
Abstract
Mitophagy is an essential cellular process that is conserved and crucial for maintaining cellular balance by selectively eliminating malfunctioning mitochondria. However, there is still limited knowledge regarding the influence of mitophagy-related genes (MRGs) on the prognosis and response to treatment of triple-negative breast cancer (TNBC). In here, the TCGA and GEO databases were used to acquire the transcriptomic and clinical information of patients with TNBC, correspondingly. Using LASSO and multivariable Cox regression analyses, a risk signature related to mitophagy was established based on the prognostic MRGs. The prognostic signature associated with mitophagy consisted of five genes (BSG, JMJD6, DNAJA3, DISC1, and SQSTM1) and independently predicted the prognosis of patients with TNBC, regardless of clinical factors (p < 0.05). Patients classified within the high-risk group demonstrated significantly lower overall survival rates when contrasted with those in the low-risk group. The model exhibited excellent performance in predicting survival and risk stratification, as evidenced by the receiver operating characteristic and C-index. The findings stayed unchanged following external validation. Moreover, we observed a notable variation in the tumor immune microenvironment among the different risk categories. Patients with a low risk of TNBC demonstrated a more favorable response to immunotherapy compared to patients with a high risk. In conclusion, our study uncovered the possible impacts of MRGs on the tumor microenvironment, clinical and pathological characteristics, and outlook of TNBC. The CRG-related signature was strongly linked to the immune response against TNBC and has the potential to serve as a valuable tool in predicting the prognosis and immunotherapy response of patients.
Similar content being viewed by others

Introduction
Breast cancer, a highly diverse disease, is the predominant form of malignant cancer in women, causing approximately 41,760 deaths worldwide each year1. Different subtypes of breast cancer can be categorized depending on the existence of progesterone receptors (PRs), estrogen receptors (ERs), and human epidermal growth factor receptor 2 (HER2) proteins, with each subtype having distinct treatment responses and prognostic outcomes2. Triple-negative breast cancer (TNBC) makes up approximately 15–20% of total breast cancer cases among these subtypes3. It displays the most aggressive biological characteristics, such as elevated proliferation rates and extensive immune infiltration, leading to the poorest prognosis4,5. TNBC, known for lacking ER, PR, and HER2 expression, exhibits insensitivity to endocrine therapy and anti-HER2 therapy6, making it a difficult subtype of breast cancer to manage. Additionally, TNBC is associated with early recurrence and unfavorable outcomes6. Hence, the discovery of novel biomarkers holds immense importance in enhancing treatment efficacy and forecasting outcomes for TNBC.
Autophagy, an essential catabolic mechanism, is crucial for upholding homeostasis in diverse biological processes7. Mitophagy, a distinct form of autophagy, is a cellular mechanism that eliminates old and impaired mitochondria via degradation in lysosomes8. Mitophagy maintains the balance of mitochondria in tumors by impacting the metabolic reprogramming and the rate of accumulation or elimination of damaged mitochondria, which is vital for the survival of cancer cells9. The rationale for prioritizing mitophagy in breast cancer research stems from its dual role in tumorigenesis and treatment resistance. Recent studies have demonstrated that dysregulated mitophagy can contribute to the progression of breast cancer by allowing tumor cells to evade apoptosis and maintain metabolic function under stress conditions, such as hypoxia and chemotherapy exposure10. Current research highlights the complex interplay between mitophagy and breast cancer biology. For instance, one study elucidated the role of Parkin-mediated mitophagy in enhancing the radiosensitivity of hypoxic breast cancer cells, suggesting that inhibiting mitophagy could sensitize these cells to radiation therapy11. Furthermore, investigations into doxorubicin resistance in breast cancer have shown that mitophagy may serve as a protective mechanism that allows cancer cells to survive treatment by eliminating dysfunctional mitochondria12. This underscores the potential of targeting mitophagy pathways as a therapeutic strategy to overcome resistance in breast cancer treatment13. Mitophagy defects are linked to multiple cancers characterized by compromised mitochondrial function, which contributes to the development of cancer and impacts the effectiveness of anti-cancer treatments14. Breast cancer has been linked to a set of genes involved in mitophagy, as discovered recently15,16,17,18. For instance, Xia and colleagues17found that an excessive amount of UCP1, a protein found in mitochondria, led to the destruction and malfunction of mitochondria. This triggered mitophagy and pyroptosis, ultimately suppressing the growth and spread of TNBC. Under the hypoxic condition, depalmitylation of GPCPD1 triggers mitophagy by modulating PRK-mediated VDAC1 ubiquitination, thereby facilitating tumor growth and metastasis in TNBC16. While earlier studies have identified a connection between genes linked to mitophagy and the outlook in TNBC, the focus had primarily been on the role of a solitary gene. Hence, an extensive examination of the primary regulator of mitophagy implicated in TNBC advancement and outlook can steer clinical decision-making and offer additional treatment alternatives for individuals with TNBC.
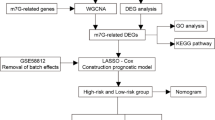
For this study, the prognostic significance of mitophagy-related genes (MRGs) in TNBC was thoroughly evaluated, utilizing the expression profiles of the TCGA and GEO datasets. After conducting LASSO regression and multivariate COX regression analyses, we successfully screened 5 MRGs. Utilizing these genes, we developed a prognostic risk model for TNBC prediction. In addition, we examined the clinical features, gene mutation patterns, tumor immune microenvironment, response to immunotherapy, and drug responsiveness of TNBC individuals with varying levels of risk. The findings indicated notable distinctions in these aspects between the two risk categories of patients. To sum up, this model is beneficial for forecasting the outcome of TNBC patients and offering guidance for immunotherapy and clinical chemotherapy.
Materials and methods
Data collection
Data from the TCGA database provided the RNA sequencing (RNA-seq) data and clinicopathological details for a total of 183 TNBC samples. Additionally, information regarding clinical features and somatic mutations with masked identities was obtained. The validation cohort of 107 TNBC patients’ clinical information and expression data were acquired from the Gene Expression Omnibus (GEO) database (ID: GSE58812). To obtain the expression of mitophagy-related genes (MRGs) in TNBC samples, we retrieved 1653 MRGs from the GeneCards database (https://www.genecards.org/) by conducting a search using the term “Mitophagy” and selecting those with a relevance score greater than 1.
Development of the mitophagy-associated gene signaturee
The impact of these genes on the prognosis in the TCGA cohort was evaluated using Univariate Cox regression with a significance level of P < 0.05. To avoid over-fitting and track the progress of each variable, the R package “glmnet” was utilized for implementing LASSO regression. Ultimately, prognostic genes were discovered through the implementation of multivariate COX regression analysis. The formula below was used by the model to export the risk score for each patient.
MRG_score =\(\:\:\sum\:\left(\text{G}\text{e}\text{n}\text{e}\:\text{E}\text{x}\text{p}\text{r}\text{e}\text{s}\text{s}\text{i}\text{o}\text{n}\text{*}\text{g}\text{e}\text{n}\text{e}\:\text{c}\text{o}\text{e}\text{f}\text{f}\text{i}\text{c}\text{i}\text{e}\text{n}\text{t}\right)\).
The risk score is derived by aggregating the products obtained by multiplying each gene expression level by its respective coefficient. Based on the median risk score in the training set, TNBC patients in each cohort were divided into groups of low-risk and high-risk. Principal component analysis (PCA) was conducted using the R packages “ggplot2” and “Rtsne”. The R packages “survival” and “survminer” were employed to generate the Kaplan-Meier (K-M) survival curve to assess the disparities in survival rate among the two groups. The R package “timeROC” was utilized to produce time-varying receiver operating characteristic (ROC) curves for assessing the prognostic model’s predictive capability. Moreover, the signature’s discriminatory ability was evaluated by calculating the C-index.
Verifying the mitophagy-associated gene signature
To confirm the predictive significance of mitophagy-associated gene signature, we utilized the GEO dataset as a validation cohort, consisting of 107 and 109 TNBC samples. The MRG_score for every patient was calculated utilizing the identical equation. PCA and t-SNE analyses were performed. We utilized Kaplan-Meier analysis to assess the predictive differentiation between the two cohorts. The model’s accuracy was evaluated by calculating the AUC value of the ROC curve.
Independent prognostic analysis of the risk score
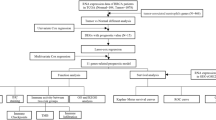
The clinical characteristics (including age, margin status, pathologic stage, and surgery) of TNBC patients in the TCGA dataset were gathered and examined alongside the risk score using univariate and multivariable Cox regression analyses.
Analysis of the immune microenvironment in tumors
To characterize the tumor immune microenvironment of the prognostic signature, a comparison was conducted between the two risk groups based on the stromal score, immune score, ESTIMATE score, and tumor purity. Next, the CIBERSORT algorithm was used to quantify the level of enrichment for 22 immune signatures in every TNBC sample. To examine the variation in immune cell infiltration between the high- and low-risk groups, box plots were utilized.
Analysis of tumor mutations and susceptibility to drugs
Using the “maftools” R package, we examined the mutation status of samples in two risk groups. Moreover, we computed the TMB score for two groups at risk. In addition, the R package “pRRophetic” was employed to forecast the IC50 of frequently utilized chemotherapeutic medications in patients with TNBC categorized as low- and high-risk groups.
Assessing the immunotherapeutic reaction disparity among different risk score categories
The immunophenoscore (IPS) for the TNBC samples was obtained from The Cancer Immunome Atlas Database. To assess the effectiveness of immune checkpoint inhibitors (ICI), we analyzed the correlations between mitophagy gene signature groups and IPS, which is widely recognized as a reliable indicator of tumor immunogenicity. Comparison was made between the levels of common immune checkpoint expressions in two risk groups. Furthermore, we acquired a cohort for immunotherapy (IMvigor210 for PD-1 treatment) and utilized the corresponding standardized information to assess if the signature could predict the response to ICIs.
Analyzing functional enrichment between different risk groups
The two groups were analyzed using GSEA (Gene Set Enrichment Analysis) to identify distinct biological processes and signaling pathways. The reference database used was “c2.cp.kegg.Hs.symbols” and “c5.go.Hs.symbols”. The threshold criteria established were Q < 0.25 and |NES| > 1.5.
Statistical analysis
The presentation of all statistical analyses was done using R 4.2.0. To compare the disparities among sets of continuous data, the Wilcoxon rank-sum test was employed. Statistical significance was determined by a two-tailed P-value less than 0.05.
Results
Development and validation of a mitophagy-related signature
We developed a prognostic risk model based on 5 MRGs associated with prognosis. To identify the MRGs linked to OS, an initial analysis of univariate Cox regression was performed on 1460 MRGs within the TCGA database. The result indicated that the 24 MRGs were significantly linked to OS, as depicted in Fig. 1A. Subsequently, the optimal prognostic variables were identified using Lasso regression and multivariate COX regression analyses. Minimum partial likelihood deviance retained a total of 17 genes related to OS, as indicated by Lasso regression (Fig. 1B and C). We conducted multivariate COX regression analysis using these 17 genes associated with OS, and ultimately identified 5 genes (BSG, JMJD6, DNAJA3, DISC1, and SQSTM1) to develop a risk model (Fig. 1D). We evaluated the expression levels of five model genes in TNBC samples, and the results showed that the expression levels of these five genes were significantly different in TNBC and normal samples (Fig. S1). The calculation of the risk_score was done in the following manner:

Development of a mitophagy-related signature. (A) The prognosis of 24 MRGs in the TCGA cohort was analyzed by univariate Cox regression analysis. (B) Ten-time cross‐validation for tuning parameter selection in the LASSO model. (C) LASSO coefficient profiles. (D) Multivariate Cox regression analysis of 5 MRGs.
Risk_score = (1.6852 * expression of BSG) + (−2.2546 * expression of JMJD6) + (1.7077 * expression of DNAJA3) + (1.2617 * expression of DISC1) + (1.3438 * expression of SQSTM1).
Utilizing the median risk score of 0.8070, patients within the TCGA cohort were classified into two distinct categories: low-risk (n = 92) and high-risk groups (n = 91). As the risk score rose, patients had a higher probability of death (Fig. 2A). The performance of this classification was good, as demonstrated by PCA and t-SNE (Fig. 2B and C). According to the Kaplan-Meier survival analysis findings, individuals identified as low-risk demonstrated significantly improved OS when contrasted with those labeled as high-risk (log-rank test, P < 0.001; Fig. 2D). The AUC values for the signature at 1 year, 3 years, and 5 years were 0.955, 0.906, and 0.977, respectively (Fig. 2E).

Landscape of risk score in the TCGA and GEO cohorts. (A) Survival time and survival status between low-risk and high-risk groups in the TCGA set. (B, C) PCA and t-SNE plots were generated using the risk score in the TCGA set. (D) Survival analysis using the Kaplan-Meier method was performed on the patients in the TCGA set. (E) ROC curve analysis for evaluating the AUC value for 1-, 3-, and 5-year survival in the TCGA set. (F) Survival time and survival status between low-risk and high-risk groups in the GEO test. (G, H) PCA and t-SNE plots were generated using the risk score in the GEO set. (I) Survival analysis using the Kaplan-Meier method was performed on the patients in the GEO set. (J) ROC curve analysis for evaluating the AUC value for 1-, 3-, and 5-year survival in the GEO set.
To validate the prognostic model, the GEO cohort was utilized to ensure its external validity and precision. Using the risk score formula mentioned earlier, we evenly categorized the 107 TNBC patients into two risk groups. Patients had a higher probability of death with the elevation of risk score escalated (Fig. 2F). According to PCA and t-SNE, the categorization was clearly separate (Fig. 2G and H). The OS of patients in the high-risk category was markedly inferior when juxtaposed with those in the low-risk category (Fig. 2I). The AUC values at 1, 3, and 5 years were recorded as 0.805, 0.851, and 0.842, respectively (Fig. 2J).
To validate the precision of our signature, we conducted a comparative analysis of the C-index, RMS, and AUC values of our signature against those of eight established risk models. Our findings indicate that our signature demonstrates significant advantages when evaluated against the C-index and RMS of these eight models (Fig. 3A and B). Additionally, we assessed the AUC values of our signature in relation to those of previously reported signatures. Notably, the AUC values obtained for the ROC curve of our signature outperformed those of the published signatures, showcasing the highest values (Fig. 3C-K).

Comparison of the accuracy of our signature with those of eight published risk models. (A-B) The C-index (A) and RMS (B) of our signature with those of eight published risk models. (C-K) The AUC values of the ROC curve of our signature with those of the published signatures.
Independent prognostic value of the risk signature
To investigate if the risk score acted as an autonomous prognostic factor, we conducted univariate and multivariable Cox regression analyses. In the TCGA cohort, the risk score was considered a risk factor (HR = 1.017, 95% CI 1.006–1.028, p = 0.003) in the univariate Cox regression analysis, indicating its ability to predict unfavorable survival compared to other features (Fig. 4A). After eliminating confounding factors, the multivariate analysis reaffirmed that the risk score remained an autonomous risk indicator (HR = 1.018, 95% CI 1.006–1.031, and p = 0.004) for TNBC patients in the TCGA cohort (Fig. 4B).

Prognostic value of the MRG_score and functional enrichment analyses. (A-B) Univariate (A) and multivariate Cox regression analyses (B) of multiple variables for the TCGA cohort. (C-D) Biological processes in high- (C) and low-risk groups (D) were implicated in the TCGA cohort. (E-F) Signaling pathways in high- (E) and low-risk groups (F) were implicated in the TCGA cohort.
Functional enrichment analyses
To explore the differences in the biological processes and signaling pathways among the groups categorized by the risk score, we utilized GSEA for analysis, adhering to the thresholds of Q < 0.25 and p < 0.05. Regarding biological processes, the high-risk group was involved in metabolic processes and transporter activity (Fig. 4C), while the low-risk group showed engagement in immune response (Fig. 4D). Regarding signaling pathways, we noticed that the high-risk category was associated with the stimulation of the interaction between butanoate metabolism, drug metabolism, metabolism of xenobiotics cytochrome P450, and tyrosine metabolism (Fig. 4E). The high-risk category showed activity in the antigen processing and presentation, cytokine-cytokine receptor interaction, DNA replication, primary immunodeficiency (Fig. 4F).
Characteristics of the tumor immune microenvironment (TIME) among different risk categories
We analyzed to determine if there were any disparities in the TIME among the two risk categories in the TCGA cohort. CIBERSORT algorithm was utilized to compute the fractions for 22 immune cells in every individual sample. In the TCGA dataset, the group at greater risk often exhibited reduced infiltrating levels of immune cells, particularly in relation to CD8 + T lymphocytes, follicular helper T cells, activated natural killer cells, and activated dendritic cells (Fig. 5A). Nevertheless, there was a notable increase in the presence of macrophage 2 (M2) infiltration within the high-risk group (Fig. 5A). Subsequently, both groups were examined for TME score using the ssGSEA algorithm with the assistance of the “estimate” R package. The low-risk group tended to elevated immune, stromal, and ESTIMATE scores, whereas the high-risk group showed a tendency towards increased tumor purity (Fig. 5B-E).

Characteristics of the tumor immune microenvironment among different risk categories. (A) The fractions of 22 immune cell types were compared between two risk groups in the TCGA cohort. (B-E) Comparison of ImmuneScore (B), StromalScore (C), and ESTIMATEScore (D), and tumor purity (E).
Analysis of the mutations and susceptibility to drugs
The genetic alterations in the low- and high-risk categories were analyzed (Fig. 6A and B). TP53 was the gene that exhibited the highest frequency of mutations in both risk groups. In the low-risk group, we observed a higher frequency of gene mutations compared to the high-risk group (Fig. 6A). Notably, genes like TNN and PYR2 exhibited this pattern. In contrast, the high-risk group exhibited a higher frequency of mutations in PIK3CA and CDH1 compared to the low-risk group (Fig. 6B). Subsequently, it was noted that TMB exhibited a greater value in the low-risk category than in the high-risk category (Fig. 6C). Additionally, we assessed the vulnerability of TNBC individuals in the high and low-risk categories to various typical treatment options. As demonstrated, the low-risk group exhibited decreased IC50 values for several therapeutic drugs, including Alisertib, Cisplatin, Epirubicin, Foretinib, and Gemcitabine (Fig. 6D-H). In contrast, the high-risk group exhibited a reduced IC50 value for Ipatasertib and Uprosertib (Fig. 6I-J).

Analysis of mutation and drug susceptibility in two groups classified by risk score. (A, B) The waterfall plot displays the characteristics of somatic mutations in the low-risk group (A) and high-risk group (B). (C) The TMB in the low- and high-risk categories. (D-J) The relationship between risk score and drug sensitivity.
Prediction of immunotherapy benefits based on mitophagy-related signature
Mounting evidence suggests that immunotherapy is a successful approach for treating tumors19. As an example, pembrolizumab has been authorized by the FDA to inhibit PD-1, while clinical trials are currently underway for TNBC with CTLA-4 inhibitors20,21. Hence, we assessed the IPS to determine the relationship between the risk score and the response to immune checkpoint inhibitors (ICIs) in individuals diagnosed with TNBC. Among patients receiving treatment with PD-1, CTLA-4 inhibitors, or a combination of both, those categorized in the low-risk group displayed a significantly elevated IPS in comparison to the high-risk group (Fig. 7A-C). Additionally, we examined the presence of immune checkpoints in two different risk groups. It can be observed that the low-risk group exhibited increased expression of crucial immune checkpoints (PD-1, PD-L1, and CTLA-4) in comparison to the high-risk group (Fig. 7D).

Prediction of immunotherapy benefits based on mitophagy-related signature. (A-C) Comparison of the immunophenoscore (IPS) in the two risk groups. (D) The presence of common immune checkpoints in low- and high-risk groups. (E) Survival analysis using the Kaplan-Meier method was conducted on patients classified into high- and low-risk categories within the IMvigor210 cohort. (F) Comparison of the percentage of patients in low- and high-risk groups who respond to immunotherapy. SD: stable disease, PD: progressive disease, CR: complete response, and PR: partial response. (G) Comparison of the risk score between CR/PR and SD/PD groups.
To assess the predictive capability of the signature for immunotherapy response, we examined patients from the IMvigor210 cohort who were treated with the Atezolizumab antibody targeting PD-L1. Our analysis revealed a lower response rate to anti-PD-L1 treatment among patients in the high-risk group, and these patients also experienced a notable survival disadvantage (Fig. 7E). Individuals classified within the high-risk group exhibited a diminished proportion of complete response (CR)/ partial response (PR) than those in the low-risk group (Fig. 7F). Patients with CR/PR have lower risk scores compared to patients with stable disease (SD)/progressive disease (PD) (Fig. 7G). Collectively, the above-mentioned discoveries indicate a link between our risk assessment and the efficacy of immunotherapy.
Discussion
Mitophagy plays a crucial role in altering metabolism and controlling aerobic glycolysis in cancer cells, offering promising potential as an innovative strategy to combat different types of cancers22. The progression of breast cancer is influenced by mitophagy, which has a dual function and is primarily determined by clinical characteristics, tumor microenvironment, and mutation status17,23,24. The selection of mitophagy as a focal point in breast cancer research is supported by its significant impact on tumor biology, therapeutic resistance, and potential as a biomarker for prognosis. Continued exploration of mitophagy’s multifaceted roles could lead to novel therapeutic approaches aimed at improving outcomes for breast cancer patients. Despite the impact of mitophagy on the development of TNBCs has been extensively studied, few studies have systematically evaluated its impact on prognosis and treatment approaches for TBNC. A recently published study focused on the role of mitophagy in TNBC and developed a mitophagy-associated gene signature that predicts patient outcomes25. Although there are some similarities between this study and ours, however, they differ in their methodologies, findings, and implications. Below are the advantages and highlights of our study. First, the genes used to construct the prognostic model of mitophagy are different. In our study, genes related to mitophagy were directly utilized to construct prognostic models. However, their study used single-cell RNA sequencing to screen genes that may be associated with mitophagy, and then built a prognostic model associated with mitophagy. The downside to this approach is that the models they build aren’t really relevant to mitophagy. Second, compared to their model, our model construction process is more concise and uses fewer genes, which may be easier to operate and implement in practical applications and suitable for rapid promotion in clinical applications. Third, our study provides a more detailed list of drugs in drug sensitivity, especially in sensitivity analysis of commonly used chemotherapy drugs, providing more specific references for clinical treatment. Fourth, this study focused on the value of this signature in predicting immunotherapy response using a variety of methods. The findings suggest that patients in the low-risk group exhibit better responses to immunotherapies, highlighting the potential for personalized treatment strategies based on mitophagy-related gene signatures. This aspect is particularly relevant given the increasing importance of immunotherapy in TNBC treatment. However, their study simply analyzed the value of this trait in predicting immunotherapy response, and the results were not good. Taken together, the advantage of our study lies in the simplicity and efficiency of its model, particularly in providing clear guidance for predicting immune therapy responses. Although they have high scientific innovation in single-cell data analysis, our model is easier to promote and apply in clinical settings, and its prediction results are more direct and practical. Therefore, this study has significant advantages and strengths in clinical application and personalized treatment.
Recent research has challenged the previous notion that immune treatments are ineffective against breast cancer, particularly TNBC. It has been discovered that TNBC has a specific subtype that possesses a fully functional immune system, suggesting the possibility of a positive response to immunotherapy26. The immune microenvironment, particularly the tumor microenvironment, has a crucial impact on the recurrence and spread of TNBC, which hinders the effectiveness of immunotherapy and chemotherapy27,28,29. The growing body of studies conducted on the tumor microenvironment has highlighted the vital importance of the infiltration of immune cells in the advancement, dissemination, and avoidance of the immune system in TNBC27,28,30. In our examination of the immune system, the high-risk group exhibited a negative correlation with immune-enhancing cells like CD8 + T lymphocytes, activated natural killer cells, and activated dendritic cells, while showing a positive correlation with immunosuppressive cells like M2 macrophages. During the progression of tumors, tumor-associated macrophages (TAMs) may serve as promoters31. In general, TAM consists of various subpopulations of macrophages, which include M1 and M2 phenotypes. M2 cells, which polarize from M0 macrophages, were the predominant tumor microenvironment (TME) component responsible for suppressing immune response within the TME32. Another potential treatment strategy for cancers could involve targeting the modulation of macrophages’ polarization to the M2 phenotype, as well as identifying key molecular regulators and therapeutic targets. NK cells, which are a group of innate lymphoid cells, play a crucial part in the immune system’s defense against infection and the development of tumors33. The formation of TME is facilitated by an increased presence of activated NK cells, leading to a decrease in tumor-infiltrating levels. Multiple human solid tumors have shown a positive prognosis when there is a high concentration of NK cells infiltrating the tumor33. NK cells regulate the growth of tumors through direct interaction with tumor cells or by influencing the activity of other groups of innate and adaptive immune cells within the TME. Furthermore, CD8 + T lymphocytes are widely recognized as the primary constituents of tumor-infiltrating lymphocytes (TLSs), playing a crucial role in the body’s defense against tumors34. Dendritic cells can enhance immune activation through antigen presentation and T-cell activation35,36. The absence of these cells may contribute to the unfavorable outlook for patients in this high-risk group.
Over the course of many years, immunotherapies that rely on immune checkpoint blockers (ICBs) have provided oncologists with the ability to predict strategies for curing tumors. Enhancing the expression of immune checkpoint genes is seen as a signal to boost the effectiveness of immunotherapy by inhibiting immune checkpoints and enhancing immune activation37. Extensive research has been conducted on the PD-1/PD-L1 pathway in immunotherapy, which obstructs the activation and proliferation of T cells. Additionally, CTLA-4 has been thoroughly investigated for its ability to provoke cell-cycle arrest and apoptosis in activated T lymphocytes38. Nevertheless, due to resistance, not all cancer patients exhibit a lasting response to ICBs. To address these issues, it is imperative to urgently study prediction models or a combination of ICBs with essential MRGs. In our investigation, we explored the relationship between the risk score and various common genes associated with ICB. The findings revealed increased expression of most immune checkpoint genes in patients classified as low-risk, suggesting that individuals in the low-risk category may experience advantages from immunotherapies through the inhibition of immune checkpoints. TNFRSF4 (OX40) belonged to the TNF receptor superfamily (TNFRSF) and was shown to possess anti-tumor properties while also regulating the function of immune cells39,40. Furthermore, the low-risk group exhibited a significant upregulation of CD44 expression. CD44 plays a crucial role in regulating the expression of PD-L1 in TNBC, potentially facilitating cancer cell growth and evading the immune system by influencing PD-L1 expression41. Simultaneously, we obtained the IPS from TCIA, which has the ability to forecast the reaction of cancer patients to immunotherapy involving anti-PD-1, anti-PD-L1, and/or anti-CTLA-4 treatment. We discovered that the IPS of the high-risk score group was considerably lower compared to the low-risk score group. This observation implies that individuals within the low-risk group are more predisposed to derive advantages from immunotherapy. The outcome was demonstrated in the IMvigor210 group. Therefore, the integration of mitophagy-related signatures could serve as a new and effective classifier to determine the characteristics of the immune microenvironment, which can be used to predict the immune response and overall survival of patients with TNBC.
Besides the infiltration of immune cells, the TMB is also a prospective indicator for forecasting prognosis and treatment in various types of cancer42,43. Based on earlier studies, TP53 is the gene that undergoes the most frequent mutations in TNBC44. Additionally, individuals with TP53 mutation display a positive profile in terms of their response to immunotherapy45. According to our analysis, TP53 exhibited the highest occurrence of mutations in the two risk categories, aligning with the findings mentioned in the aforementioned literature. In TNBC, TTN mutations are commonly found and research has indicated that these mutations may enhance TMB and enhance the effectiveness of immunotherapy46. Furthermore, individuals with TTN gene mutations exhibit improved progression-free survival (PFS) or OS compared to patients without these mutations47. High TMB is strongly associated with improved tumor treatment effectiveness and prognosis, leading to better survival outcomes48,49. In our examination, the group with lower risk exhibited elevated TMB.
The findings regarding the prognostic signature associated with mitophagy in TNBC underscore the importance of personalized medicine in oncology. The identification of five key genes that independently predict patient outcomes highlights the potential for developing tailored therapeutic strategies based on individual risk profiles. This stratification could guide clinical decision-making, particularly in determining the appropriateness of immunotherapy, chemotherapy, and targeted therapies for patients with varying risk levels. In clinical practice, the observed differences in tumor immune microenvironment among risk groups suggest that patients with low-risk TNBC are more likely to benefit from immunotherapy. This insight calls for healthcare providers to consider the risk stratification of TNBC patients when formulating treatment plans. For instance, low-risk patients may be prioritized for immunotherapeutic approaches, while high-risk patients might require more aggressive treatment regimens, including chemotherapy or novel targeted therapies. Furthermore, the implications for nursing practice are significant. Nurses play a crucial role in patient education, management of treatment side effects, and monitoring of therapeutic responses. Understanding the prognostic factors associated with mitophagy and their influence on treatment efficacy can empower nurses to provide more informed care and support to TNBC patients. They can facilitate discussions about treatment options, help manage expectations regarding outcomes, and advocate for patients’ needs based on their individual risk profiles. In conclusion, the integration of mitophagy-related prognostic signatures into clinical practice not only enhances the precision of treatment approaches for TNBC but also necessitates a collaborative effort among healthcare providers, including nurses, to optimize patient outcomes and improve the overall quality of care. Future research should focus on validating these findings and exploring the underlying mechanisms that drive the differential responses to therapies based on expression profiles of MRGs.
In addressing the selection of appropriate treatment plans based on the signature, it is crucial to consider the heterogeneity of mitophagy across different tumor types and individual patient profiles. Recent studies have highlighted the potential of mitophagy-related gene signatures to be integrated into existing treatment modalities, particularly in the context of immunotherapy and targeted therapies. For instance, a study by Wang et al. identified distinct mitophagy-related subtypes in hepatocellular carcinoma, demonstrating that patients with higher expression of mitophagy-related genes exhibited poorer survival outcomes but better response to immune-checkpoint blockade therapy. This suggests that mitophagy signatures could serve as valuable biomarkers to stratify patients for immunotherapy, thereby optimizing treatment efficacy and minimizing unnecessary exposure to ineffective therapies50. Moreover, the application of machine learning algorithms to develop prognostic models based on mitophagy-related genes has been explored in various cancers, including colon adenocarcinoma and lung adenocarcinoma. These models not only predict patient outcomes but also inform treatment strategies by identifying patients who may benefit from specific chemotherapeutic agents51,52. However, the implementation of such models in clinical practice faces challenges, including the need for larger, multicenter studies to validate these findings and address sample size limitations. Data heterogeneity remains a significant concern, particularly when integrating genomic data from diverse populations and tumor types. Variability in gene expression profiles and treatment responses can complicate the interpretation of mitophagy-related signatures. To overcome this, standardization of data collection and analysis methods is essential, alongside the establishment of comprehensive databases that encompass a wide range of demographic and clinical variables. In conclusion, while the integration of mitophagy-related gene signatures into treatment planning shows promise, further research is necessary to validate these approaches in larger cohorts and address the challenges of data heterogeneity. By doing so, we may enhance personalized treatment strategies and improve outcomes for patients with various malignancies.
The well-established prognostic model demonstrated outstanding performance in both internal and external cohorts. Nevertheless, it exhibited subsequent limitations. To begin with, the retrospective nature of our investigation inherently introduces selection bias, which can skew results and limit the generalizability of the findings. This bias arises from the reliance on existing data, which may not comprehensively represent the entire population of interest. Future studies should consider adopting a prospective design to mitigate these biases, thereby enhancing the reliability of the data collected. The limited number of TNBC cases in our cohort presents another significant challenge. The scarcity of TNBC patients restricts the statistical power of our analyses, making it difficult to draw robust conclusions. To address this limitation, multi-center collaborations should be encouraged to amass a larger and more diverse dataset. Such efforts would not only increase the sample size but also allow for a more nuanced understanding of the clinical characteristics and outcomes associated with TNBC. Moreover, the absence of critical clinical details, particularly regarding surgical interventions and neoadjuvant therapies, further complicates the interpretation of our results. These factors are known to significantly influence the prognosis of TNBC. Future research should prioritize the collection of comprehensive clinical data to facilitate more thorough analyses. Implementing standardized data collection protocols across institutions could enhance the quality of the information gathered, thereby providing clearer insights into the factors that affect patient outcomes. Furthermore, experimental validation is necessary to support our findings. Experimental studies, particularly those conducted in vivo and in vitro, are vital for establishing causative relationships and elucidating the mechanisms underlying TNBC progression. Future research should prioritize the development of robust experimental frameworks that can validate the findings of retrospective studies. This could involve the use of animal models that closely mimic human TNBC pathology, thereby providing insights that are directly translatable to clinical settings. In conclusion, while our study has provided valuable insights, it is essential to recognize and analyze its limitations comprehensively. By addressing the issues of selection bias, sample size, and the lack of critical clinical information, as well as emphasizing the need for thorough experimental validation, future research can build upon our findings and contribute to a more nuanced understanding of TNBC. Implementing these strategies will not only enhance the rigor of subsequent studies but also pave the way for improved therapeutic approaches and patient outcomes in this challenging disease landscape.
Conclusions
A prognostic model, utilizing 5 genes related to mitophagy, was created and verified to stratify TNBC patients for prognosis. Moreover, patients with a lower risk score are more prone to experiencing positive outcomes from immune therapy. The findings of this research offer a fresh point of reference for delving deeper into the mechanisms of mitophagy and tumor immune response. Additionally, it provides valuable insights to steer personalized therapeutic approaches for individuals diagnosed with TNBC.
Data availability
The data used to support the results of this study can be obtained from the Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and Gene Expression Omnibus (GEO) database (accession number: GSE58812, https://www.ncbi.nlm.nih.gov/gds/).
References
DeSantis C. E. et al. Breast cancer statistics, 2019. CA Cancer J. Clin. 69, 438–451. https://doi.org/10.3322/caac.21583 (2019).
Heer, E. et al. Global burden and trends in premenopausal and postmenopausal breast cancer: a population-based study. Lancet Global Health. 8, e1027–e1037. https://doi.org/10.1016/s2214-109x(20)30215-1 (2020).
Carey, L., Winer, E., Viale, G., Cameron, D. & Gianni, L. Triple-negative breast cancer: disease entity or title of convenience? Nat. Rev. Clin. Oncol. 7, 683–692. https://doi.org/10.1038/nrclinonc.2010.154 (2010).
Denkert, C., Liedtke, C., Tutt, A. & von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 389, 2430–2442. https://doi.org/10.1016/s0140-6736(16)32454-0 (2017).
Torre, L. A. et al. Global cancer statistics, CA Cancer J. Clin. 65, 87–108 (2015). (2012). https://doi.org/10.3322/caac.21262
Won, K. A. & Spruck, C. Triple–negative breast cancer therapy: current and future perspectives (Review). Int. J. Oncol. 57, 1245–1261. https://doi.org/10.3892/ijo.2020.5135 (2020).
Chung, C., Seo, W., Silwal, P. & Jo, E. K. Crosstalks between inflammasome and autophagy in cancer. J. Hematol. Oncol. 13, 100. https://doi.org/10.1186/s13045-020-00936-9 (2020).
Ma, K. et al. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell. Dev. Biology. 8, 467. https://doi.org/10.3389/fcell.2020.00467 (2020).
Doblado, L. et al. Mitophagy in human diseases. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms22083903 (2021).
Chen, C. et al. Mitophagy: insights into its signaling molecules, biological functions, and therapeutic potential in breast cancer. Cell. Death Discovery. 10, 457. https://doi.org/10.1038/s41420-024-02226-6 (2024).
Ahmadpour, S. T. et al. Doxorubicin-Induced autophagolysosome formation is partly prevented by mitochondrial ROS elimination in DOX-Resistant breast Cancer cells. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms22179283 (2021).
Zhou, Y., Wei, X., Li, W., Zhang, S. & Zhao, Y. Comprehensive analysis of mitophagy-related subtypes of breast cancer and the association with immune related characteristics. Heliyon 9, e23267. https://doi.org/10.1016/j.heliyon.2023.e23267 (2023).
Fang, J. et al. Acid ground nano-realgar processed product inhibits breast cancer by inducing mitophagy via the p53/BNIP3/NIX pathway. J. Cell. Mol. Med. 27, 3478–3490. https://doi.org/10.1111/jcmm.17917 (2023).
Denisenko, T. V., Gogvadze, V. & Zhivotovsky, B. Mitophagy in carcinogenesis and cancer treatment. Discover Oncol. 12, 58. https://doi.org/10.1007/s12672-021-00454-1 (2021).
Malik, N. et al. Dysregulation of mitochondrial translation caused by CBFB deficiency cooperates with mutant PIK3CA and is a vulnerability in breast Cancer. Cancer Res. 83, 1280–1298. https://doi.org/10.1158/0008-5472.Can-22-2525 (2023).
Liu, Y. et al. Hypoxia-induced GPCPD1 depalmitoylation triggers mitophagy via regulating PRKN-mediated ubiquitination of VDAC1. Autophagy 19, 2443–2463. https://doi.org/10.1080/15548627.2023.2182482 (2023).
Xia, J. et al. Mitochondrial protein UCP1 inhibits the malignant behaviors of Triple-negative breast Cancer through activation of mitophagy and pyroptosis. Int. J. Biol. Sci. 18, 2949–2961. https://doi.org/10.7150/ijbs.68438 (2022).
Xie, X. Q. et al. Targeting ATAD3A-PINK1-mitophagy axis overcomes chemoimmunotherapy resistance by redirecting PD-L1 to mitochondria. Cell. Res. 33, 215–228. https://doi.org/10.1038/s41422-022-00766-z (2023).
He, X. & Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell. Res. 30, 660–669. https://doi.org/10.1038/s41422-020-0343-4 (2020).
Kwapisz, D. Pembrolizumab and Atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 70, 607–617. https://doi.org/10.1007/s00262-020-02736-z (2021).
Killock, D. Pembrolizumab can delay progression of TNBC. Nat. Rev. Clin. Oncol. 18, 64. https://doi.org/10.1038/s41571-020-00465-x (2021).
Xie, Y., Liu, J., Kang, R. & Tang, D. Mitophagy in pancreatic Cancer. Front. Oncol. 11, 616079. https://doi.org/10.3389/fonc.2021.616079 (2021).
Deng, R. et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy 17, 3011–3029. https://doi.org/10.1080/15548627.2020.1850609 (2021).
Li, Q. et al. The oncoprotein MUC1 facilitates breast cancer progression by promoting Pink1-dependent mitophagy via ATAD3A destabilization. Cell. Death Dis. 13, 899. https://doi.org/10.1038/s41419-022-05345-z (2022).
Ding, P. et al. Single-cell sequencing unveils mitophagy-related prognostic model for triple-negative breast cancer. Front. Immunol. 15, 1489444. https://doi.org/10.3389/fimmu.2024.1489444 (2024).
Oner, G. et al. Triple-negative breast cancer-Role of immunology: A systemic review. Breast J. 26, 995–999. https://doi.org/10.1111/tbj.13696 (2020).
Kudelova, E. et al. Genetic heterogeneity, tumor microenvironment and immunotherapy in Triple-Negative breast Cancer. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms232314937 (2022).
So, J. Y., Ohm, J., Lipkowitz, S. & Yang, L. Triple negative breast cancer (TNBC): Non-genetic tumor heterogeneity and immune microenvironment: emerging treatment options. Pharmacol. Ther. 237, 108253. https://doi.org/10.1016/j.pharmthera.2022.108253 (2022).
Deepak, K. G. K. et al. Tumor microenvironment: challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 153, 104683. https://doi.org/10.1016/j.phrs.2020.104683 (2020).
Asleh, K., Riaz, N. & Nielsen, T. O. Heterogeneity of triple negative breast cancer: current advances in subtyping and treatment implications. J. Exp. Clin. Cancer Res. 41, 265. https://doi.org/10.1186/s13046-022-02476-1 (2022).
De Palma, M. & Lewis, C. E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 23, 277–286. https://doi.org/10.1016/j.ccr.2013.02.013 (2013).
Locati, M., Curtale, G., Mantovani, A. & Diversity Mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 15, 123–147. https://doi.org/10.1146/annurev-pathmechdis-012418-012718 (2020).
Melaiu, O., Lucarini, V., Cifaldi, L. & Fruci, D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front. Immunol. 10, 3038. https://doi.org/10.3389/fimmu.2019.03038 (2019).
Farhood, B., Najafi, M. & Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 234, 8509–8521. https://doi.org/10.1002/jcp.27782 (2019).
Peng, X., He, Y., Huang, J., Tao, Y. & Liu, S. Metabolism of dendritic cells in tumor microenvironment: for immunotherapy. Front. Immunol. 12, 613492. https://doi.org/10.3389/fimmu.2021.613492 (2021).
Marciscano, A. E. & Anandasabapathy, N. The role of dendritic cells in cancer and anti-tumor immunity. Semin Immunol. 52, 101481. https://doi.org/10.1016/j.smim.2021.101481 (2021).
Sehgal, K. et al. Clinical and pharmacodynamic analysis of Pomalidomide dosing strategies in myeloma: impact of immune activation and cereblon targets. Blood 125, 4042–4051. https://doi.org/10.1182/blood-2014-11-611426 (2015).
Liu, J. et al. A comprehensive prognostic and immune analysis of enhancer RNA identifies IGFBP7-AS1 as a novel prognostic biomarker in uterine Corpus endometrial carcinoma. Biol. Proced. Online. 24, 9. https://doi.org/10.1186/s12575-022-00172-0 (2022).
Jensen, S. M. et al. Signaling through OX40 enhances antitumor immunity. Semin Oncol. 37, 524–532. https://doi.org/10.1053/j.seminoncol.2010.09.013 (2010).
Wu, J., Wang, Y. & Jiang, Z. TNFSF9 is a prognostic biomarker and correlated with immune infiltrates in pancreatic Cancer. J. Gastrointest. cancer. 52, 150–159. https://doi.org/10.1007/s12029-020-00371-6 (2021).
Kong, T. et al. CD44 promotes PD-L1 expression and its Tumor-Intrinsic function in breast and lung cancers. Cancer Res. 80, 444–457. https://doi.org/10.1158/0008-5472.Can-19-1108 (2020).
Bi, F., Chen, Y. & Yang, Q. Significance of tumor mutation burden combined with immune infiltrates in the progression and prognosis of ovarian cancer. Cancer Cell. Int. 20, 373. https://doi.org/10.1186/s12935-020-01472-9 (2020).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Annals Oncology: Official J. Eur. Soc. Med. Oncol. / ESMO. 30, 44–56. https://doi.org/10.1093/annonc/mdy495 (2019).
Shah, S. P. et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486, 395–399. https://doi.org/10.1038/nature10933 (2012).
Cheng, J., Ding, X., Xu, S., Zhu, B. & Jia, Q. Gene expression profiling identified TP53(Mut)PIK3CA(Wild) as a potential biomarker for patients with triple-negative breast cancer treated with immune checkpoint inhibitors. Oncol. Lett. 19, 2817–2824. https://doi.org/10.3892/ol.2020.11381 (2020).
Gao, C. et al. Tumor mutation burden and immune invasion characteristics in triple negative breast cancer: genome High-Throughput data analysis. Front. Immunol. 12, 650491. https://doi.org/10.3389/fimmu.2021.650491 (2021).
Jia, Q., Wang, J., He, N., He, J. & Zhu, B. Titin mutation associated with responsiveness to checkpoint Blockades in solid tumors. JCI Insight. 4 https://doi.org/10.1172/jci.insight.127901 (2019).
Goodman, A. M., Sokol, E. S., Frampton, G. M., Lippman, S. M. & Kurzrock, R. Microsatellite-Stable tumors with high mutational burden benefit from immunotherapy. Cancer Immunol. Res. 7, 1570–1573. https://doi.org/10.1158/2326-6066.Cir-19-0149 (2019).
Sha, D. et al. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 10, 1808–1825. https://doi.org/10.1158/2159-8290.Cd-20-0522 (2020).
Wang, Y. et al. Characterization of immune features and immunotherapy response in subtypes of hepatocellular carcinoma based on mitophagy. Front. Immunol. 13, 966167. https://doi.org/10.3389/fimmu.2022.966167 (2022).
Tang, Y. et al. Development and validation of a prognostic model for mitophagy-related genes in colon adenocarcinoma: A study based on TCGA and GEO databases. PLoS One. 18, e0284089. https://doi.org/10.1371/journal.pone.0284089 (2023).
Dai, D., Liu, L., Guo, Y., Shui, Y. & Wei, Q. A comprehensive analysis of the effects of key mitophagy genes on the progression and prognosis of lung adenocarcinoma. Cancers (Basel). 15. https://doi.org/10.3390/cancers15010057 (2022).
Acknowledgements
Not applicable.
Funding
This work was financially supported by Science and Technology Program of Hubei Province (No. 2022BCE027 and No. 2022CFD065).
Author information
Authors and Affiliations
Contributions
Gang Liu and Jianying Ma designed the study. Gang Liu, Guozheng Yu, and Dongzhi Yin downloaded the data; Gang Liu, Guozheng Yu, and Dongzhi Yin carried out statistical analysis and graph making; All authors wrote and edited the manuscript. The final manuscript was reviewed and approved for publication by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, G., Yu, G., Yin, D. et al. Discovery of a new mitophagy-related gene signature for predicting the outlook and immunotherapy in triple-negative breast cancer. Sci Rep 15, 6794 (2025). https://doi.org/10.1038/s41598-025-91613-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91613-9
