Our results revealed intriguing patterns in the expression of various functionally associated gene sets. Notably, gene sets related to inflammation, such as IL6 JAK STAT3 SIGNALING and INFLAMMATORY RESPONSE, displayed distinct “V-shaped” or inverted “V-shaped” patterns (Suppl. Figure 5B, C, and Suppl. Table 2). Furthermore, similar patterns were observed in gene sets associated with apoptosis and metabolism among the Hallmark gene sets. Additionally, the KEGG gene sets exhibited a “V-shaped” pattern in gene sets linked to tumors and metabolism, such as P53 SIGNALING PATHWAY and PANCREATIC CANCER (Suppl. Figure 5D, E, and Suppl. Table 3). The findings suggest the presence of distinct triggering mechanisms underlying the development and progression of MASLD.
The aforementioned discoveries robustly substantiate the outcomes acquired through the comparative expression analysis of disease severity in progressive liver samples via “mfuzz” and “STEM”, affirming that the “V” expression pattern is not a random occurrence but rather indicates the presence of significant underlying influences worthy of further exploration.

Expression pattern cluster analysis of 62 DEGs. (A) Six distinct clusters were identified by Mfuzz clustering analysis using the R package “mfuzz”. (B) STEM clustering was performed using the Short Time series Expression Miner (STEM) software package. (C) Line graphs of significant profile eigengenes identified through the STEM software package.
GSEA for the progression of MASLD
We utilized closely correlated gene information associated with hepatic diseases, as compiled by I. Graupera et al.23, to identify nine functionally associated gene sets: Genetic Factors, Oxidative Stress, Fibrosis and Resolution, Inflammatory Response, Apoptosis of hepatocytes, Apoptosis of Hepatic Stellate Cells, Angiogenesis, Cellular Senescence, and Hepatocellular Carcinoma. Detailed information about these gene sets is provided in Table 3. Our objective was to investigate the alterations in these functions throughout the development and progression of MASLD. Subsequently, we applied the GSEA method to analyze MASLD in five different states and retained all significant normalized enrichment scores (NES) (Fig. 5). This analysis revealed a distinct “V-shaped” pattern within the aforementioned gene sets related to liver diseases. This pattern, characterized by a turning point at SS stage, unveiled a gradual decline in Fibrosis and Resolution, Inflammatory Response, Hepatocyte Apoptosis, Hepatic Stellate Cell Apoptosis, Angiogenesis, Cellular Senescence, and Hepatocellular Carcinoma before the turning point, followed by a complete reversal of these changes after the turning point. Conversely, Genetic Factors and Oxidative Stress exhibited a consistent downward trend throughout the entire progression of MASLD. The findings also demonstrated the distinct functional expression between the stages of MASLD development (pre-SS state) and MASLD progression (post-SS state), thereby providing further substantiation for the disparate triggering mechanisms underlying MASLD occurrence and development.
Furthermore, we observed that the nine gene sets under consideration did not exhibit significant enrichment when comparing HO and SS, suggesting a higher functional similarity between the samples from HO and SS. This observation was further supported by the disease stage-dependent molecular expression patterns, as the clustering result of the 62 DEGs consistently demonstrated a smaller slope of the line segment between HO and SS (Fig. 4A).

Enrichment analyses of MASLD related networks. Gene sets that exhibit significant enrichment are color-coded based on their normalized enrichment scores (NES). Only NES values from tests with a p-value less than 0.05 and a False Discovery Rate (FDR) below 0.25 are presented. Upregulated gene sets are prominently in red, while downregulated sets are distinctly highlighted in blue.
Development of a predictive model for MASLD incidence (from HC to HO to SS)
Our study focuses on establishing a predictive model for the incidence of MASLD using the HC-HO-SS dataset. Observing a distinctive “V-shaped” expression pattern within gene clusters, we propose that the development and progression of MASLD involve fundamentally distinct gene expression profiles. Discriminating between these two processes and constructing predictive or diagnostic models is crucial for improving accuracy and reducing errors. To address these challenges, we employed the Least Absolute Shrinkage and Selection Operator (LASSO) regression method, known for preventing overfitting and screening more representative parameters through regularization. Therefore, we applied LASSO regression to develop a risk prediction model for 62 DEGs, with a particular focus on refining the selection to identify key genes pivotal to MASLD progression.
In the process of MASLD development, we employed a cross-validation with λ value of 0.03830797, identifying 27 genes significantly associated with this progression (Fig. 6A, B). These 27 genes exhibited significant associations with the development of MASLD (Fig. 6C), along with notable correlations among them (Fig. 6D). For detailed information on these 27 genes, including their presence in relevant literature reports, are provided in Suppl. Table 4. Subsequently, we utilized the “predict” function within the “glmnet” package to calculate a risk score based on the LASSO model, effectively distinguishing between HC, HO, and SS samples (Fig. 6E). This model holds significant promise for predicting MASLD incidence accurately and facilitating early intervention.
Using the area under the ROC curve as a metric for predictive accuracy, we observed that the relative accuracy of HC samples compared to HO samples was 0.972, SS samples compared to HC samples was 0.988, and HO samples compared to SS samples was 0.879 (Fig. 6F). Notably, despite our prior findings indicating a high similarity between the HO and SS states, our model demonstrated a significant level of discrimination between these two states. To evaluate the predictive capability of our model, we collected HC and SS samples from the GSE66676 cohort, resulting in a moderate predictive performance with an accuracy rate of 0.617 (Fig. 6G, H). Additionally, when examining HC and HO samples from the GSE126848 cohort, our model successfully differentiated patients with HC from those with HO based on their risk scores (Fig. 6I), achieving a prediction accuracy rate of 0.732 (Fig. 6J).

The development and validation process of prognostic risk score model for predicting the occurrence of MASLD. (A, B) MASLD onset-related genes were identified through LASSO logistic regression analysis. The optimal penalty parameter lambda was selected using 10-fold cross-validation. (C) A heatmap visualizes the variation in risk scores across different samples based on the expression of 27 genes. (D) Heatmaps display the correlations in gene expression among a selected set of 27 genes. (E) Risk scores were compared across various MASLD stages, including healthy controls (HC), healthy obese individuals (HO), and those with simple steatosis (SS). (F) ROC analysis was conducted to assess the predictive value of risk score. (G) A comparison of risk scores between HC and SS samples was performed using the GSE66676 dataset. (H) ROC analysis evaluated the predictive value of risk score in the GSE66676 dataset. (I) Risk scores were compared between HC and HO samples using the GSE126848 dataset. (J) ROC analysis assessed the predictive value of risk score in the GSE126848 dataset.
Developing a predictive model for the progression of MASLD (from SS to MASH to HCC)
Our research is dedicated to understanding the progression of MASLD, particularly the transition from SS to MASH, a critical step associated with a heightened risk of HCC. To predict the risk of MASLD progression, we employed LASSO logistic regression analysis, focusing on the identification of DEGs pivotal in this process. Through cross-validation and selection of λ = 0.04551661 (Fig. 7A, B), we have identified 14 genes that exhibit robust associations with the progression of MASLD. For more comprehensive understanding of these 14 genes, along with references in relevant literature, please refer to Suppl. Table 5.
The expression levels of these 14 genes exhibited a continuous distribution pattern across the spectrum of MASLD progression (Fig. 7C), and a significant correlation was observed among them (Fig. 7D). Through the application of risk score calculation, our model effectively differentiated samples representing SS, MASH, and HCC (Fig. 7E). The accuracy for distinguishing SS samples from MASH samples reached 0.878, while the accuracy for distinguishing SS samples from HCC samples was notably high at 0.984. Furthermore, the accuracy for distinguishing MASH samples from HCC samples was measured at 0.856 (Fig. 7F).
To evaluate the predictive capability of our model for MASH risk, we applied it to compute risk scores for SS and MASH samples in the GSE126848 and GSE167523 cohorts (Fig. 7G, I), achieving prediction accuracies of 0.671 and 0.845, respectively (Fig. 7H, J). These findings substantiate that our model exhibits a robust ability to predict MASLD progression.
While we were unable to obtain additional patient samples for assessing our model’s capacity to predict the progression of MASLD to HCC, our analysis of STAM mouse (a mice model of MASH induced by streptozotocin and high-fat diet24) samples from the GSE83596 cohort revealed a significantly elevated risk score in samples from mice that progressed from MASH to HCC, compared to those that did not (Suppl. Figure 6). Studies revealed that the fibrosis grade in MASLD serves as a highly reliable indicator of its potential for carcinogenesis25. Applying our risk-prediction model to calculate risk scores for samples from cohorts GSE162694, GSE130970, and GSE135251, we observed a significant correlation between the model and fibrosis grade. Specifically, patients with higher risk scores exhibited a greater likelihood of having higher fibrosis grades (Fig. 7K).
Upon conducting correlation analysis with additional clinical information from the GSE130970 and GSE135251 cohorts, a significant association emerged between the risk score and the NAFLD activity score (NAS), signifying those patients with higher risk scores exhibited elevated NAS levels (Fig. 7L). Furthermore, within the GSE130970 cohort, a correlation was identified between risk scores and cytological ballooning grades, suggesting that patients with higher risk scores displayed more severe cytological ballooning grades (Fig. 7M). These findings demonstrate the effectiveness of our risk prediction model in accurately predicting disease progression in MASLD patients.

Construction and validation of prognostic risk score model of MASLD progression. (A, B) MASLD progression related genes were identified through LASSO logistic regression analysis, and the optimal penalty parameter lambda was determined using 10-fold cross-validation (C) A heatmap depicts the variation in risk scores among samples based on the expression of 14 genes. (D) Heatmaps display gene-gene expression correlations for the chosen set of 14 selected genes. (E) Risk scores were compared across different stages of MASLD, including simple steatosis (SS), metabolic dysfunction-associated steatohepatitis (MASH), and hepatocellular carcinoma (HCC). (F) ROC analysis was conducted to evaluate the predictive value of the risk score. (G) The comparison of risk scores between SS and MASH samples was performed in the GSE126848 dataset. (H) ROC analysis assessed the predictive value of the risk score in the GSE126848 dataset. (I) The comparison of risk scores between SS and MASH samples was conducted in the GSE167523 dataset. (J) ROC analysis evaluated the predictive value of the risk score in GSE167523 dataset. (K) The risk score was examined in different fibrotic stages of three datasets (left: GSE162694; middle: GSE130970; right: GSE135251). (L) Risk score in various NAS were explored in two datasets (left: GSE130970; right: GSE135251). (M) Risk score was investigated in different cytological ballooning grades within the GSE130970 dataset.
The risk score model is associated with the prognosis of hepatocellular carcinoma (HCC)
In our subsequent investigation, our objective was to explore the potential significance of the risk prediction model for MASLD progression, established through LASSO logistic regression analysis, in the context of HCC progression. We evaluated the risk profiles of 424 HCC patients from the TCGA dataset, classifying them into high and low-risk groups based on their respective risk scores at the median threshold. Following this categorization, we analyzed the prognostic survival data for these two patient cohorts. However, our findings revealed no statistically significant difference in prognosis between the high-risk and low-risk groups (Fig. 8A).
The severity of liver fibrosis plays a crucial role in determining the prognosis of individuals with liver disease. Initially, we observed a consistent association between patient risk score and the grade of fibrosis in both the HCC and MASLD cohorts. Specifically, patients with higher risk scores in the HCC cohort exhibited elevated levels of fibrosis (Fig. 8B). Subsequently, we interrogated whether the severity of fibrosis in TCGA-HCC samples was correlated with the prognosis of patients. When comparing the prognoses of HCC patients with varying degrees of fibrosis, we discovered no significant differences (Suppl. Figure 7 A). Consistent with our earlier findings, it is evident that the MASLD progression model plays a pivotal role in determining fibrosis grades in both MASLD and HCC samples. However, it is noteworthy that fibrosis grade alone does not serve as an effective predictor for prognosis in HCC patients. Nevertheless, we have identified that combining a patient’s risk score with their fibrosis grade provides a robust prognosis prediction. Notably, patients with low-risk scores and low grades of fibrosis exhibit better prognoses compared to other individuals (Fig. 8C).
The predictive model for the progression of MASLD comprised 14 genetic features. Notably, our analysis revealed a significant correlation between the expression levels of CYP7A1 and TNFRSF12A and the prognosis of HCC patients (Suppl. Figure 7B). Specifically, decreased expression of CYP7A1 were associated with poor prognosis in HCC patients (Fig. 8D), while elevated expression levels of TNFRSF12A were linked to unfavorable outcomes in HCC patients (Fig. 8E). Importantly, a discernible trend emerged during the progression of MASLD, there was a declining in CYP7A1 expression level (Fig. 8F), whereas TNFRSF12A exhibited an increasing trend in expression (Fig. 8G). These findings suggest that CYP7A1 acts as a tumor suppressor while TNFRSF12A functions as a carcinogenic factor in MASLD progression, and their respective expression levels appear to have a significant impact on the prognosis of HCC patients.

The performance evaluation of the prognostic prediction model risk score in the TCGA-HCC cohort. (A) Overall survival analysis was performed to compare low-risk patients with high-risk patients. (B) The risk score was examined across different fibrotic stages of HCC patients. (C) Overall survival analysis was performed, comparing samples characterized by both low risk and low fibrotic stage against other groups. (D) Expression analysis of CYP7A1 during MASLD progression. (E) Expression analysis of TNFRSF12A during the progression of MASLD. (F) Comparative analysis of overall survival in patients with low and high expression levels of CYP7A1. (G) Comparative analysis of overall survival in patients with low and high expression levels of TNFRSF12A.
Discussion
We acknowledge a notable limitation in our study: MASLD does not exclusively result from obesity. However, given the rising global prevalence of obesity, there is a likelihood that MASLD induced by obesity may progress into more severe forms26. Consequently, our research predominantly focuses on the progression of MASLD primarily attributed to obesity.
The progression from MASLD to MASH could be a prolonged journey, during which dietary enhancements or pharmaceutical interventions might have the potential to halt or even reverse the condition. Nevertheless, our emphasis is on understanding the changes in gene expression that may occur within liver tissue throughout the development of MASLD, as well as uncovering the potential triggers that drive MASLD to advance into HCC. Therefore, we have a distinct interest in the ongoing deterioration or progression of MASLD, recognizing that it may not strictly adhere to an ideal continuous process in reality.
Through a comparative analysis of MASLD stages and healthy normal samples, coupled with the application of WGCNA, we have successfully identified 62 genes exhibiting differential expression. These genes not only exhibit distinct expression patterns across various MASLD stages when contrasted with healthy liver samples but also display notable correlations within the scale-free network identified via WGCNA screening. This screening strategy, known for its efficiency in selecting representative genes, has been widely employed in previous studies27,28. Subsequent functional enrichment analysis has provided insights into involvement of these 62 genes in critical processes, including inflammatory response, apoptosis, carcinogenesis, metabolism, and others closely tied to the development of MASLD.
The exploration of genetic data can significantly enhance the design of novel therapeutic strategies for MASLD and facilitate the identification of diagnostic and prognostic biomarkers in clinical applications18. Initially, we hypothesized that the progression of HCC induced by MASLD might undergo five stages: healthy, obesity, simple steatosis, metabolic dysfunction-associated steatotic liver disease (MASH), and hepatocellular carcinoma (HCC). Our molecular expression analysis spanning these disease stages revealed a crucial divergence in the expression patterns of the 62 differentially expressed genes, specifically occurring at the simple steatosis (SS) stage. This finding suggests the presence of two distinct expression patterns during the initiation and progression of MASLD. This result indicates that predicting events further into the future may lead to an exponential decline in accuracy. Based on this understanding, we adopted a compromise approach in constructing the prediction models by shortening the prediction time horizon to enhance precision. Specifically, we developed two independent prediction models. One pattern of continuous molecular expression was evident during the HC-HO-SS stage, while another continuous molecular expression pattern emerged during the SS-MASH-HCC stage. Despite this, predicting the progression from SS to HCC remains a highly challenging task. Consequently, we incorporated non-genetic factors, such as fibrosis grade and inflammation level, to risk-adjust the model. Subsequently, we observed similar alterations in the functions related to the development and progression of MASLD, indicating distinct triggering mechanisms for its occurrence and development. Interestingly, the GSEA results revealed no significant differences between HO and SS, indicating that stages prior to SS are indeed less problematic. This finding may be of considerable importance. Consequently, in our subsequent studies, we focused more on the progression from SS to HCC. Additionally, this result suggests that future research on MASLD progression should place greater emphasis on stages following SS, while potentially de-emphasizing the pre-SS stages.
Our findings intriguingly support the widely acknowledged “two-hit theory”: The initial “first hit” involves a disruption in liver lipid metabolism, leading to an imbalance in lipid influx or clearance. During this phase, steatosis may be reversible and not necessarily result in permanent liver damage. The subsequent “second hit” entails an inflammatory storm, potentially triggered by oxidative stress, lipid peroxidation, and cytokine activity29. Although the occurrence of secondary hepatitis is low, its impact is highly toxic and irreversible. Lobular inflammation directly precipitates ballooning degeneration and perisinusoidal fibrosis, promoting cell apoptosis and hepatocyte death. This cascade ultimately culminating in scar formation and the development of MASH30,31. We postulate that the “first hit” occurs during the onset of MASLD, during which lipid metabolism dysfunction gradually intensifies, while the extent of inflammatory reactions and cell apoptosis tends to decrease, contrary to expectations (Suppl. Figure 5). The concept of the “Second hit” appears to serve as a marker for the progression of MASLD. Our results reveal that the activation of inflammatory responses, apoptosis pathways, tumor signaling, and disease progression initiates after the Simple Steatosis (SS) stage. This indicates the potential triggering factors for the development of MASLD and eventually leads to MASH (Suppl. Figure 5 and Fig. 5). Therefore, our findings are significant in shedding light on the factors that initiate and propel the onset and progression of MASLD. Nevertheless, further research efforts are needed to uncover the precise changes involved in this process.
During the screening for key genes, we did not identify any genes exhibiting consistent trends throughout the development of MASLD. To construct a precise linear risk prediction model, we segmented entire process of MASLD occurrence and progression based on the overall distribution trends of genes. Based on the establishment of the LASSO regression risk prediction model, we derived the prediction models for the occurrence and progression of MASLD. The prediction model for the progression of MASLD, in particular, constitutes the core of our research. It is capable of assessing the risk of disease progression for patients in accordance with gene signatures and also determining whether patients are at risk of developing hepatocellular carcinoma. Simultaneously, the risk prediction model for MASLD can adopt individualized treatment regimens for MASLD patients at diverse risk levels through risk stratification. The significance of our research lies in identifying appropriate biomarkers, which is of paramount importance for the early detection and diagnosis of MASLD as well as its progression. Nevertheless, this has resulted in the predictive model we constructed not attaining the originally intended broader predictive capacity. Prior to the application of our predictive model, it is essential to ascertain whether the predicted group is at the stage prior to SS or subsequent to it. Once this is determined, our model can achieve a certain level of risk prediction efficacy. In comparison to existing models outlined in the literature, some of which can only forecast a single disease (as demonstrated in a previous article19), our model not only attains superior accuracy but also offers refinement. The advantage of our MASLD occurrence prediction model lies in its ability to predict or distinguish among obese patients and adapt to individuals with simple steatosis or both conditions.
The degree of liver fibrosis and inflammation and morphology of hepatocytes serve as crucial indicators of the transition from fatty liver to HCC25. Zhang et al. employed WGCNA to predict HCC in MASLD and discovered that the differentially expressed genes (DEGs) were significantly enriched in the inflammation-related pathway32. Intriguingly, we also identified that the inflammatory signaling pathway could exert an effect during the transformation from MASLD to HCC. To assess disease severity, the fibrosis degree, the NAFLD Activity Score (NAS), and the extent of cytological ballooning are commonly utilized33. However, clear definitions of key histopathological components have been lacking, resulting in significant interobserver variations in diagnosis9. Therefore, objective and quantifiable gene expression indicators will be a better choice for clinical diagnosis and prognosis prediction.
We have carried out more work on the stage of the transformation from MASLD to HCC. Although the number of SS patients developing HCC is relatively small, these patients have to confront greater treatment pressure. If appropriate methods can be identified to predict the risk of cancer, early intervention measures can be provided for patients as soon as possible. Regarding the predictive model we established for MASLD progression, we observed its robust discriminative capability in evaluating the degree of fibrosis, NAS, and cytological ballooning. This suggests its potential effectiveness in predicting cancer risk among MASLD patients, possibly offering a more objective and comprehensive risk assessment than some complex clinical indicators. When assessing the predictive performance of the model, we discovered that for STAM mice, the risk score of mice with tumors was significantly higher than that of mice without tumors, demonstrating the applicability of our predictive model in the mouse model and the potential for its future application in clinical trials. Although cirrhotic MASLD patients belong to the high-risk group of progressing to HCC, non-cirrhotic patients also require a low-cost screening protocol to predict their risk of cancer. The SS-MASH-HCC risk prediction model constructed by us based on 14 gene signatures also possesses a certain predictive ability for this part of patients, and it is expected that a cost-effective risk prediction tool can be developed in the future.
The MASLD progression model, which was constructed using a gene signature containing 14 genes, did not exhibit strong predictive capabilities for patient prognosis within the TCGA liver cancer sample cohort. This may be because not all patients in the sample cohort are MASH-induced HCC patients. Nevertheless, our model was successfully validated for its capacity to distinguish the extent of fibrosis in liver cancer patients. The existing literature indicates that the degree of fibrosis is correlated with the prognosis outcomes of patients with liver cancer; however, when interrogating the TCGA cohort, we were not able to observe their relationship very well. Additionally, we observed that patients with both low risk and minimal liver fibrosis levels had more favorable prognoses compared to other patient groups. The fibrosis-hypoxia-glycolysis-immune prognostic model constructed by Li et al. indicates a correlation with the prognosis of HCC34. This implies that establishing models combining other factors with fibrosis might be more beneficial for the prognosis correlation of patients with liver cancer. When examining the individual 14 genes with the gene signature of the MASLD progression model, we discovered that CYP7A1 and TNFRSF12A displayed significant associations with the prognosis of HCC.
CYP7A1, also known as cholesterol 7α-hydroxylase, serves as the limiting enzyme in the classical bile acid synthesis pathway. Previous research has highlighted its essential role in enterohepatic circulation and its close association with cholesterol-bile acid metabolism and various liver conditions35,36. Bioinformatics studies have noted distinct expression patterns of CYP7A1 in both MASLD and HCC20,37. Furthermore, Increased CYP7A1 expression and bile acid synthesis ameliorated hepatic inflammation and fibrosis, indicating that CYP7A1 has anti-tumor effects38. CYP7A1 increases the accumulation of primary bile acids, which in turn promotes the expression of CXCL16, NKT immune cell aggregation, and has an inhibitory effect on tumors39. Our research indicates that in the HC-HO-SS stage, the expression level of CYP7A1 gradually rises, which is in accordance with the results discovered by Govaere et al.40. Moreover, we have also identified the expression tendency of CYP7A1 during the progression from MASLD to HCC. During the development of MASLD to HCC, the expression of CYP7A1 gradually decreases, and HCC patients with higher CYP7A1 levels often have a better prognosis (Fig. 8). These results all indicate that CYP7A1 plays an important role in MASLD and HCC, but the mechanism is still uncertain. Therefore, further investigations are needed to fully understand the role of CYP7A1 in the context of MASLD and MASLD-induced HCC.
On the other hand, TNFRSF12A, or tumor necrosis factor receptor superfamily member 12 A, has been implicated in the disease severity due to its abnormal overexpression41. This suggests TNFRSF12A’s potential as an indicator of disease aggressiveness and poorer prognosis. Studies have shown that knocking down TNFRSF12A inhibits the proliferation and migration of hepatocellular carcinoma cells42. Our findings are consistent with these results, demonstrating an increase in TNFRSF12A expression as MASLD progresses to HCC. HCC patients with lower levels of TNFRSF12A have better prognosis (Fig. 8). Therefore, we propose that TNFRSF12A, along with CYP7A1, may play a crucial role in the development of HCC resulting from MASLD.
Our study highlighted the significant involvement of CYP7A1 and TNFRSF12A in the MASLD-induced HCC. These findings suggest that these two genes hold promise as targets for future therapeutic interventions in MASLD and liver cancer.
Materials and methods
Dataset acquisition and preprocessing
Datasets related to MASLD were retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), including GSE89632, GSE48452, GSE164760, GSE66676, GSE126848, GSE162694, GSE130790, GSE135251, and GSE167523. Gene annotation for microarray data was performed using the GEO query package in R (version 4.2.0). For multiple probes corresponding to a single gene, average values were calculated. High-throughput sequencing data in FPKM-formatted data were selected for statistical analysis, with data standardization applied using the “limma” package. The HCC data was obtained from the Cancer Genome Atlas (TCGA) database via the UCSC Xena website. Dataset details, including sample information, are presented in Table 1.
Sample consolidation included 14 healthy liver samples, 27 obese patient liver samples, 14 SS liver samples, and 18 MASH liver samples from GSE48452; 24 healthy liver samples, 20 SS liver samples, and 19 MASH liver samples from GSE89632; 6 healthy liver samples, 74 MASH liver samples, and 53 HCC samples from GSE164760. Compared to the SS sample, healthy obese individuals are generally not considered to be in a state of disease. In this group, the liver does not show fibrosis or fatty degeneration. Batch effects in the merged dataset were removed using the sva tool package43.
Differential expression analysis and UMAP analysis
Differential Expressed Genes (DEGs) were identified using the “limma” package44, filtering for absolute Log2 fold-change (Log2FC) > 0.5 and adjusted p-value < 0.05. Visualization was performed with ggplot for a volcano plot. Uniform Manifold Approximation and Projection (UMAP) of combined expression profiles utilized the “umap” package in R (version 4.2.0), and results were graphically presented with the ggplot2 plotting tool (https://ggplot2.tidyverse.org/). A Venn diagram depicting dataset overlap was generated using the online tool Venny 2.1 (https://bioinfogp.cnb.csic.es/).
Weighted gene co-expression network analysis (WGCNA)
WGCNA was applied to identify gene clusters associated with MASLD development and progression, using the WGCNA package45,46. Genes were filtered based on median absolute deviation (MAD) in the top 75% and MAD greater than 0.01, resulting in 12,489 genes. The beta parameter value was assessed to establish a scale-free network topology, choosing a beta value of 5. The dynamic tree cut algorithm was applied to the dendrogram for module identification, configuring each module to contain 60 genes and implementing a height cutoff of 0.2 to merge similar modules. For a detailed explanation of the WGCNA algorithm, refer to Zhang Bin et al.47.
Enrichment analysis (GO, KEGG, GSVA, GSEA, gene set enrichment analysis)
To elucidate the functional implications of the gene set within cellular and pathway contexts, DAVID (https://david.ncifcrf.gov/) was utilized for Gene Ontology (GO) term enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway functional enrichment analysis48. Statistical significance was set at a p-value threshold of < 0.05 and the result was corrected using Bonferroni. Visualization of the outcomes was accomplished through histogram plots using the R package “ggplot2.” Enrichment chord diagrams were created using the R package “GOplot.”
Gene Set Enrichment Analysis (GSEA) was employed to evaluate statistically significant differences in gene sets between two biological states. Gene sets in GMT format were obtained from the MsigDB database (https://www.gsea-msigdb.org/gsea/). Additionally, Gene Set Variation Analysis (GSVA) computed enrichment scores for pathways from the MSigDB database, based on the gene expression matrix of each sample. The gene set enrichment analysis utilized the “limma” package’s “gene-SetTest” function. We employed this function to analyze the enrichment extent of differential genes between the WGCNA module and HC as well as the four disease states, where the differential genes originated from the analysis outcomes of “limma”.
Expression pattern cluster analysis
Expression patterns across MASLD stages were analyzed using the “mfuzz” R package for gene clustering49, and relationships were further explored with the “STEM” software50.
Analysis of the LASSO logistic regression model
The LASSO model was constructed and validated using the “glmnet” package in R. Leveraging the coefficients (β) from the LASSO regression model, a linear combination with mRNA expression levels was established to create predictive or diagnostic gene features51. In the LASSO regression analysis, the Lambda value influences the selection of variables in the model. A smaller Lambda leads to a relatively complex model, while a larger Lambda results in an overly simplistic model. Hence, an appropriate Lambda value needs to be selected through suitable methods (such as cross-validation). The risk score computation is as follows: Risk Score = (β1 * Gene1) + (β2 * Gene2) + (β3 * Gene3) + . + (βN * GeneN). A model for distinguishing various MASLD states is established based on this risk score. Classifier effectiveness was assessed using metrics such as mean squared error (MSE), accuracy, sensitivity (Se), specificity (Sp), positive predictive value (PPV), negative predictive value (NPV), and the area under the receiver operating characteristic (ROC) curve52. ROC curves were generated and compared utilizing the “pROC” package in R (version 4.2.0).
Statistical analysis
Statistical analysis was carried out using R (version 4.2.0). Gene expression levels were compared between groups using an unpaired t-test. Survival analysis utilized the “survival” and “survminer” R packages (https://rpkgs.datanovia.com/survminer/index.html). Survival curves were generated using the Kaplan-Meier method, and the log-rank test was employed to ascertain significant differences in survival. Gene correlations were assessed using Pearson’s correlation test. A significance level of P < 0.05 was deemed as statistically significant.
Conclusions
Bioinformatics methodologies were employed to analyze expression patterns related to MASLD onset and progression. Distinct molecular expression patterns were identified, elucidating fundamental principles governing MASLD emergence and progression. A predictive model for MASLD onset and progression was developed and validated, showing promise for aiding clinical diagnoses of MASLD and HCC.
Data availability
The datasets generated and/or analysed during the current study are available in the GEO and TCGA repository, https://www.ncbi.nlm.nih.gov/geo/, https://www.cancer.gov/ccg/research/genome-sequencing/tcga. The accession numbers of the GEO datasets used in this study are: GSE48452, GSE89632, GSE164760, GSE66676, GSE126848, GSE167523, GSE162694, GSE130970, GSE135251 and GSE83596.
References
Zeigerer, A. NAFLD–A rising metabolic disease. Mol. Metab. 50, 101274 (2021).
Younossi, Z. et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15(1), 11–20 (2018).
Younossi, Z. M. et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64(1), 73–84 (2016).
Estes, C. et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 69(4), 896–904 (2018).
Polyzos, S. A., Kountouras, J. & Mantzoros, C. S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 92, 82–97 (2019).
Milic, S., Lulic, D. & Stimac, D. Non-alcoholic fatty liver disease and obesity: Biochemical, metabolic and clinical presentations. World J. Gastroenterol. 20(28), 9330–9337 (2014).
van Diepen, J. A. et al. Hepatocyte-specific IKK-beta activation enhances VLDL-triglyceride production in APOE*3-Leiden mice. J. Lipid Res. 52(5), 942–950 (2011).
Wu, Y. et al. Insulin-like growth factor-I regulates the liver microenvironment in obese mice and promotes liver metastasis. Cancer Res. 70(1), 57–67 (2010).
Burt, A. D., Lackner, C. & Tiniakos, D. G. Diagnosis and Assessment of NAFLD: Definitions and histopathological classification. Semin. Liver Dis. 35(3), 207–220 (2015).
Schuster, S., Cabrera, D., Arrese, M. & Feldstein, A. E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 15(6), 349–364 (2018).
Anstee, Q. M., Reeves, H. L., Kotsiliti, E., Govaere, O. & Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 16(7), 411–428 (2019).
Swinburn, B. A. et al. The global obesity pandemic: Shaped by global drivers and local environments. Lancet 378(9793), 804–814 (2011).
Fukunaga, S., Mukasa, M., Nakano, D., Tsutsumi, T. & Kawaguchi, T. Changing from NAFLD to MASLD: Similar cumulative incidence of reflux esophagitis between NAFLD and MASLD. Clin. Mol. Hepatol. 30(1), 121–123 (2024).
Yang, J. D. et al. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 16(10), 589–604 (2019).
Paternostro, R. & Trauner, M. Current treatment of non-alcoholic fatty liver disease. J. Intern. Med. 292(2), 190–204 (2022).
Powell, E. E., Wong, V. W. & Rinella, M. Non-alcoholic fatty liver disease. Lancet 397(10290), 2212–2224 (2021).
Huang, D. Q., El-Serag, H. B. & Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 18(4), 223–238 (2021).
Eslam, M., Valenti, L. & Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 68(2), 268–279 (2018).
Zeng, F. et al. Predicting non-alcoholic fatty liver disease progression and immune deregulations by specific gene expression patterns. Front. Immunol. 11, 609900 (2020).
Wu, C. et al. Bioinformatics analysis explores potential hub genes in nonalcoholic fatty liver disease. Front. Genet. 12, 772487 (2021).
Zou, Z. Y., Wong, V. W. & Fan, J. G. Epidemiology of nonalcoholic fatty liver disease in non-obese populations: Meta-analytic assessment of its prevalence, genetic, metabolic, and histological profiles. J. Dig. Dis. 21(7), 372–384 (2020).
Cobbina, E. & Akhlaghi, F. Non-alcoholic fatty liver disease (NAFLD) - pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 49(2), 197–211 (2017).
Graupera, I. et al. Molecular characterization of chronic liver disease dynamics: From liver fibrosis to acute-on-chronic liver failure. JHEP Rep. 4(6) (2022).
Takakura, K. et al. Characterization of non-alcoholic steatohepatitis-derived hepatocellular carcinoma as a human stratification model in mice. Anticancer Res. 34(9), 4849–4855 (2014).
Wong, V. W. S., Adams, L. A., de Lédinghen, V., Wong, G. L. H. & Sookoian, S. Noninvasive biomarkers in NAFLD and NASH—current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 15(8), 461–478 (2018).
Grohmann, M. et al. Obesity drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 175(5), 1289–1306e1220 (2018).
Hong, S-Y. et al. Identification of the pivotal role of SPP1 in kidney stone disease based on multiple bioinformatics analysis. BMC Med. Genom. 15(1) (2022).
Du, J. et al. Identification of prognostic model and biomarkers for cancer stem cell characteristics in glioblastoma by network analysis of multi-omics data and stemness indices. Front. Cell. Dev. Biol. 8(2020).
Borrelli, A. et al. Role of gut microbiota and oxidative stress in the progression of non-alcoholic fatty liver disease to hepatocarcinoma: Current and innovative therapeutic approaches. Redox Biol. 15, 467–479 (2018).
Brunt, E. M. Histopathology of non-alcoholic fatty liver disease. Clin. Liver Dis. 13(4), 533–544 (2009).
Day, C. P. & James, O. F. W. Steatohepatitis: A tale of two hits? Gastroenterology 114, 842–845 (1998).
Zhang, Z., Wang, S., Zhu, Z. & Nie, B. Identification of potential feature genes in non-alcoholic fatty liver disease using bioinformatics analysis and machine learning strategies. Comput. Biol. Med. 157, 106724 (2023).
Kleiner, D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41(6), 1313–1321 (2005).
Li, Q. et al. The impact of liver fibrosis on the progression of hepatocellular carcinoma via a hypoxia-immune-integrated prognostic model. Int. Immunopharmacol. 125(Pt A), 111136 (2023).
Chambers, K. F., Day, P. E., Aboufarrag, H. T. & Kroon, P. A. Polyphenol effects on cholesterol metabolism via bile acid biosynthesis, CYP7A1: A review. Nutrients 11(11) (2019).
Farooqui, N., Elhence, A. & Shalimar. A current understanding of bile acids in chronic liver disease. J. Clin. Exp. Hepatol. 12(1), 155–173 (2022).
Bao, H. et al. Integrated bioinformatics and machine-learning screening for immune-related genes in diagnosing non-alcoholic fatty liver disease with ischemic stroke and RRS1 pan-cancer analysis. Front. Immunol. 14(2023).
Liu, H., Pathak, P., Boehme, S. & Chiang, J. L. Cholesterol 7α-hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J. Lipid Res. 57(10), 1831–1844 (2016).
Ma, C. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360(6391) (2018).
Govaere, O. et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 12(572) (2020).
Wang, Y. et al. Association of TNFRSF12A methylation with prognosis in hepatocellular carcinoma with history of alcohol consumption. Front. Genet. 10(2020).
Wang, T. et al. Knockdown of the differentially expressed gene TNFRSF12A inhibits hepatocellular carcinoma cell proliferation and migration in vitro. Mol. Med. Rep. 15(3), 1172–1178 (2017).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28(6), 882–883 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
Langfelder, P. & Horvath, S. Fast R functions for robust correlations and hierarchical clustering. J. Stat. Softw. 46(11) (2012).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Zhang, B. & Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17 (2005).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51(D1), D587–d592 (2023).
Kumar, L. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2(1), 5–7 (2007).
Ernst, J. & Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 7(1) (2006).
Wu, T. T., Chen, Y. F., Hastie, T., Sobel, E. & Lange, K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics 25(6), 714–721 (2009).
Chambless, L. E. & Diao, G. Estimation of time-dependent area under the ROC curve for long-term risk prediction. Stat. Med. 25(20), 3474–3486 (2006).
Funding
This work was supported by the National Natural Science Foundation of the People’s Republic of China (No. 81572333, 81772707, 81972732); Shanxi Provincial Clinical Research Center for Interventional Medicine, No. 202204010501004; Natural Science Foundation of Shanxi Province (No. 20210302123258).
Author information
Authors and Affiliations
Contributions
JH, YL, and RS designed the experiments and analyzed data. YL, and JH prepared the manuscript. YL, RS, and DF reviewed and revised of the manuscript. JH, and DF conceived and supervised the project. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Institutional review board statement
No ethical approval nor informed consent was required in this study due to the public availability of data in the GEO and TCGA databases.
Informed consent
Statement: Informed consent was obtained from all subjects involved in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lan, Y., Song, R., Feng, D. et al. Bioinformatic analysis of molecular expression patterns during the development and progression of metabolic dysfunction-associated steatotic liver disease (MASLD). Sci Rep 15, 7294 (2025). https://doi.org/10.1038/s41598-025-90744-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-90744-3
