
Abstract
Head and neck squamous cell carcinoma (HNSCC) remains a prevalent and lethal malignancy, with a five-year survival rate of just 50% for cases of locally advanced disease. Chromosomal aberrations, particularly the deletion of the short arm of chromosome 3 (3p), have been strongly associated with poor prognosis and more aggressive tumor phenotypes. The tumor microenvironment (TME) plays a pivotal role in tumor progression and resistance to therapy. This study aims to elucidate the impact of 3p deletion on the TME, immune cell infiltration, and treatment response in HNSCC, to identify novel therapeutic targets to improve patient outcomes. We analyzed single-cell RNA sequencing (scRNA-seq) data from the Gene Expression Omnibus (GEO) and bulk transcriptome data from The Cancer Genome Atlas (TCGA). Pseudo-time trajectory and cell-cell communication analyses were performed with the Monocle and CellChat packages. The Wilcoxon test was used to evaluate the differential gene expression between wild-type (wt) and mutant (mut) groups. Prognostic models were developed using the Least Absolute Shrinkage and Selection Operator (LASSO) algorithm and Cox regression analyses to find the genes related to survival, with survival analysis conducted via Kaplan-Meier curves. Gene set enrichment analysis (GSEA) was employed to investigate pathway dysregulation, and immune cell infiltration was assessed using various immune scoring methodologies to explore the differences immune environment. The Tumor Immune Dysfunction and Exclusion (TIDE) database was utilized to predict the potential efficacy of immune checkpoint inhibitors. mRNA and protein expression levels of SPP1 were examined by RT-qPCR and Western blotting, while cell proliferation was assessed using the CCK8 assay. The mut group demonstrated significant alterations in cellular composition, characterized by increased endothelial cells and macrophages and decreased fibroblasts and CD8 + T cells, indicative of an immunosuppressive TME. Differential expression analysis revealed downregulation of immune pathways, including antigen processing and presentation, T cell receptor signaling, and B cell receptor signaling pathways in the mut group, along with enhanced metabolic activity in glycolysis and lipid metabolism. The prognostic model identified nine key genes associated with poor survival in HNSCC. The mut group exhibited poorer overall survival and a more immunosuppressive microenvironment compared to the wt group, which correlated with the outcomes observed in high-risk versus low-risk groups. High-risk patients also showed a diminished response to immunotherapy compared to low-risk patients. Additionally, SPP1 emerged as a critical gene associated with chemotherapy resistance and macrophage M2 polarization. This study demonstrates that 3p deletion significantly reshapes the TME, contributing to poor prognosis in HNSCC by fostering an immunosuppressive environment and enhancing chemoresistance. These findings highlight the potential for developing targeted therapies that address the genetic and immunological landscape of HNSCC.
Similar content being viewed by others

Introduction
Head and neck squamous cell carcinoma (HNSCC) is a highly prevalent and aggressive form of cancer, ranking as the eighth most common malignancy worldwide1,2. Despite advances in therapeutic strategies, including surgery, radiation, and chemotherapy, the prognosis for patients with HNSCC remains poor, particularly for those with locally advanced disease, where the five-year survival rate hovers around 50%3,4,5,6. This bleak outcome highlights the urgent need for an improved understanding of the molecular and cellular mechanisms driving HNSCC progression and therapeutic resistance7.
Genomic instability is a hallmark of cancer, and in HNSCC, chromosomal aberrations are frequent and closely associated with poor clinical outcomes8. Among these, the deletion of the short arm of chromosome 3 (3p) stands out as a significant genetic event linked to more aggressive tumor phenotypes and worse prognosis9,10. The 3p region contains numerous tumor suppressor genes, and its loss is thought to contribute to tumorigenesis and disease progression11. However, the mechanisms by which 3p deletion influences the tumor microenvironment (TME) and therapy response in HNSCC remain incompletely understood.
The TME, which comprises a complex network of non-malignant cells, extracellular matrix components, and signaling molecules, plays a crucial role in cancer development and metastasis12. It not only supports tumor growth but also contributes to immune evasion and resistance to therapy. In HNSCC, an immunosuppressive TME has been linked to poor prognosis and reduced effectiveness of immunotherapies13,14, especially macrophage15,16. Therefore, elucidating the impact of 3p deletion on the TME is critical for identifying novel therapeutic targets and improving treatment outcomes for patients with HNSCC.
In this study, we employed integrative single-cell RNA sequencing (scRNA-seq) to dissect the cellular and molecular alterations associated with 3p deletion in HNSCC. By analyzing the transcriptomic profiles of individual cells from HNSCC tumors, we aimed to uncover the specific changes in cell composition, gene expression, and signaling pathways that are driven by 3p deletion. We further investigated the implications of these alterations for chemoresistance and immune cell infiltration, with the goal of identifying potential biomarkers and therapeutic targets. Our findings also identified SPP1 as a key biomarker associated with chemotherapy resistance and macrophage polarization, further highlighting its role in tumor progression and as a potential therapeutic target in HNSCC.
Materials and methods
Data sources and preprocessing
Single-cell RNA sequencing (scRNA-seq) data for head and neck squamous cell carcinoma (HNSCC) were sourced from the Gene Expression Omnibus (GEO) database, specifically from the GSE103322 and GSE139324 datasets. The GSE103322 dataset served as the training set, comprising 18 primary tumor samples, including 5 samples with available 3p chromosomal deletion (mut group) and 5 samples with available 3p chromosomal wild type data (wt group) which were retained. The GSE139324 dataset, comprising 26 samples of tumor-infiltrating immune cells, was utilized to investigate the role of SPP1 in macrophage polarization. Additionally, bulk transcriptome data from 501 HNSCC samples were obtained from The Cancer Genome Atlas (TCGA) database, with 428 samples exhibiting 3p characteristics retained for further analysis (detailed clinical characteristics are provided in Supplementary Table 1). The GSE41613 dataset, serving as a validation cohort, was also retrieved from the GEO database. Batch effects between the TCGA and GEO datasets were corrected using the “ComBat” algorithm from the R package sva. The sva package contains functions for removing batch effects and other unwanted variation in high-throughput experiment.
Integration of scRNA-seq data and dimensionality reduction
To integrate the normalized scRNA-seq data, the “IntegrateData” function from the Seurat package was utilized. Seurat is an R package designed for QC, analysis, and exploration of single-cell RNA-seq data. It is used for integrating GSE103322 datasets across different 3p characteristics. Cell quality was evaluated based on two metrics: the number of genes detected per cell and the proportion of mitochondrial genes. Cells with fewer than 200 detected genes or mitochondrial gene content exceeding 10% were excluded. Genes expressed in fewer than three cells were filtered out. Doublets were identified using Scrublet17, with an expected doublet rate of 0.05, and cells with a doubletScore above 0.3 were removed. The “SelectIntegrationFeatures” function was employed to identify the 2000 most highly variable genes (HVGs). The values in the single-cell level expression matrix are normalized. We employed the global-scaling normalization method (‘NormalizeData’ function) in Seurat to scale the raw counts (UMI) in each cell to 10,000, and then log-transformed the results. Following data scaling, principal component analysis (PCA) was performed to reduce dimensionality. Cluster analysis of HNSCC cells was conducted using the “FindNeighbors” and “FindClusters” functions at a resolution of 0.8. Visualization of the clustering results and validation of cluster assignments were achieved using t-distributed Stochastic Neighbor Embedding (t-SNE).
Cell type identification and pseudo-time trajectory analysis
Cell annotation followed the original study18 and TISCH (http://tisch.comp-genomics.org/home/). For immune cells, TISCH automatically annotated the cell clusters with a marker-based annotation method employed in MAESTRO. MAESTRO is a computational framework for single-cell RNA-seq data analysis, which integrates gene expression and regulatory information to infer cell types and states. The annotation process utilized differentially expressed genes (DEGs) between clusters, and the results were manually corrected based on cell-type annotations provided by the original studies. Based on the major-lineage level annotations, marker gene expression across all the cell-types and malignant cell identity, all the cells were classified into three types, malignant cells, immune cells (CD8Tex, CD4Tconv, Treg, Macrophage, Mast, and B cells) and stromal cells (Myofibroblast, Fibroblast and Endothelial) which was defined as malignancy level annotation. To facilitate comparison between the mut group and wt group, malignant cells were categorized into three subtypes: malignant-mut cells (corresponding to the mut group), malignant-wt cells (corresponding to the wt group), and malignant-cc cells (malignant cells present in both groups). Pseudotime analysis was conducted to reconstruct cellular lineage development and infer differentiation, growth, or transitional trajectories based on gene expression changes across different cellular subtypes. Monocle 2 (v2.8.0) was used to determine the malignant subgroups pseudo-time trajectory (http://cole-trapnell-lab.github.io/monocle-release/tutorials/). The Monocle object was formed using the Monocle implemented “newCellDataSet” function from the Seurat object with a lowerDetectionLimit = 0.5. The parameters celltype, pseudotime, and state were applied to evaluate the differentiation trajectories of malignant cells.
Cell-Cell communication analysis
To investigate cell-cell communication, we first employed RankNet, a machine learning algorithm for learning to rank, to map the relative information flow between the mut and wt groups. RankNet is based on a neural network model that predicts the relative order of items based on their features, enabling the prioritization of interactions between cell groups. Next, we utilized the CellChat R package, a tool designed for intercellular interaction analysis, to explore potential communication among malignant-mut subsets, malignant-wt subsets, and other cell types. The CellChatDB human database was imported to construct ligand-receptor interactions, and secreted signaling was used as the basis for cell-cell communication analysis17,19. For the analysis, we configured 1,000 permutations to evaluate reciprocal exchanges and adhered to the software’s default settings for other parameters. Interaction pairs with a P value below 0.05 and an average log-fold change greater than 0.1 were considered significant. Finally, differences in cell-cell communication between the mut and wt groups were visualized using circular plots, bar charts, dot plots, and heatmaps.
Transcription factor analysis and metabolic activity quantification in each cell type
Transcription factors (TFs) were predicted from the 1657 cells between the malignant-mut subsets and malignant-wt subsets using the SCENIC package, which was applied to assess transcription factor regulation strength based on the 20-thousand motifs database for RcisTarget and GRNboost. A volcano plot was used to visualize the TF activity scores the malignant-mut subsets and malignant-wt subsets. The PROGENy package was utilized to infer the activity of 14 signaling pathways, including Androgen, Estrogen, EGFR, Hypoxia, JAK-STAT, MAPK, NF-κB, PI3K, p53, TGF-β, TNF-α, TRAIL, VEGF, and WNT, based on gene expression data20. By default pathway activity inference is based on gene sets comprising the top 100 most responsive genes upon corresponding pathway perturbation, which we refer to as footprint genes of a pathway. Metabolic activity at the single-cell level was quantified using the scMetabolism R package which quantifies and analyzes the metabolic activity of cells using scRNA-seq data. Metabolic pathway scores were estimated using the VISION method, which is a computational framework for analyzing scRNA-seq data, focusing on extracting and visualizing biologically meaningful patterns.
Multi-Dimensional analysis of mut and wt groups
Patients from the TCGA dataset were stratified into mut and wt groups based on the status of 3p chromosomal deletion. Kaplan-Meier survival curves were generated using the “survival” package in R, a widely used tool for survival analysis that provides functions for fitting survival models, estimating survival curves, and performing statistical tests such as the log-rank test. The log-rank test was employed to determine the significance of survival differences, with a threshold of P < 0.05. The enrichment of 14 oncogenic pathways, six metabolic pathways, and immune-related cells and pathways was assessed using the single-sample Gene Set Enrichment Analysis (ssGSEA) method. ssGSEA is a non-parametric method that calculates enrichment scores for each sample based on the rank of genes within a predefined gene set, allowing for the quantification of pathway activity at the individual sample level21. Immune cell infiltration was evaluated using multiple immune scoring methodologies with immune and ESTIMATE scores calculated using the “ESTIMATE” R package. ESTIMATE is a computational tool that infers the fraction of stromal and immune cells in tumor samples based on gene expression signatures, providing insights into the tumor microenvironment22.
Prognostic model construction and validation
DEGs between malignant-mut subsets and malignant-wt subsets in the scRNA-seq were identified using the “FindAllMarkers” function. DEGs were selected based on |fold change (FC)| > 1 and an adjusted p-value < 0.05. Prognosis-associated genes were further refined using univariate Cox regression and the Least Absolute Shrinkage and Selection Operator (LASSO) algorithm from the DEGs. The refined gene set was used to construct a prognostic prediction model within the TCGA HNSCC cohort. The risk score for each patient was calculated using the formula:
where ‘exp’ denotes gene expression value and ‘coef’ represents the gene coefficient derived from LASSO analysis. The model included the following genes: CAV1, FDCSP, MAGEB2, ME1, MT2A, SPINK6, SPP1, TSPAN7, and TXNIP. Patients were categorized into high- and low-risk groups based on the median risk score. The prognostic significance of the model and other clinical variables was evaluated through univariate and multivariate Cox regression analyses. The model’s performance was assessed using receiver-operating characteristic (ROC) curves and overall survival data from both the training and validation cohorts. A clinical nomogram using riskscore and clinical characteristics based on the coefficients of Cox regression was established to predict the OS of patients at 1, 3, and 5 years. The clinical utility of this nomogram was assessed by calibration curve. Decision curve analysis (DCA) was conducted to determine the model’s accuracy and the area under decision curve (AUDCA) calculated.
Gene set enrichment analysis (GSEA)
GSEA was performed to ascertain whether specific gene sets were differentially expressed between the defined groups, using protocols from the MSigDB Collection. Risk categories were used as phenotypic labels. For each analysis, gene sets were permuted 1000 times to ensure robust statistical inference. The enrichment of Gene Ontology (GO) terms and Hallmark pathways was quantified using the false discovery rate (FDR) and normalized enrichment score (NES).
Immune checkpoint and immunotherapy efficacy analysis
Differential expression of immune checkpoints across various risk categories was evaluated. The Tumor Immune Dysfunction and Exclusion (TIDE) database was used to predict the potential efficacy of immune checkpoint inhibitor therapy. Violin plots were generated to illustrate differences in BCR, TCR, cytolytic activity, and TIDE predictions between high- and low-risk groups, as well as between responders (R) and non-responders (NR) in the IMvigor210 and Melanoma-PRJEB23709-PD1 cohorts.
Construction of a 3p deletion diagnostic model
To identify patients with 3p deletions among HNSCC cohorts, LASSO and Support Vector Machine-Recursive Feature Elimination (SVM-RFE) feature selection methods were applied to DEGs identified between malignant-mut cells and malignant-wt cells (see Sect. 2.7 for details; |log2 fold change| > 1 and adjusted p-value < 0.05). This resulted in the identification of 22 genes significantly associated with 3p deletion. The TCGA cohort was randomly divided into training and testing sets in a 7:3 ratio. In the training set, considering the large number of identified genes, univariate logistic regression analysis was performed to further narrow down the candidates, selecting 14 genes with a p-value < 0.01. These genes (GPX2, ABCC5, EPCAM, FDCSP, IGFBP3, PDPN, COL17A1, S100A8, KLK6, LUM, WNT5A, EID3, SPP1 and CYP4F3) were then used to construct a diagnostic model via logistic regression analysis. The model’s performance and effectiveness were validated in the testing set using ROC curve analysis. The intersection of these genes with prognostically relevant candidates further supported their significance.
Identification of key gene of 3p deletion
To further identify key genes influencing 3p deletion, we compared the genes associated with the prognostic and diagnostic models. SPP1 was identified as a common gene in both models, suggesting its potential role as a key gene in 3p deletion. Expression levels of the SPP1 gene across various cancer types were examined using the TCGA database. Student’s t-test was used to compare SPP1 gene transcription levels between cancer samples and normal controls, with cutoff p-values and fold changes set at 0.05 and 1.5, respectively. The impact of SPP1 expression on clinical outcomes was assessed through survival analysis using the “survival” package. Additionally, the “pRRophetic” R package, a computational tool that predicts drug sensitivity based on gene expression profiles, was used to calculate the half-maximal inhibitory concentration (IC50) values for different chemotherapeutic agents (e.g., cisplatin, paclitaxel) in patients. These calculations were based on data from the Genomics of Drug Sensitivity in Cancer (GDSC) database (www.cancerrxgene.org)32. Pairwise correlations between chemotherapeutic agents were calculated using Spearman’s rank correlation coefficient, and these correlations were visually represented in a correlation matrix. Additionally, the relationship between the biomarker SPP1 and glycolysis levels was examined using Spearman’s correlation. The relationship between SPP1 expression and M2 macrophage infiltration was analyzed using TIMER2.0, employing five distinct algorithms. To discover the role of SPP1 in macrophage polarization, we apply GeneSwitches23, a tool that can process scRNA-seq data together with pseudotime trajectories to identify the genes (named switching genes) that act as on/off switches between cell states and importantly the ordering at which these switches take place.
Cell culture and CCK-8 assay (Cell counting Kit-8)
The parental HN30 cell line and the paclitaxel-resistant HN30 cell line were purchased from Zhejiang Meisen Technology Co. Ltd. (Zhejiang, China). Complete medium containing 10% FBS (Biological Industries, Beit-Haemek, Israel), 100 µg/mL streptomycin and 100 U/mL penicillin in high-sugar DMEM (Solarbio, Beijing, China) was prepared as a suitable medium for all cell lines in this study. The CCK-8 assay was utilized to evaluate the impact of paclitaxel on cell viability in control and SPP1 knockdown groups. Cells were seeded at a density of 5 × 10³ per well in 96-well plates and treated with varying concentrations of paclitaxel (0, 20 nM, 40 nM, 60 nM, and 80 nM) after an initial 24-hour incubation period. Following treatment, 100 µL of medium was replaced with the respective paclitaxel dilutions and incubated for 48 h. Subsequently, 10 µL of CCK-8 reagent was added to each well and incubated for 2 h in the dark at 37 °C. Absorbance was measured at 450 nm with a reference wavelength of 630 nm using a microplate reader. p value < 0.05 is considered statistically significant.
Lentivirus transfection and Western blot analysis
The SPP1 knockdown plasmid (shSPP1) and control (shNC) were ordered from GeneChem (Shanghai, China). Western blot analysis was conducted to quantify SPP1 expression levels across various experimental groups. Cell lysis and protein extraction were followed by protein quantification, mixing with Laemmli sample buffer, denaturation, and loading equal amounts of protein onto a 10% SDS-PAGE gel for electrophoresis. The separated proteins were transferred onto a nitrocellulose membrane, blocked for 1 h at room temperature, and incubated with a primary antibody specific to SPP1(Abcam, ab214050) overnight at 4 °C. Following three washes with TBST, the membrane was incubated with an HRP-conjugated secondary antibody for 1 h. After further washes, protein bands were detected using an ECL substrate, with exposure to X-ray film. Tubulin was used as a loading control.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR) assay
The RT-qPCR assay was employed to quantify relative mRNA expression levels of SPP1, CXCL10, CXCL9, TGFβ1, and CCL22 across various experimental groups. Total RNA was extracted from cell samples using a commercial kit, with RNA integrity and purity assessed via spectrophotometry. cDNA synthesis was performed from 1 µg of RNA using a high-capacity reverse transcription system. qPCR reaction mixtures, containing the synthesized cDNA, gene-specific primers, and SYBR Green Master Mix, were prepared and subjected to real-time PCR analysis. The thermal cycling protocol included an initial denaturation, followed by two-step cycling of denaturation and annealing/extension, and a final melt curve analysis to verify amplification specificity. Gene expression data were normalized to the endogenous reference gene GAPDH to account for variations in sample loading and amplification efficiency. Fold changes in gene expression relative to the control group were calculated using the comparative ΔΔCt method. p value < 0.05 is considered statistically significant.
Results
Single-cell landscape for 3p deletion in the HNSCC
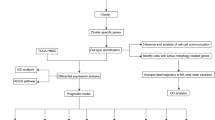
Figure 1 presents a schematic workflow of our study. In this study, we analyzed a total of 10 samples from the GSE103322 dataset. After preliminary screening to eliminate low-quality data, 4200 cells were retained for subsequent analysis. Clustering analysis divided these 4200 cells into 20 clusters (Fig. 2A). We identified 13 cell types specific to HNSCC among these 20 clusters: Malignant, Myofibroblast, Fibroblast, CD8Tex, CD4Tconv, Endothelial, Treg, Macrophage, Unknown, Mast, and B cells. These were visualized using tSNE plots, categorized by wt or mut groups (Fig. 2B). An analysis of the cellular composition across the wt and mut groups revealed a decrease in the proportion of Fibroblasts, CD8Tex, CD4Tconv, Treg cells, B cells, and Myofibroblasts in the mut group, while the proportion of Endothelial cells and Macrophages was increased (Fig. 2D). To further explore cellular development, we analyzed the developmental trajectories of the mut and wt groups using the Monocle 2 package. The results indicated that Malignant-cc cells could differentiate into malignant-mut and malignant-wt subsets (Fig. 2E), suggesting that the malignant-wt subsets represents an earlier stage of cellular development, while the malignant-mut subsets corresponds to a later stage of differentiation. Additionally, we investigated cell-cell communication networks between mut and wt groups using CellChat. Active pathways were observed to vary in the mut and wt groups, like LIFR, TWEAK, CCL, BTLA, APRIL, and SPP1 pathways being active in the mut group while TNF, NMU, and IL1 pathways being vibrant in the wt group (Supplementary Fig. 1A). Within the SPP1 and TNF signaling pathway identifying ligand-receptor interactions among various cell types, particularly within the malignant subsets (malignant-wt and malignant-mut subsets) (Fig. 2F, Supplementary Fig. 1B-C). Our findings indicate that ligand-receptor-mediated interactions are predominantly active in the SPP1 and TNFA signaling pathways, suggesting activation of SPP1 in the mut group.

Flowchat.

Single-cell RNA sequencing analysis of HNSCC samples for 3p deletion. (A) t-distributed Stochastic Neighbor Embedding (t-SNE) representation of cells classified into 20 distinct clusters. (B–C) Identification of 11 major cell types using canonical markers: Malignant, Myofibroblast, Fibroblast, CD8Tex, CD4Tconv, Endothelial, Treg, Macrophage, Unknown, Mast, and B cells. (D) Bar plot illustrating the proportional differences of cell types between mutant (Mut) and wild-type (Wt) groups. Decreases in Fibroblasts, CD8Tex, CD4Tconv, Treg cells, B cells, and Myofibroblasts were observed in the Mut group, alongside increases in Endothelial cells and Macrophages. (E) Trajectory analysis demonstrating differentiation states within malignant cells. The malignant-mut subsets indicates later differentiation stages. (F) Cell-cell communication analysis identifying ligand-receptor interactions within the SPP1 and TNF signaling pathways, particularly among wt and mut groups.
Differences in transcription factors and signaling pathways between malignant-wt and malignant-mut subsets
To examine the differences in transcription factor expression between the malignant-wt and malignant-mut subsets, we conducted SCENIC analysis, which revealed a potential upregulation of NFE2L2 in the malignant-mut subsets (Fig. 3A). Subsequent analyses confirmed that NFE2L2 expression was primarily localized to the malignant-mut subsets, with minimal expression in the malignant-wt subsets (Fig. 3B). PROGENy (500) analysis indicated that, compared to the malignant-wt subsets, the malignant-mut subsets exhibited elevated activity in the Estrogen and PI3K pathways, while activity in the P53, NFκB, JAK-STAT, and TNFa pathways was reduced (Fig. 3C). Given the critical role of metabolic pathways in tumor cell survival, we employed the scMetabolism package to assess the activity of 30 metabolic pathways across 13 different cell types. The analysis revealed significantly increased activity of Glycolysis/Gluconeogenesis, Glycerophospholipid metabolism, Glycerolipid metabolism, Galactose metabolism, and Fructose and mannose metabolism in the malignant-mut subsets compared to the malignant-wt subsets (Fig. 3D).

Comparison transcription factors and signaling pathways between malignant-Wt and malignant-mut subsets. (A) SCENIC analysis of transcription factor activity showing upregulation of NFE2L2 in malignant-mut cells compared to malignant-wt subsets. (B) Feature plot of NFE2L2_32g expression, predominantly observed in the malignant-mut subsets with minimal expression in the malignant-wt subsets. (C) PROGENy analysis of 14 tumor-associated signaling pathways, showing elevated Estrogen and PI3K pathway activity in the Malignant-Mut group, with reduced activity in P53, NFκB, JAK-STAT, and TNFα pathways. (D) scMetabolism analysis of 30 metabolic pathways, revealing significantly increased activity of Glycolysis/Gluconeogenesis, Glycerophospholipid, Glycerolipid, Galactose, and Fructose/Mannose metabolism in the malignant-mut subsets.
Transcriptome analysis between wt and mut groups in TCGA HNSCC cohort
To investigate survival differences between HNSCC patients in the wt and mut groups, we downloaded bulk RNA-seq and clinical data from the TCGA HNSCC cohort. Kaplan-Meier survival analysis demonstrated that patients in the wt group had significantly better survival outcomes compared to those in the mut group (Fig. 4A). GSEA analysis further identified that HEDGEHOG_SIGNALING_PATHWAY, GLUTATHIONE_METABOLISM, WNT_SIGNALING_PATHWAY, and GLYCEROLIPID_METABOLISM were upregulated in the mut group, while ANTIGEN_PROCESSING_AND_PRESENTATION, NATURAL_KILLER_CELL_MEDIATED_CYTOTOXICITY, T_CELL_RECEPTOR_SIGNALING, B_CELL_RECEPTOR_SIGNALING_PATHWAY, and PRIMARY_IMMUNODEFICIENCY were downregulated (Fig. 4B). Additionally, we observed significant differences in the activity of fourteen oncogenic pathways and six metabolic pathways between the two groups (Fig. 4C-D). Notably, within the TCGA cohort, the wt and mut groups exhibited significant differences in Immunity and ESTIMATE scores, with the wt group showing higher immune cell infiltration and enhanced immune-related pathway activity compared to the mut group (Fig. 4E-G). These findings suggest that the mut group is characterized by an immunosuppressive microenvironment.

Differential survival disparities and immune infiltration between Wt and Mut groups. (A) Kaplan–Meier survival analysis in the TCGA cohort showing significantly better outcomes in the wt group compared to the mut group. (B) GSEA of differentially expressed genes (DEGs) highlighting upregulated pathways in the mut group, including Hedgehog Signaling, Glutathione Metabolism, WNT Signaling, and Glycerolipid Metabolism, with downregulation of immune-associated pathways such as Antigen Processing, NK Cell Cytotoxicity, and T Cell Receptor Signaling. (C–D) ssGSEA comparison of oncogenic and metabolic pathway activities between wt and mut groups. (E) Immune and ESTIMATE score comparisons showing higher immune infiltration and immune pathway activity in the wt group. (F–G) Differential expression of immune-related genes and immune cell infiltration analysis between mut and wt groups.
Construction and validation of a prognostic model for 3p deletion
To develop a prognostic model for predicting survival in HNSCC patients, we utilized the TCGA cohort as the training set and HNSCC patient data from the GEO database as the validation set. We initially selected 101 DEGs from single-cell data for comparison between malignant-wt and malignant-mut subsets. Subsequently, LASSO regression analysis reduced the number of DEGs in the risk score model, identifying nine prognosis-related genes (CAV1, FDCSP, MAGEB2, ME1, MT2A, SPINK6, SPP1, TSPAN7, and TXNIP )(Fig. 5A). Patients were stratified into high-risk and low-risk groups, with the high-risk group demonstrating poorer prognosis in both the training and test cohorts (Fig. 5B). ROC analysis confirmed the significant predictive value of these DEGs in the TCGA cohort, with 1-year, 3-year, and 5-year AUCs of 0.65, 0.66, and 0.7, respectively (Fig. 5C). In the validation cohort, the AUCs for 1-year, 3-year, and 5-year predictions were 0.66, 0.69, and 0.73, respectively (Fig. 5C). The model was constructed using univariate and multivariate Cox regression analysis, incorporating factors such as sex, age, 3p deletion, clinical stage, alcohol use, and HPV infection status. Among these, 3p deletion, age, and HPV infection status were identified as independent predictors of overall survival (OS), with the risk score remaining independent of other clinical prognostic variables in both the training and test cohorts (Fig. 5D-E). A nomogram was developed to estimate 1-, 3-, and 5-year survival probabilities based on these independent prognostic variables (Fig. 5F-G). Decision curve analysis (DCA) indicated that the model outperformed the use of risk scores alone (Fig. 5H).

Prognostic model construction and clinical characteristics analysis. (A) LASSO regression for prognostic signature development, identifying nine key genes associated with patient survival. (B) Kaplan–Meier survival analysis in TCGA and GEO cohorts stratifying patients into high- and low-risk groups, with poorer survival outcomes observed in high-risk groups. (C) ROC curves validating the predictive accuracy of the prognostic signature in the TCGA and GEO cohorts. (D–E) Multivariate Cox regression analysis identifying independent predictors of overall survival, including age, 3p deletion, HPV infection status, and the risk score, which remained independent of clinical variables. (F–G) Nomogram development incorporating independent prognostic factors to predict 1-, 3-, and 5-year survival probabilities. (H) Decision curve analysis (DCA) demonstrating the model’s superior predictive performance compared to risk scores alone.
Immune infiltration and immunotherapy response across risk levels
We assessed risk scores for immune cell subsets, cell functions, associated pathways, and immunotherapy responses to explore the relationship between risk groups, immunological status, and immunotherapy outcomes. GSEA analysis revealed that EPITHELIAL_MESENCHYMAL_TRANSITIONS, GLYCOLYSIS, HYPOXIA, and ANGIOGENESIS pathways were upregulated in the high-risk group, while T_CELL_RECEPTOR and B_CELL_RECEPTOR_SIGNALING_PATHWAYS and B_CELL_MEDIATED_IMMUNITY were downregulated (Fig. 6A-C). Additionally, a significant correlation was observed between the risk score in HNSCC and both CYT and GEP, with higher levels in the low-risk group (Fig. 6D). The mean TCR and BCR diversity values varied with risk score, with the lowest-risk group exhibiting the greatest diversity (Fig. 6E). Immune cell infiltration was predominantly negatively correlated with risk levels (Fig. 6F), and the expression of immune-related genes was downregulated in the high-risk group (Fig. 6G). Regarding immune checkpoint blockade (ICB) response, high-risk patients exhibited higher TIDE scores compared to low-risk patients in the GEO database, suggesting a poorer response to immunotherapy (Fig. 6H). This finding was further validated in the IMvigor210 and Melanoma-PRJEB23709-PD1 cohorts, where a higher proportion of non-responders was observed in the high-risk group (Fig. 6I). Collectively, these results indicate that the prognostic model is effective in predicting immunotherapy responses.

Immune infiltration and immunotherapy responses across different risk levels. (A–C) GSEA analysis comparing high- and low-risk groups, showing upregulation of Epithelial-Mesenchymal Transition, Glycolysis, Hypoxia, and Angiogenesis pathways in the high-risk group, and downregulation of immune-related pathways. (D–E) Differential CYT and GEP scores, as well as TCR and BCR diversity, with lower-risk groups exhibiting higher levels and diversity. (F) Correlation analysis of immune infiltration with risk scores using multiple algorithms, revealing predominantly negative associations. (G) Expression analysis of immune-related genes, showing reduced expression in the high-risk group. (H) TIDE analysis indicating a poorer immunotherapy response in the high-risk group. (I) Proportions of responders and non-responders to immunotherapy in the IMvigor210 and Melanoma-PRJEB23709-PD1 cohorts, with higher non-responder rates in the high-risk group.
Construction and validation of a diagnostic model for 3p deletion
We applied RMSE and LASSO regression algorithms to select genes capable of effectively diagnosing patients with 3p deletion from a set of 101 genes, identifying 40 and 31 candidate genes, respectively. Overlapping these two sets resulted in the selection of 22 pivotal genes. We then employed a logistic regression model to screen 14 genes (GPX2, ABCC5, EPCAM, FDCSP, IGFBP3, PDPN, COL17A1, S100A8, KLK6, LUM, WNT5A, EID3, SPP1, CYP4F3) for the construction of a diagnostic model, which was internally validated using a 7:3 ratio in the HNSCC cohorts (Fig. 7A-C). The diagnostic efficiency was evaluated using ROC curves, with the AUC exceeding 0.906 and 0.954 in the test and validation cohorts, respectively, indicating high diagnostic value (Fig. 7D).

Diagnostic model construction and screening of key genes. (A–B) Identification of diagnostic genes associated with 3p deletion using RMSE and LASSO algorithms, yielding 40 and 31 candidate genes, respectively. (C) Venn diagram showing the overlap between two algorithms, identifying 22 common genes. (D) ROC curves of diagnostic signatures in training and validation cohorts, with AUC values exceeding 0.9, indicating high diagnostic accuracy. (E) Venn diagram of prognostic and diagnostic models, identifying SPP1 as a common gene. (F) TIMER2.0 analysis of SPP1 expression across cancer types, showing significant upregulation in 15 cancers and downregulation in KICH, KIRC, and SKCM. (G) Higher SPP1 expression in HNSCC compared to normal tissues. (H) Survival analysis linking high SPP1 expression with poorer prognosis.
Identification of key gene associated with 3p deletion
Comparison between the nine candidate genes from the prognostic model and the 14 potential genes from the diagnostic model revealed two overlapping pivotal genes, including SPP1(Fig. 7E). We further analyzed SPP1 mRNA expression across pan-cancer and corresponding normal tissues using TIMER2.0, finding that SPP1 mRNA was significantly upregulated in 15 cancer types (BLCA, BRCA, CHOL, COAD, ESCA, HNSC, KIRP, LIHC, LUAD, LUSC, PRAD, READ, STAD, THCA, and UCEC) and notably downregulated in KICH, KIRC, and SKCM (Fig. 7F). HNSCC tissues exhibited higher SPP1 expression compared to normal tissues (Fig. 7G), a finding consistent across both GEO and TCGA data, indicating a worse prognosis for groups with high SPP1 expression(Fig. 7H).
SPP1 knockdown enhances chemotherapy sensitivity and is associated with macrophage M2 polarization
We further analyzed the Genomics of Drug Sensitivity in Cancer (GDSC) dataset and explored the association between SPP1 expression and drug sensitivity. The analysis revealed a significant correlation between SPP1 expression and Paclitaxel sensitivity (Fig. 8A). To validate this observation, SPP1 was knocked down in HNSCC cell lines, and RT-qPCR and Western blot analyses confirmed the effective reduction of SPP1 expression (Fig. 8B-C). Subsequent CCK8 assays demonstrated that SPP1 knockdown significantly enhanced the sensitivity of HNSCC cell lines to Paclitaxel compared to control cells (Fig. 8D). Additionally, SPP1 expression was found to be upregulated in Paclitaxel-resistant cell lines (Fig. 8E). Focusing on macrophages, we utilized multiple algorithms within TIMER2.0 to analyze the correlation between macrophage infiltration levels and SPP1 expression, identifying a strong association in HNSC, HNSC-HPV-negative, and HNSC-HPV-positive groups (Fig. 8F). Furthermore, we analyzed single-cell data from the GSE139324 dataset, selecting macrophage subpopulations and categorizing them into M0, M1, and M2 subtypes. Pseudotime analysis indicated that SPP1 expression was predominantly associated with M2 polarization (Fig. 8G). To further elucidate this relationship, we examined the expression of proteins associated with M1 and M2 polarization. The results demonstrated that SPP1, TGF1, and CCL22 were upregulated in M2-polarized macrophages, while CXCL9 and CXCL10 were upregulated in M1-polarized macrophages, providing further evidence that SPP1 is linked to macrophage M2 polarization (Fig. 8H).

Correlation between SPP1, paclitaxel resistance and M2 polarization. (A) GDSC analysis showing significant association between SPP1 expression and paclitaxel sensitivity. (B–C) RT-qPCR and Western blot confirming effective SPP1 knockdown in experimental groups. (D) CCK8 assay demonstrating enhanced paclitaxel sensitivity in HNSCC cells with SPP1 knockdown. (E) Western blot analysis showing elevated SPP1 expression in paclitaxel-resistant cell lines. (F) TIMER2.0 analysis correlating SPP1 expression with macrophage infiltration in HNSC subgroups. (G) Pseudotime analysis associating SPP1 expression with macrophage M2 polarization. (H) Expression analysis of macrophage polarization markers, showing elevated SPP1, TGFβ1, and CCL22 in M2 macrophages, with higher CXCL9 and CXCL10 in M1 macrophages.
Discussion
In this study, we conducted an in-depth exploration of the cellular heterogeneity and molecular characteristics of HNSCC utilizing scRNA-seq data. A particular focus was placed on elucidating the impact of 3p chromosomal deletion on tumor biology. The findings from our analysis reveal distinct differences in cellular composition, molecular pathways, and the immune microenvironment between the wt and mut groups, providing insights that could inform the development of targeted therapies and prognostic assessments.
Our results demonstrate significant variations in the cellular makeup of HNSCC between the wt and mut groups, with notable differences observed in the proportions of immune and stromal cells. Specifically, the mut group exhibited a decrease in fibroblasts, CD8 + T cells, and regulatory T cells (Tregs), alongside an increase in endothelial cells and macrophages. This shift suggests the presence of an immunosuppressive tumor microenvironment associated with 3p deletions, a finding consistent with previous research linking chromosomal deletions in cancer to immune evasion and a more aggressive tumor phenotype24. Additionally, pseudo-time trajectory analysis using Monocle indicated that the mut subgroup represents a more differentiated cellular state, which may contribute to its poorer prognosis. Differential expression analysis between the wt and mut groups identified the upregulation of key oncogenic pathways, including P53, VEGF, and MAPK, in the mut group, while pathways related to immune responses, such as WNT and NF-κB, were downregulated. These findings correlate with the observed immunosuppressive TME and suggest that targeting these dysregulated pathways could restore immune function and inhibit tumor progression in patients with 3p deletions. Furthermore, scMetabolism analysis indicated enhanced metabolic activity in the mut group, particularly in glycolysis25 and lipid metabolism26, processes that are known to support tumor growth and survival in the hypoxic conditions typical of solid tumors like HNSCC. Survival analysis reinforced the prognostic significance of 3p deletion status, with patients in the mut group showing significantly worse outcomes compared to those in the wt group. More, the wt group showed higher immune cell infiltration and enhanced immune-related pathway activity compared to the mut group, suggesting that the mut group is inhibited by an immunosuppressive microenvironment. This could contribute to a worse survival among the patients. The construction of a prognostic model based on DEGs identified from scRNA-seq data further validated the independent prognostic relevance of genes associated with 3p deletion. The model demonstrated robustness across multiple cohorts, consistently identifying high-risk patients with poorer survival outcomes. This prognostic tool, which integrates clinical variables with genetic markers, has the potential to aid in the stratification of HNSCC patients and the personalization of treatment strategies. The analysis of immune infiltration patterns revealed a reduced presence of immune cells in high-risk patients, which was associated with poorer responses to immunotherapy, as predicted by TIDE scores. These results suggest that patients with 3p deletions may derive limited benefit from current immune checkpoint inhibitors, underscoring the need for alternative therapeutic strategies. Combination therapies that target both metabolic pathways and immune checkpoints could potentially enhance treatment efficacy for these patients.
Our findings highlight significant distinctions in immune-related pathways, cellular functions, and immunotherapy responses across risk groups, underscoring the prognostic model’s ability to stratify patients based on their immunological profiles. The enrichment of EPITHELIAL_MESENCHYMAL_TRANSITIONS, GLYCOLYSIS, HYPOXIA, and ANGIOGENESIS pathways in the high-risk group suggests a tumor microenvironment conducive to cancer progression, invasion, and resistance to therapy. Conversely, the downregulation of T_CELL_RECEPTOR and B_CELL_RECEPTOR_SIGNALING_PATHWAYS in high-risk patients implies an impaired adaptive immune response, which is further supported by reduced BCR and TCR diversity in these individuals. The negative correlation between immune cell infiltration and risk levels, coupled with the reduced expression of immune-related genes in the high-risk group, suggests a suppressed immune landscape in these patients, potentially contributing to poorer clinical outcomes. Higher TIDE scores in the high-risk group reflect diminished responsiveness to immune checkpoint blockade (ICB) therapies, as corroborated by the higher proportion of non-responders in the IMvigor210 and Melanoma-PRJEB23709-PD1 cohorts. These results emphasize that the high-risk group’s immunosuppressive environment poses a barrier to effective immunotherapy. In contrast, the low-risk group, characterized by higher CYT and GEP scores, presents a more favorable immune profile, indicative of robust anti-tumor immunity and better ICB responsiveness. This stratification capability demonstrates the prognostic model’s utility in guiding personalized immunotherapy strategies. The identification of 22 pivotal genes using RMSE and LASSO regression algorithms, and the subsequent selection of 14 diagnostic genes, constructs a diagnostic model using logistic regression algorithm. The diagnostic model, validated internally with AUC values exceeding 0.9 in both test and validation cohorts, demonstrates high predictive accuracy in identifying patients with 3p deletion. This achievement highlights the model’s potential as a diagnostic tool for clinical application. SPP1 emerged as a critical overlapping gene between the prognostic and diagnostic models, exhibiting significant upregulation in 15 cancer types, including HNSCC. Its consistent overexpression in both GEO and TCGA datasets, along with its association with poor prognosis, underscores its role as a pivotal oncogene in HNSCC. Functionally, SPP1 may contribute to tumor progression through its involvement in angiogenesis, immune evasion, and extracellular matrix remodeling, aligning with the enriched pathways observed in the high-risk group. These findings not only validate the biological relevance of SPP1 but also position it as a potential therapeutic target.
Additionally, our study identified SPP1 as a key gene linked to chemotherapy resistance and macrophage polarization towards the M2 phenotype, which is associated with tumor promotion and immune suppression. It has been reported that SPP1 promotes the resistance of breast cancer cells to paclitaxel through the JNK pathway27. The knockdown of SPP1 in HNSCC cell lines increased sensitivity to paclitaxel, indicating that SPP1 inhibition may enhance the efficacy of chemotherapy in patients with high SPP1 expression. The strong correlation between SPP1 expression and macrophage M2 polarization further supports the potential of targeting SPP1 to modulate the TME and improve patient outcomes. These findings are consistent with previous research, which has highlighted the critical role of SPP1 in various cancers. For instance, Qi et al. demonstrated that disrupting interactions between FAP + fibroblasts and SPP1 + macrophages could enhance immunotherapy efficacy in colorectal cancer28. Similarly, studies by Anuja Sathe et al. found that intercellular networking between macrophages and fibroblasts, as well as the presence of SPP1 + macrophages, support tumor growth and metastasis in colorectal cancer29. The pancreatic tumor microenvironment factor promotes cancer stemness via the SPP1-CD44 axis30. Liu et al. found that SPP1 + macrophages promote metastasis of colorectal cancer31. Additionally, research by Baniela Brina et al. revealed that SPP1 is regulated by the AKT/mTOR and MNK/EIF4E pathways to promote tumor growth in prostate cancer32. Further, Yu et al. found that SPP1 + macrophages may be associated with cell senescence, leading to poor prognosis in colorectal cancer33. The expression of SPP1 could serve as a diagnostic marker in gastrointestinal cancer34 reported by Jiang et al. Eri Matsubara reviewed that SPP1 expression on TAMs is correlated with poor prognosis and chemoresistance in lung adenocarcinoma35. Zou et al. reported that the SPP1 gene might be an effective biomarker for predicting the prognosis and the efficacy of immunotherapy in PSCC (Squamous cell carcinoma of the penis) patients36.
However, this study has certain limitations. The cross-sectional design does not capture the dynamic changes in the TME over time or in response to treatment. Longitudinal studies are needed to assess how the TME evolves and its role in therapeutic resistance and disease progression. Furthermore, the in vitro functional validation of SPP1 was limited to chemoresistance assays, and additional experiments are required to explore its broader role in modulating the immune microenvironment and its potential as a therapeutic target. Finally, while the prognostic model showed good predictive performance, its validation was limited to retrospective cohorts, necessitating prospective clinical validation to confirm its utility in real-world settings. In conclusion, this study provides a comprehensive analysis of the molecular and cellular alterations associated with 3p chromosomal deletion in HNSCC. The distinct TME and pathway dysregulations observed in the mut group highlight the potential for targeted therapies that address both the genetic and immunological landscape of HNSCC. Further research is warranted to explore the therapeutic potential of targeting pathways such as SPP1 and to validate the prognostic model in larger, independent cohorts.
Conclusion
This study reveals that 3p chromosomal deletion in HNSCC leads to an immunosuppressive tumor microenvironment, contributing to poor prognosis and reduced immunotherapy efficacy. The identification of SPP1 as a key gene linked to chemoresistance and macrophage M2 polarization suggests it as a potential therapeutic target. These findings advocate for targeted therapies addressing both genetic and immunological factors in HNSCC.
Data availability
The datasets generated and analyzed during the current study are available at https://xenabrowser.net/ and https://www.ncbi.nlm.nih.gov/.
References
Johnson, D. E. et al. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 6(1), 92 (2020).
Tang, X. et al. Development and validation of an ADME-Related gene signature for survival, treatment outcome and immune cell infiltration in head and neck squamous cell carcinoma. Front. Immunol. 13, 905635 (2022).
Chi, H. et al. Natural killer cell-related prognosis signature characterizes immune landscape and predicts prognosis of HNSCC. Front. Immunol. 13, 1018685 (2022).
Bhatia, A. & Burtness, B. Treating head and neck cancer in the age of immunotherapy: A 2023 update. Drugs 83(3), 217–248 (2023).
Kitamura, N. et al. Current trends and future prospects of molecular targeted therapy in head and neck squamous cell carcinoma. Int. J. Mol. Sci. 22(1). (2020).
Cramer, J. D. et al. The changing therapeutic landscape of head and neck cancer. Nat. Rev. Clin. Oncol. 16(11), 669–683 (2019).
Gu, Z. et al. Pharmacogenomic landscape of head and neck squamous cell carcinoma informs precision oncology therapy. Sci. Transl Med. 14(661), eabo5987 (2022).
Gross, A. M. et al. Multi-tiered genomic analysis of head and neck cancer ties TP53 mutation to 3p loss. Nat. Genet. 46(9), 939–943 (2014).
Kim, H. A. J. et al. 3p arm loss and survival in head and neck cancer: An analysis of TCGA dataset. Cancers (Basel) 13(21), (2021).
Shaikh, M. H. et al. Chromosome 3p loss in the progression and prognosis of head and neck cancer. Oral Oncol. 109, 104944 (2020).
Yagyu, T. et al. Human chromosome 3p21.3 carries TERT transcriptional regulators in pancreatic cancer. Sci. Rep. 11(1), 15355 (2021).
Chen, S. M. Y. et al. Tumor immune microenvironment in head and neck cancers. Mol. Carcinog. 59(7), 766–774 (2020).
Ruffin, A. T. et al. Improving head and neck cancer therapies by Immunomodulation of the tumour microenvironment. Nat. Rev. Cancer 23(3), 173–188 (2023).
Kürten, C. H. L. et al. Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing. Nat. Commun. 12(1), 7338 (2021).
Lu, T. et al. CD73 in small extracellular vesicles derived from HNSCC defines tumour-associated immunosuppression mediated by macrophages in the microenvironment. J. Extracell. Vesicles 11(5), e12218 (2022).
Yan, Q. et al. Tumor-associated macrophage-derived Exosomal miR21-5p promotes tumor angiogenesis by regulating YAP1/HIF-1α axis in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 81(1), 179 (2024).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell. Syst. 8(4), 281–291e9 (2019).
Puram, S. V. et al. Single-Cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171(7), 1611–1624e24 (2017).
Efremova, M. et al. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15(4), 1484–1506 (2020).
Schubert, M. et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 9(1), 20 (2018).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462(7269), 108–112 (2009).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Cao, E. Y., Ouyang, J. F. & Rackham, O. J. L. GeneSwitches: ordering gene expression and functional events in single-cell experiments. Bioinformatics 36(10), 3273–3275 (2020).
Taylor, A. M. et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 33(4), 676–689e3 (2018).
Zhu, W. et al. LncRNA CYTOR promotes aberrant Glycolysis and mitochondrial respiration via HNRNPC-mediated ZEB1 stabilization in oral squamous cell carcinoma. Cell. Death Dis. 13(8), 703 (2022).
Yuan, L. Y. et al. Bioinformatic analysis and verification of a lipid metabolism-related long noncoding RNA prognostic signature for head and neck squamous cell carcinoma. Cell. Signal. 112, 110903 (2023).
Insua-Rodríguez, J. et al. Stress signaling in breast cancer cells induces matrix components that promote chemoresistant metastasis. EMBO Mol. Med. 10(10). (2018).
Qi, J. et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 13(1), 1742 (2022).
Sathe, A. et al. Colorectal cancer metastases in the liver establish immunosuppressive spatial networking between tumor-associated SPP1 + macrophages and fibroblasts. Clin. Cancer Res. 29(1), 244–260 (2023).
Nallasamy, P. et al. Pancreatic tumor microenvironment factor promotes cancer stemness via SPP1-CD44 Axis. Gastroenterology 161(6), 1998 (2021).
Liu, X. et al. ANGPTL2 + cancer-associated fibroblasts and SPP1 + macrophages are metastasis accelerators of colorectal cancer. Front. Immunol. 14, 1185208 (2023).
Brina, D. et al. The Akt/mTOR and MNK/eIF4E pathways rewire the prostate cancer translatome to secrete HGF, SPP1 and BGN and recruit suppressive myeloid cells. Nat. Cancer 4(8), 1102–1121 (2023).
Yu, S. et al. A senescence-based prognostic gene signature for colorectal cancer and identification of the role of SPP1-positive macrophages in tumor senescence. Front. Immunol. 14, 1175490 (2023).
Jiang, N., Jin, L. & Li, S. Role of SPP1 in the diagnosis of gastrointestinal cancer. Oncol. Lett. 26(3), 411 (2023).
Matsubara, E. et al. The significance of SPP1 in lung cancers and its impact as a marker for protumor tumor-associated macrophages. Cancers (Basel) 15(8). (2023).
Zou, Y. et al. SPP1 is associated with adverse prognosis and predicts immunotherapy efficacy in penile cancer. Hum. Genomics. 17(1), 116 (2023).
Funding
This work was supported by the grants of Fujian Provincial Clinical Research Center for Cancer Radiotherapy and Immunotherapy (2020Y2012); Supported by the National Clinical Key Specialty Construction Program (2021); Fujian Clinical Research Center for Radiation and Therapy of Digestive, Respiratory and Genitourinary Malignancies (2021Y2014). National Natural Science Foundation of China (82473376, 12374405); Major Scientific Research Program for Young and Middle-aged Health Professionals of Fujian Province, China (2021ZQNZD010); Joint Funds for the Innovation of Science and Technology, Fujian province (2021Y9196), Natural Science Foundation of Fujian Province (2023J011267,2024J011108, 2024J011086), and High-level Talent Training Program of Fujian Cancer Hospital (2022YNG07), Subsidy for Young and Middle-aged Experts with Outstanding Contributions in Fujian Province’s Health System in 2021–2022 (F23R-TG01-01).
Author information
Authors and Affiliations
Contributions
The authors declare their contribution as follows. C.X.C. and X.S.Q. conceived and drafted of the manuscript. P.J.P. assisted with data curation. X.W.Q., Y.H.X. and C.X. analyzed the data. C.R.H. participated in the formal analysis of the study. W.Y. and Q.S.F. provided project administration and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The data of our study came from open-access databases and did not involve data of human participants. All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, X., Xu, S., Pan, J. et al. Integrative single cell transcriptomic analysis reveals 3p deletion associated tumor microenvironment and chemoresistance in head and neck squamous cell carcinoma. Sci Rep 15, 8224 (2025). https://doi.org/10.1038/s41598-025-92078-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-92078-6
