
Abstract
Cutaneous melanoma (CM) is an aggressive skin cancer with high metastatic potential and poor prognosis. Splicing factors, which regulate pre-mRNA alternative splicing (AS) events, have been suggested as potential therapeutic targets in CM. The objective of this study was to identify candidate splicing factors involved in CM through a systems biology approach and to elucidate their roles in CM progression. 390 AS events associated with patient survival were identified using bivariate Cox regression and receiver operating characteristic (ROC) analyses. 121 splicing factors significantly associated with patient prognosis were screened by univariate Cox regression analysis. A bipartite association network between AS events and splicing factors was constructed using Spearman correlation analysis. Based on the network topology, five candidate splice factors were identified. Among them, U2SURP, a poorly characterized serine/arginine-rich protein family member, was selected for further analysis in CM. Results indicated that U2SURP gene expression was significantly negatively correlated with the Immune Infiltration Score, the infiltration levels of dendritic cells, gamma-delta T cells, natural killer (NK) cells, and cytotoxic cells, as well as the expression of the immune checkpoint gene PD-1, suggesting that U2SURP may serve as a potential target for CM immunotherapy. Experimental validation showed that U2SURP mRNA and protein were overexpressed in CM cells, and silencing of U2SURP using siRNA significantly reduced CM cell survival, proliferation and migration. Furthermore, single-cell functional analysis showed that U2SURP gene expression was positively correlated with CM cell proliferation and differentiation. This study systematically identified candidate splicing factors involved in CM and provided new insights into the role of U2SURP in CM progression. These findings contribute to a deeper understanding of the pathogenesis of CM and establish new approaches for identifying splicing-related cancer therapeutic targets.
Similar content being viewed by others
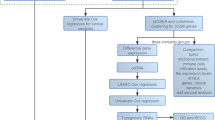


Introduction
Cutaneous melanoma (CM) is one of the most lethal forms of skin cancer, accounting for approximately 75% of skin cancer-related deaths1. In recent years, CM has become one of the malignancies with the fastest-growing incidence2. CM is characterized by high aggressiveness, early metastasis, and poor prognosis3. Especially in advanced stages, the five-year survival rate of stage IV patients is only 4.6%4,5. Despite the availability of various clinical treatment options, most patients are diagnosed at middle or late stages, leading to missed opportunities for effective intervention and overall poor outcomes6. Thus, there is an urgent need to explore new molecular mechanisms and potential therapeutic targets for CM to improve treatment strategies and patient prognosis.
Alternative splicing (AS) is a fundamental and widespread regulatory mechanism of gene expression, with aberrant AS often being a hallmark of cancer development7. Dysregulated AS has been closely linked to tumorigenesis, progression, metastatic potential, and treatment resistance8. Splicing factors play crucial roles in recognizing pre-mRNA splice sites and assembling the spliceosome9. Increasing evidence suggests that splicing factors regulate AS events, thereby driving cancer development and progression across multiple cancer types10. For example, mutations in splicing factor 3b subunit 1 (SF3B1), one of the most commonly mutated RNA splicing factors in uveal melanoma, lead to aberrant splicing of the BRD9 gene, which in turn affects cell proliferation and apoptosis11. Overexpression of serine and arginine-rich splicing factor 3 (SRSF3) has been shown to inhibit renal cell carcinoma progression by regulating the AS of Sp4 transcription factor (SP4)12. U2 snRNP-associated SURP motif-containing protein (U2SURP), a poorly defined member of the serine/arginine rich protein family, which is notably upregulated in triple-negative breast cancer (TNBC) and is associated with poor prognosis, promotes TNBC progression by regulating alternative splicing of SAT113. Knockdown of U2SURP expression inhibited the proliferation, migration and invasion of esophageal carcinoma cells14. For CM, the splicing factor PHD finger protein 5A (PHF5A) is upregulated in most CM cell lines, and is closely related to the ERK signaling pathway. Knockdown of PHF5A affects splicing defects in tumor-related genes, inhibits proliferation and promotes apoptosis in CM cell lines15. The oncoprotein SRSF1 regulates circMYC expression, which impacts melanoma cell proliferation16. Additionally, U2 small nuclear RNA auxiliary factor 2 (U2AF2) has been implicated in the AS of CD44v8-10, which influences melanoma metastasis17. Given the important role of splicing factors, we hypothesized that there are more splicing factors that play important doing roles in CM to be discovered. The investigation of abnormal AS events and their regulatory splicing factors may offer novel insights into tumorigenesis and lay a foundation for drug target identification and clinical therapeutic interventions.
In recent years, Cox regression analysis has been widely used for the identification of AS events, splicing factors18,19,20, and the prediction of cancer prognosis21,22. However, biological processes are complex, and more synergistic interactions between genes play a role in organisms, affecting cancer development and patient prognosis. Traditional univariate Cox regression analysis focuses on individual genes or AS events, while some AS events that have a low correlation with patient survival time but show a significant survival correlation when combined with other events may be overlooked23.
In this study, we adopted a systems biology approach to identify CM candidate splicing factors at the whole transcriptome level. Bivariate Cox regression analysis combined with ROC analysis was first applied to identify AS events associated with patient survival. Subsequently, an association network between splicing factors and AS events was constructed using Spearman correlation analysis, and candidate splicing factors were identified based on degree centrality ranking. The roles of the candidate splicing factors in CM were evaluated through literature review and experimental validation, demonstrating the reliability and accuracy of the screening approach. Finally, U2SURP was selected for further analysis in CM through molecular biology and cell biology experiments. This study aims to provide new perspectives and methods for investigating the molecular mechanisms underlying tumorigenesis, and provided new insights into the role of U2SURP in CM.
Results
Clinical Characteristics of the CM Cohort
Data from the TCGA database were used to analyze 98 cutaneous melanoma (CM) patient samples, which included patient survival data and AS events to identify splicing events associated with CM survival. The cohort comprised 58 male and 40 female patients, with a median age of 63 years. The mortality rate was 21.89%, highlighting the high lethality of CM. According to the American Joint Committee on Cancer (AJCC) staging criteria, 2 patients (2.04%) were in stage I, 62 (63.26%) in stage II, 27 (27.55%) in stage III, and 3 (3.06%) in stage IV. The clinical information of CM cohort from TCGA dataset is summarized in supplemental Table S1.
Screening of AS events based on Cox regression and ROC analysis
A total of 70,084 AS events related to CM were retrieved from the TCGASpliceSeq database. After excluding events with “null” PSI values and those with variance below 0.001, 8486 AS events were retained. Likelihood ratio testing and proportional hazards (ph) assumption testing were conducted using Cox regression, followed by ROC analysis, resulting in 808 two-factor combination models. The flow chart for screening candidate splicing factors was shown in supplemental Figure S1. From these, 390 AS events were selected for further analysis (supplemental Table S2).
Screening of splicing factors with Cox regression analysis and association network
A total of 404 splicing factors were initially identified (supplemental Table S3). Using Cox proportional hazards regression analysis, 121 splicing factors were found to be significantly associated with patient prognosis (likelihood ratio test p < 0.05, proportional hazards model assumption test p > 0.05). Spearman correlation analysis between AS events and these splicing factors (p < 0.05) revealed 6577 significant correlation pairs. Spearman correlation is a measure of the strength of the monotonic relationship between two variables, that is, the extent to which they keep pace with a tendency to become larger or smaller. Based on these correlations, an association network linking AS events and splicing factors was constructed (Fig. 1). From this network, the top five splicing factors were identified by degree centrality as candidate factors for further study (Table 1). Meanwhile, we selected 60% of the samples and carried out 100 analyses using the bootstrap method to construct a bipartite graph correlation network of splicing factors with AS events and calculate the degree centrality of splicing factors. The degree centrality of the five candidate splicing factors in the bootstrap analysis is shown in supplemental Figure S2.

Screen of candidate splicing factors based on bipartite graph correlation network. (A) Diagram of the association network between top five splicing factors and AS events. The red circle represents the splicing factor and other colored circles represent AS events. Green represents degree 1 for AS events, light blue represents degree 2, blue represents degree 3, light purple represents degree 4, and purple represents degree 5. (B) Degree centrality of top five candidate splicing factors.
Characterization of Candidate Splicing Factors
The role of the five candidate splicing factors in CM was investigated through univariate Cox proportional hazards regression analysis, which revealed significant associations (p < 0.05) between these splicing factors and patient survival (Fig. 2A). To validate their expression levels, two independent datasets (GSE15605 and GSE46517) were analyzed (Fig. 2B, C). The raw data of GSE15605 and GSE46517 were shown in the supplemental Table S4 and Table S5. Compared to normal skin tissues, MATR3, SRSF10, and U2SURP genes were found to be significantly overexpressed in tumor tissues, while PRPF38B showed elevated expression specifically in the GSE15605 dataset (p < 0.05). Literature evidence indicates the involvement of these five splicing factors in cancer development. MATR3 encodes a nuclear matrix protein, which is proposed to stabilize certain messenger RNA species24,25. MATR3 is overexpressed in malignant melanoma, and its knockdown inhibits CM cell proliferation both in vitro and in vivo, induces G1 phase arrest, and increases apoptosis30. SRSF10 is a member of the SR family of proteins, which are involved in constitutive and regulated RNA splicing26. SRSF10 has been implicated in multiple cancers, including liver, colorectal, and glioma31,32,33. PRPF38B enables RNA binding activity, and participates in mRNA splicing as a splicing factor27, which plays significant roles in gastric34 and ovarian cancers35. ZNF131 is predicted to regulate gene transcription and translation processes, as well as cytokine production and immune system process28. Activation of ZNF131 has been reported to regulate the expression of APEX1, thereby promoting the proliferation and migration of CM cells36. U2SURP, a key component of the spliceosome37, that enables RNA binding activity and participates in RNA processing29, is notably upregulated in TNBC and is associated with poor prognosis13. Considering that U2SURP gene has the highest significance (p = 0.002) with patient survival among the five splicing factors, and its biological function and regulatory mechanism in CM remain unexplored, we selected U2SURP gene for further study through molecular biology and cell biology experiments.

Characterization of five candidate splicing factors. (A) Results of univariate Cox regression analysis of candidate splicing factors. (B) Expression of five splicing factors in the GSE15605 dataset. (C) Expression of candidate splicing factors in the GSE46517 dataset. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Functional analysis of candidate splicing factors U2SURP
Given that U2SURP is a splicing factor that may have a broad impact in general on splicing, the 86 U2SURP-related AS events were analyzed (supplemental Table S6). The correlation between U2SURP and AS events was shown in supplemental Figure S3. These AS events correspond to 65 genes. Gene Ontology (GO) pathway analysis showed that these genes are significantly enriched in protein binding, positive regulation of gluconeogenesis, negative regulation of cell growth (Fig. 3A). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses revealed that these genes are mainly involved in metabolic pathways, protein processing in endoplasmic reticulum, fatty acid metabolism, PPAR signaling pathway (Fig. 3B). These results suggested that U2SURP may affect the splicing of these genes, which in turn affects the regulation of cellular metabolism, protein processing, and cell growth control.

Functional enrichment analysis of genes corresponding to U2SURP-related AS events. (A) GO enrichment analysis of genes. (B) KEGG enrichment analysis of genes.
Tumor-infiltrating immune cells are potential biomarkers for cancer and may aid in the development of novel diagnostic and therapeutic strategies38. To explore the association between U2SURP gene and immune cell infiltration, U2SURP gene expression levels were analyzed alongside immune cell infiltration scores. Results indicated that U2SURP gene expression was significantly negatively correlated with the immune infiltration score (Spearman’r = -0.254, FDR = 1.31E-07), as well as with the infiltration levels of dendritic cells, gamma delta T cells, natural killer (NK) cells, and cytotoxic cells. In contrast, U2SURP gene expression showed a significant positive correlation with the infiltration of iTreg, CD8 naive, and CD4 naive cells (Fig. 4A). Additionally, U2SURP gene was negatively correlated with immune checkpoint gene (ICG) PD-1 (Spearman’r = -0.13, p < 0.01), and positively correlated with PD-L1 (Spearman’r = 0.13, p < 0.01) and PD-L2 (Spearman’r = 0.14, p < 0.01). It is speculated that U2SURP gene may regulate the activity of immune system by inhibiting or activating these genes and enhance anti-tumor immune response (p < 0.05) (Fig. 4B). These findings suggest that U2SURP gene may serve as a potential target for tumor immunotherapy.

The Correlation of U2SURP expression with the level of immune cell infiltration and ICGs expression in CM. (A) Correlation analysis of U2SURP gene and the immune cell infiltration in CM. (B) Correlation analysis of U2SURP gene and ICGs PD-1, PD-L1, PD-L2, CTLA4 in CM.
To elucidate the potential role of U2SURP gene in CM, its function was assessed at the single-cell level using CancerSEA tool. The analysis revealed that U2SURP gene expression was positively associated with key cellular processes, including cell cycle progression, proliferation, differentiation, stemness, and DNA damage (Fig. 5). These results suggest that U2SURP gene may enhance proliferation and differentiation of CM cells and play a role in the development of CM.

Correlation analysis of U2SURP gene expression and functional status at single cell level in MEL-EXP0071 by CancerSEA tools. (A) Correlation between U2SURP gene expression and cell proliferation functional status in CM. (B) Functional correlation analysis of U2SURP gene expression with cell stemness and cell cycle in CM. (C) Functional correlation analysis of U2SURP gene expression with differentiation and DNA damage in CM. * p < 0.05, ** p < 0.01.
To further clarify the role of U2SURP gene in various biological pathways, Pearson correlation analysis was performed to identify genes significantly associated with U2SURP gene expression in CM, resulting in 2343 genes (r ≥ 0.6). GO and KEGG enrichment analyses were subsequently conducted. GO analysis showed that U2SURP-associated genes were mainly enriched in processes such as RNA binding, RNA splicing, transcription regulation by RNA polymerase II, and protein ubiquitination, supporting its role as a splicing factor (Fig. 6A). KEGG analysis indicated significant enrichment in the spliceosome, RNA transport, p53 signaling, mTOR signaling, and the PD-L1/PD-1 checkpoint pathway (Fig. 6B). Previous studies have highlighted similar pathways; for example, Li et al. demonstrated that WDR74 regulates melanoma initiation and metastasis via the RPL5-MDM2-p53 pathway39, while Qi et al. found that Sanggenon G inhibits melanoma invasion and migration by targeting the FAK/PI3K/AKT/mTOR pathway40. These findings imply that U2SURP-associated genes may play critical roles in CM progression.

Functional enrichment analysis of U2SURP-related genes. (A) GO enrichment analysis of U2SURP-related genes. (B) KEGG pathway enrichment analysis of U2SURP-related genes.
Expression levels of U2SURP in the CM cell lines
The expression of U2SURP gene in CM was further examined utilizing quantitative Real-Time PCR (qRT-PCR) and Western blot analyses. Compared to normal human melanocytes (PIG1), U2SURP mRNA and protein levels were significantly elevated in CM cells (A2058, A375, SK-MEL-2) (p < 0.05) (Fig. 7). The raw data of qRT-PCR in cells were shown in the supplemental Table S7. The Western blot analyses results of three biological replicates are shown in supplemental Figure S4. These results suggest that U2SURP gene may contribute to CM pathogenesis through its overexpression.

U2SURP gene expression level in CM cell lines. (A) The expression of U2SURP mRNA in PIG1 and CM cell lines was detected by qRT-PCR. (B) The expression of U2SURP protein in PIG1 and CM cell lines was detected by Western blot. (C) U2SURP protein gray analysis statistical map. **p < 0.01, ***p < 0.001, ****p < 0.0001. Biological replicates: n = 3.
Knockdown of U2SURP inhibits the proliferation and migration of CM cells
To determine the role of mRNA gene in CM progression, three siRNA sequences targeting U2SURP mRNA were designed for specific knockdown. The two most effective sequences, along with a non-targeting control (siNC), were transfected into A375 cells to assess the impact of U2SURP mRNA knockdown on cell proliferation, migration, and colony formation. qRT-PCR analysis confirmed that targeted transfection of siU2SURP could significantly reduce U2SURP mRNA expression compared with control group (Fig. 8A). The raw data of qRT-PCR in A375 cell were shown in the supplemental Table S8. The CCK8 assay revealed that U2SURP mRNA knockdown significantly decreased A375 cell viability (Fig. 8B). Further evaluation through EdU (Fig. 8C) and colony formation (Fig. 8D) assays showed that U2SURP mRNA knockdown substantially impaired cell proliferation and colony formation abilities in A375 cells. Similarly, Transwell assays demonstrated a marked reduction in cell migratory capacity following U2SURP mRNA silencing (Fig. 8E). These results indicate that U2SURP gene is essential for the proliferation and migration of CM cells, highlighting its potential as a therapeutic target in CM treatment.

The siU2SURP inhibited the proliferation and migration of A375 cells. (A) The effect of siU2SURP on the expression of U2SURP mRNA in A375 cells was detected by qRT-PCR. (B) CCK8 assay was used to detect the effect of siU2SURP on the survival of A375 cells. (C) The effect of siU2SURP on the proliferation of A375 cells was detected by EdU. (D) Clone formation assay was used to detect the effect of siU2SURP on A375 cell cloning. (E) The effect of siU2SURP on the migration of A375 cells was detected by Transwell assay. *p < 0.05, **p < 0.01, ****p < 0.0001.
Discussion
Cutaneous melanoma (CM) is a highly aggressive form of cancers41. Alternative splicing (AS) is a key molecular feature of cancers, and its dysregulation is often a hallmark of tumorigenesis. As a crucial mechanism of post-transcriptional regulation, RNA splicing plays a fundamental role in various biological processes42,43. AS is governed by splicing factors that bind to cis-elements in pre-mRNA, and their dysregulated expression contributes significantly to cancer progression44. For instance, several splicing factors are overexpressed in cancer and promote tumor growth. Wang et al. reported that BUD31, a spliceosome component, is overexpressed in ovarian cancer, promoting exon inclusion in BCL2L12 and enhancing tumorigenesis45. Similarly, Liu et al. demonstrated that PQBP1 overexpression promotes tumor progression by facilitating BAX exon skipping, leading to reduced apoptosis in ovarian cancer46. Despite the established relevance of splicing mechanisms, many aspects of these processes and their role in CM remain poorly understood. A deeper exploration of oncogenic AS events and related splicing factors could offer novel therapeutic targets for cancer. However, the existing literature mostly focuses on individual splicing factors45,46. In this study, a whole-transcriptome level systems biology approach was used to identify and characterize candidate splicing factors involved in CM. By taking a whole transcriptome approach, we are able to capture a more comprehensive view of the splicing factor landscape in relation to CM. This allows us to potentially identify a broader range of splicing events and factors that may be involved in CM. Five critical splicing factors were identified, among which U2SURP gene was selected for further investigation in CM. Our findings, for the first time, demonstrated U2SURP gene overexpression in CM and confirmed its role in promoting CM cell proliferation and migration.
Here bivariate Cox regression combined with ROC analysis was used to screen the survival-associated AS events. Compared to the traditional univariate Cox regression, this method took into account the potential synergistic effects among factors rather than independent effects. It could screen the AS events that were ignored by the traditional univariate Cox regression. Also, it was able to discover new factors from another perspective, which had not been found by traditional methods. Meanwhile, splicing factors were screened based on a bipartite graphical association network of AS events and splicing factors. Although the correlation network is still unable to completely characterize the regulatory pathway, splicing factors can be screened out based on the fact that the more important the position of a node in the network, the higher the possibility of its significant influence. Literature evidence confirmed the involvement of these splicing factors in various cancers. For U2SURP gene, 86 AS events were found to be significantly associated with U2SURP gene (supplemental Figure S3). Among them, the 4th exon-skipping in PARPBP was found to promote the proliferation, migration and invasion of ovarian cancer cells, and inhibit cell apoptosis47. Meanwhile, U2SURP gene can regulate the alternative splicing of UPF3A, generate the full-length subtype of UPF3A, and participate in the occurrence of colorectal cancer48. The loss of U2SURP leads to the formation of the intron-3 retained SAT1 isoform, which in turn promotes the carcinogenic properties of TNBC cells13. Although the relationship between U2SURP and AS events and their role in CM still need to be further verified by experiments, it still suggests that U2SURP may play an important role in CM. Further experimental validation demonstrated that both mRNA and protein levels of U2SURP were significantly upregulated in CM. Silencing U2SURP led to reduced survival, proliferation, and migration of CM cells. The effect of U2SURP on the proliferation and migration of CM cells may be related to the regulation of alternative splicing of cell-cycle genes by U2SURP49 and the influence of U2SURP on the lactylation level, as well as the interaction with immune cells in tumor microenvironment14. Single-cell analysis also indicated positive correlations between U2SURP gene and cell cycle, differentiation, and proliferation in CM. These results validate the accuracy and efficacy of the screening approach employed in this study. Moreover, the screening strategy used here is not limited to CM, and can be extended to explore splicing factors in other types of cancers, and its applicability is not restricted to the screening of splicing factors, can also be utilized to screen for regulatory factors such as transcription factors, miRNAs or lncRNAs.
The tumor immune microenvironment, which contains numerous components influencing immune responses, plays a pivotal role in cancer progression and patient prognosis50. We investigated the relationship between U2SURP gene expression and immune cell infiltration, and found a significant correlation between the U2SURP gene and the infiltration levels of CD8 + T cells, macrophages, neutrophils, iTreg, CD8 naïve cells, and CD4 naïve cells. Study has shown that tumor-specific cytotoxic effectors derived from naive CD8 + T cells are a potential means of overcoming TGF-β immunosuppression in the tumor microenvironment51. Also, regulating the mutual communication between naive CD4 T cells and M0 macrophages may improve the immunosuppressive microenvironment of cervical cancer and prevent immune escape52. This suggests that the U2SURP gene may contribute to the immune escape of CM tumor cells. Moreover, U2SURP gene expression was positively correlated with immune score in CM. Immune scores have been reported as indicators of patient survival, recurrence, metastasis, and response to immunotherapy53. In recent years, more and more studies have reported that immune checkpoint genes are involved in the development of a variety of cancers54,55. Here, we found that U2SURP gene is significantly correlated with the immune checkpoint genes PD-1, PD-L1 and PD-L2 in CM. PD-1 is an inhibitor of T cell proliferation and function and plays a critical role in immune tolerance56. PD-1 immune checkpoint blockade has significant clinical benefits for CM patients57. High expression of PD-L1 in tumor cells is one of the important reasons for drug resistance of PD-L1 inhibitors58. U2SURP may evade the attack of the immune system by decreasing the expression of PD-1, or by promoting the aberrant activation of certain signaling pathways in CM cells, resulting in decreased PD-1 expression and increased PD-L1 expression in CM. In addition, U2-related genes were significantly enriched in p53 signal, mTOR signal and PD-L1/PD-1 checkpoint pathway. The mTOR signaling pathway plays an important role in cell growth, metabolism and immune regulation40. U2SURP may affect the metabolic status and function of immune cells by regulating mTOR signaling, and then influence the tumor immune response. These results provide a new direction for the identification of potential CM immunotherapeutic targets. In future, we will further explore the relationship between U2SURP and immune checkpoint genes through experiments, as well as the influence on the biological function of CM cells.
With the rapid development of biotechnology, CITE-Seq technology plays an important role in the biomedical field because of its ability to analyze cell populations in fine detail59. In the future, we can use this technology to analyze CM samples to detect tumor cells and a variety of different immune cells infiltrating tumor tissues, and conduct in-depth analysis of tumor heterogeneity, which will promote the progress of immunotherapy to a certain extent. In addition, in recent years, the rapid development of spatial omics has provided an important solution to the problem of spatial structural characteristics related to the prognosis and treatment sensitivity of cancer patients60,61. DBIT-based spatial 3D technology can be used to further investigate how splicing factors regulate the functions of tumor cells and immune cells in specific spatial environments, which helps to reveal the role of splicing factors in tumorigenesis and development from the spatial dimension, and provides new ideas for improving patient prognosis.
Although this study has verified the effect of U2SURP gene on the biological function of CM cells, there are still some limitations. First, we performed functional experiments on just one cell line and stayed with in vitro experimental studies only. In vitro models may not fully replicate the complex in vivo environment. For example, in vitro models lack the interaction with the immune system, stromal cells, and the complex physiological and biochemical gradients present in living organisms. This may lead to differences in the observed effects of U2SURP on CM compared to what would be seen in vivo. Second, the expression and role of U2SURP in a true clinical cohort has not been confirmed. Again, the upstream regulators, as well as downstream AS events and pathways of U2SURP gene in CM still need to be further investigated. Finally, the role of U2SURP gene in guiding disease perception and clinical practice needs to be further explored. In the future, we will focus on the in-depth study of the oncogenic mechanism of U2SURP gene on CM by conducting tumor-carrying experiments in nude mice to enhance the credibility of the experimental results. We will also utilize the latest technologies, such as CITE-Seq and DBIT-based spatial 3D technology, to further study the mechanism of U2SURP gene on larger sample sets and clinical samples, so as to provide new research directions and theoretical basis for the treatment of CM patients.
Conclusions
In summary, this study used systems biology methods combined with molecular cell biology experiments to identify candidate splicing factors associated with CM, revealing a previously unrecognized role of U2SURP in CM, suggesting that it may be a potential therapeutic target for CM. Although it is necessary to further explore the role and mechanism of U2SURP gene in CM on larger sample sets and in vivo studies, this study provides a reliable method for screening AS events and splicing factors in different cancer types, and lays a foundation for further research on the mechanism of tumor-related AS events and splicing factors.
Materials and methods
Data collection and preprocessing
Alternative splicing (AS) event data for cutaneous melanoma (CM) were retrieved from the TCGA SpliceSeq database62, including AS event types, exon locations, corresponding gene names, and Percent Spliced In (PSI) values for different samples. The PSI value, ranging from 0 to 1, quantifies the proportion of transcripts resulting from a specific AS pattern. Considering that we intended to analyze survival-related AS events, we selected AS data from 98 patients and 1 normal samples that contained survival information. Each sample contained a total of 70,084 AS events, including seven types: exon skipping (ES), intron retention (RI), alternative promoter (AP), alternative terminator (AT), alternative donor site (AD), alternative acceptor site (AA), and reciprocal exon (ME). Considering the reliability of the data, AS events with with “null” PSI values were excluded because a null PSI may imply that the event was not detected in the sample with missing cargo data. In addition, AS events with a variance of PSI values < 0.001 were also removed as it indicates that the variable does not vary much across conditions and may not be biologically significant. A total of 8486 candidate AS events were obtained for further analysis.
Splicing factors associated with CM were identified using the SpliceAid2 database63 and relevant literature64. Expression data for splicing factors were obtained from the TCGA-SKCM dataset and normalized using log2(FPKM + 1).
Screening method for AS events
The Cox proportional hazards regression was performed on all possible pairs of AS events using the “survival” package in R, and the ph assumption was tested. Combinations with a likelihood ratio test p-value ≤ 0.01 and a ph test p-value > 0.05 were selected. Subsequently, ROC curves were used to evaluate the predictive capability of the selected two-factor combinations for CM patient survival. Combinations with an area under the ROC curve (AUC) > 0.85 were retained, and the corresponding AS events were extracted for further analysis.
Probing methods for candidate splicing factors
Univariate Cox proportional hazards regression analysis was conducted to identify splicing factors significantly associated with survival (likelihood ratio test p ≤ 0.05, ph assumption test p > 0.05). The Spearman correlation coefficients between AS events and splicing factors were calculated using the PSI values of AS events and the expression levels of the selected splicing factors. Based on significant correlations (p < 0.05), an association network was constructed to represent the relationships between AS events and splicing factors. The top five splicing factors with the highest degree centrality65 were identified as candidate splicing factors. Degree centrality is recognized as one of the most intuitive measures of node centrality. The higher the degree centrality of the node, the more important that node is likely to be in the network. By focusing on splicing factors with high degree centrality, we are essentially identifying those factors that are highly interconnected within the network and that may have a broad influence on the splicing events. In biological terms, a high degree centrality for a splicing factor implies that it is likely to be involved in multiple regulatory pathways related to splicing.
Validation of candidate splicing factors
To validate the accuracy and reliability of the candidate splicing factor selection, their functions and mechanisms were further investigated. The association between splicing factors and patient survival was re-evaluated using univariate Cox regression analysis. Independent Gene Expression Omnibus (GEO) datasets (GSE1560566 and GSE4651767) (https://www.ncbi.nlm.nih.gov/geo/) were used to assess differences in the expression levels of splicing factors. A literature review was conducted to determine the relationship between candidate splicing factors and cancer, with a focus on U2SURP gene, whose function has not been experimentally confirmed. The correlation between U2SURP gene and immune cell infiltration was analyzed using the Gene Set Cancer Analysis (GSCA) database68. While its association with immune checkpoint genes was examined via Spearman correlation analysis. The functional role of U2SURP gene in cancer cells at the single-cell level was explored using the Cancer Single-Cell State Atlas (CancerSEA, http://biu.edu.cn/CancerSEA/). CancerSEA is the pioneering resource that investigates the comprehensive functional states of cancer cells at a single-cell level69. The relevance between U2SURP gene expression level and functional states in MEL-EXP0071 was obtained. Co-expressed genes with U2SURP gene in CM (Pearson r ≥ 0.6) were identified through expression correlation analysis, and GO and KEGG enrichment analyses were performed on these genes using the “ClusterProfiler” R package. Finally, the impact of U2SURP gene on CM was verified through cellular experiments.
Cell culture
The PIG1 and A375 cell lines were obtained from EK-Bioscience Inc. (Shanghai, China), while A2058 and SK-MEL-2 cell lines were purchased from iCell Bioscience Inc. (Shanghai, China). Cells were cultured in high-glucose medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). Cultures were maintained in a humidified incubator at 37 °C with 5% CO₂, and passaged every 3–4 days to ensure healthy growth and prevent over confluence.
Cell transfection
Three siRNA sequences targeting U2SURP mRNA and a negative control sequence were designed and synthesized by Junji Biotechnology (Suzhou, China) (Table 2). A375 cells were transfected with siRNAs targeting U2SURP to downregulate its expression. The efficiency of U2SURP mRNA knockdown was assessed using qRT-PCR, and the two most effective siRNAs were selected for further experiments. Cells were divided into two groups: the negative control group (transfected with siNC) and the treatment group (transfected with siU2SURP). Transfection conditions were as follows: Dilute Lip8000 (5 μl) with 125 μl pure medium and mix well, dilute siRNA (3 μl) with 125 μl pure medium and mix well, mix the two well and leave for 20 min. After 24–48 h post-transfection, the culture medium was discarded, and cells were collected for subsequent experiments.
qRT-PCR and Western blot analysis
Total RNA was extracted using Solarbio reagents (Solarbio, China) according to the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized using the Kermey cDNA Reverse Transcription Kit (Kermey, Zhengzhou, China). qRT-PCR was performed on the LightCycler 480 II system (Roche, Switzerland) with the 2 × SYBR Green qPCR Premix (Kermey, Zhengzhou, China) (Table 3). GAPDH was used as an internal control for normalization.
The qRT-PCR reaction system was prepared and added into 384-well plates, with 3 multiple holes set in each group, and then the plate membrane was sealed and centrifuged at 2000 rpm for 1 min. At the same time, qRT-PCR self-test was turned on, and the plate was put into the 384-well plate after the self-test. qRT-PCR reaction conditions were as follows: 95 ℃ for 30 s; 40 cycles (95 ℃ for 5 s; 60 ℃ for 30 s, read fluorescence); the reaction conditions for the lysis curve were: 95 ℃ for 15 s (Ramp Rate 4.4 °C/s), 60 °C for 60 s (Ramp Rate 2.2 °C/s), and 95 °C for 15 s (Ramp Rate 0.11 °C/s) for 1 cycle. The qRT-PCR reaction was performed according to the set program. At the end of the reaction, the corresponding Cp values were derived according to the solubilization and amplification curves, and then the relative expression of U2SURP mRNA was detected by 2-ΔΔCT analysis.
Total protein was extracted from cells using RIPA lysis buffer (RIPA lysate: protease inhibitor = 100:1) (Solarbio, China), followed by Western blot analysis to detect U2SURP protein levels. Antibody dilution ratio: Beta-tubulin 1: 10,000 (Hangzhou HuaAn Biotechnology Company, item No. ET1602-4), U2SURP 1: 4000 (Proteintech, item No. 21399–1-AP). The bands were visualized and analyzed using the Azure C300 Imaging System (Azure, USA). Finally, Western blot analysis of protein band gray values with Image J.
Cell Counting Kit 8 (CCK8) assay
The CCK8 assay was used to assess cell proliferation. Cells were seeded in 96-well plates at a density of 3000 cells per well, with three replicates per group. At 24, 48, and 72 h post-transfection, fresh medium containing 10% CCK8 reagent was added to each well. Plates were incubated for 2 h, after which absorbance at 450 nm was measured using a microplate reader to determine cell viability.
EdU assay
Cells were seeded in 96-well plates at a density of 15,000 cells per well and incubated at 37 °C with 5% CO₂ for 2 days. EdU staining was performed according to the manufacturer’s instructions, and stained cells were observed and imaged using a fluorescence inverted microscope.
Transwell migration assay
Migration assays were conducted using 24-well plates. Each well received 600 µL of high-glucose medium supplemented with 20% FBS. A total of 20,000 cells were seeded into Transwell chambers containing 200 µL of serum-free cell suspension. After 48 h of incubation, cells were washed twice, fixed with fixative for 40 min, and rinsed with phosphate-buffered saline (PBS). Cells were then stained with 0.1% crystal violet, rinsed, and gently removed from the top surface of the membrane. Migrated cells were observed and photographed using an inverted microscope.
Cell colony formation assay
Cells were seeded in 6-well plates at a density of 1000 cells per well and incubated for 2 weeks. After incubation, cells were washed with PBS and fixed with universal tissue fixative (Servicebio, China) for 40 min. Colonies were stained with 0.1% crystal violet, washed with PBS, and imaged. Colony counting was performed using ImageJ software.
Statistical analysis
In this experiment, Prism8 (GraphPad software) was used for statistical analysis and mapping t test was used to analyze the statistical difference between the two groups. One-way ANOVA was used to analyze the differences between the three groups and more than three groups. The protein gray scale was analyzed by Image J software. p < 0.05 is considered significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Data availability
The data used in this publication have been deposited in TCGA and NCBI’s Gene Expression Omnibus and are accessible through TCGA SpliceSeq database [35] and GEO Series accession number GSE15605 [39] and GSE46517 [40]. The data sets analyzed in this study can be obtained by contacting the corresponding authors.
References
Eggermont, A. M., Spatz, A. & Robert, C. Cutaneous melanoma. Lancet 383(9919), 816 (2014).
Arnold, M. et al. Global burden of cutaneous melanoma in 2020 and projections to 2040. JAMA Dermatol. 158(5), 495 (2022).
Gosman, L. M., Țăpoi, D. A. & Costache, M. Cutaneous melanoma: A review of multifactorial pathogenesis, immunohistochemistry, and emerging biomarkers for early detection and management. Int. J. Mol. Sci. 24(21), 15881 (2023).
Long, G. V. et al. Cutaneous melanoma. Lancet 402(10400), 485 (2023).
Wang, J. Y., Wang, E. B. & Swetter, S. M. What is melanoma?. Jama 329(11), 948 (2023).
Villani, A. et al. The treatment of advanced melanoma: Therapeutic update. Int. J. Mol. Sci. 23(12), 6388 (2022).
Bradley, R. K. & Anczuków, O. RNA splicing dysregulation and the hallmarks of cancer. Nat. Rev. Cancer 23(3), 135 (2023).
Takeiwa, T. et al. Roles of splicing factors in hormone-related cancer progression. Int. J. Mol. Sci. 21(5), 1551 (2020).
Du, J. X. et al. Splicing factors: Insights into their regulatory network in alternative splicing in cancer. Cancer Lett. 501, 83 (2021).
Anczuków, O. & Krainer, A. R. Splicing-factor alterations in cancers. RNA 22(9), 1285 (2016).
Tanaka, A. et al. Understanding and therapeutic targeting of aberrant mrna splicing mechanisms in oncogenesis. Rinsho Ketsueki 61(6), 643 (2020).
Zhang, L. et al. Srsf3 suppresses rcc tumorigenesis and progression via regulating sp4 alternative splicing. Biochim. Biophys. Acta 1871(8), 119841 (2024).
Deng, L. et al. Myc-driven u2surp regulates alternative splicing of sat1 to promote triple-negative breast cancer progression. Cancer Lett. 560, 216124 (2023).
Zheng, X. et al. Integrative bioinformatics and experimental analyses identify u2surp as a novel lactylation-related prognostic signature in esophageal carcinoma. Immunol. Res. 73(1), 45 (2025).
Meißgeier, T. et al. Splicing control by phf5a is crucial for melanoma cell survival. Cell Prolif. 58, e13741 (2024).
Jin, C. et al. Circmyc regulates glycolysis and cell proliferation in melanoma. Cell Biochem. Biophys. 78(1), 77 (2020).
Zhang, P. et al. Cd82 suppresses cd44 alternative splicing-dependent melanoma metastasis by mediating u2af2 ubiquitination and degradation. Oncogene 35(38), 5056 (2016).
Jiang, Y. et al. Prognostic risk assessment model for alternative splicing events and splicing factors in malignant pleural mesothelioma. Cancer Med. 12(4), 4895 (2023).
Feng, H. et al. Identification and validation of critical alternative splicing events and splicing factors in gastric cancer progression. J. Cell Mol. Med. 24(21), 12667 (2020).
Zhang, D. et al. Systematic analysis of the relationship between ovarian cancer prognosis and alternative splicing. J. Ovarian Res. 14(1), 120 (2021).
Ben-Assuli, O. et al. Utilizing shared frailty with the cox proportional hazards regression: Post discharge survival analysis of chf patients. J. Biomed. Inform. 140, 104340 (2023).
Zhang, H. et al. Comprehensively analysis of splicing factors to construct prognosis prediction classifier in prostate cancer. J. Cell Mol. Med. 27(18), 2684 (2023).
Zhu, R. et al. A systems biology-based approach to screen key splicing factors in hepatocellular carcinoma. Mol. Carcinog. 62(8), 1107 (2023).
Erazo, A. & Goff, S. P. Nuclear matrix protein matrin 3 is a regulator of zap-mediated retroviral restriction. Retrovirology 12, 57 (2015).
Salem, A. et al. Matrin3: Disorder and als pathogenesis. Front. Mol. Biosci. 8, 794646 (2021).
Meng, X., Yang, S. & Camp, V. J. A. The interplay between the DNA damage response, RNA processing and extracellular vesicles. Front. Oncol. 9, 1538 (2019).
Abdel-Fatah, T. M. A. et al. The localization of pre mrna splicing factor prpf38b is a novel prognostic biomarker that may predict survival benefit of trastuzumab in patients with breast cancer overexpressing her2. Oncotarget 8(68), 112245 (2017).
Lai, W. S. et al. The tandem zinc finger RNA binding domain of members of the tristetraprolin protein family. Wiley Interdiscip. Rev. RNA 10(4), e1531 (2019).
De Maio, A. et al. Rbm17 interacts with u2surp and cherp to regulate expression and splicing of RNA-processing proteins. Cell Rep. 25(3), 726 (2018).
Kuriyama, H. et al. Matrin-3 plays an important role in cell cycle and apoptosis for survival in malignant melanoma. J. Dermatol. Sci. 100(2), 110 (2020).
Liu, X. et al. Srsf10 stabilizes cdc25a by triggering exon 6 skipping to promote hepatocarcinogenesis. J. Exp. Clin. Cancer Res. 41(1), 353 (2022).
Sohail, M. et al. A novel class of inhibitors that target srsf10 and promote p53-mediated cytotoxicity on human colorectal cancer cells. NAR Cancer 3(2), zcab019 (2021).
Liu, X. et al. Srsf10 inhibits biogenesis of circ-atxn1 to regulate glioma angiogenesis via mir-526b-3p/mmp2 pathway. J. Exp. Clin. Cancer Res. 39(1), 121 (2020).
Zong, Z. et al. Integrative bioinformatics analysis of prognostic alternative splicing signatures in gastric cancer. J. Gastrointest. Oncol. 11(4), 685 (2020).
Ouyang, Y. et al. Alternative splicing acts as an independent prognosticator in ovarian carcinoma. Sci. Rep. 11(1), 10413 (2021).
Huang, T. et al. Linc00470 accelerates the proliferation and metastasis of melanoma through promoting apex1 expression. Cell Death Dis. 12(5), 410 (2021).
An, J. et al. Identification of spliceosome components pivotal to breast cancer survival. RNA Biol. 18(6), 833 (2021).
Tay, C., Tanaka, A. & Sakaguchi, S. Tumor-infiltrating regulatory t cells as targets of cancer immunotherapy. Cancer Cell 41(3), 450 (2023).
Li, Y. et al. Wdr74 modulates melanoma tumorigenesis and metastasis through the rpl5-mdm2-p53 pathway. Oncogene 39(13), 2741 (2020).
Qi, X. et al. Sanguinarine inhibits melanoma invasion and migration by targeting the fak/pi3k/akt/mtor signalling pathway. Pharm. Biol. 61(1), 696 (2023).
Schadendorf, D. et al. Melanoma. Lancet 392(10151), 971 (2018).
Inoue, D. et al. Spliceosomal disruption of the non-canonical baf complex in cancer. Nature 574(7778), 432 (2019).
Leclair, N. K. et al. Poison exon splicing regulates a coordinated network of sr protein expression during differentiation and tumorigenesis. Mol. Cell 80(4), 648 (2020).
Wan, L. et al. Splicing factor srsf1 promotes pancreatitis and krasg12d-mediated pancreatic cancer. Cancer Discov. 13(7), 1678 (2023).
Wang, Z. et al. Splicing factor bud31 promotes ovarian cancer progression through sustaining the expression of anti-apoptotic bcl2l12. Nat. Commun. 13(1), 6246 (2022).
Liu, X. et al. Splicing factor pqbp1 curtails bax expression to promote ovarian cancer progression. Adv. Sci. (Weinh) 11(15), e2306229 (2024).
Chen, S. et al. Snora70e promotes the occurrence and development of ovarian cancer through pseudouridylation modification of rap1b and alternative splicing of parpbp. J. Cell Mol. Med. 26(20), 5150 (2022).
Wang, Q. et al. U2-related proteins cherp and sr140 contribute to colorectal tumorigenesis via alternative splicing regulation. Int. J. Cancer 145(10), 2728 (2019).
Martín, E. et al. Alternative splicing regulation of cell-cycle genes by spf45/sr140/cherp complex controls cell proliferation. RNA 27(12), 1557 (2021).
Sadeghi Rad, H. et al. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 41(3), 1474 (2021).
Nguyen, H. H. et al. Naïve cd8(+) t cell derived tumor-specific cytotoxic effectors as a potential remedy for overcoming TGF-β immunosuppression in the tumor microenvironment. Sci. Rep. 6, 28208 (2016).
Chen, H. et al. Integrated immunological analysis of single-cell and bulky tissue transcriptomes reveals the role of interactions between m0 macrophages and naïve cd4(+) t cells in the immunosuppressive microenvironment of cervical cancer. Comput. Biol. Med. 163, 107151 (2023).
Schwab, C. L. et al. Past, present and future targets for immunotherapy in ovarian cancer. Immunotherapy 6(12), 1279 (2014).
Wang, S. et al. Enpp1 is an innate immune checkpoint of the anticancer cgamp-sting pathway in breast cancer. Proc. Natl. Acad. Sci. USA 120(52), e2313693120 (2023).
Yi, M. et al. Exploiting innate immunity for cancer immunotherapy. Mol. Cancer 22(1), 187 (2023).
Mahoney, K. M., Rennert, P. D. & Freeman, G. J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 14(8), 561 (2015).
Hamid, O. et al. Safety and tumor responses with lambrolizumab (anti-pd-1) in melanoma. N. Engl. J. Med. 369(2), 134 (2013).
Gide, T. N. et al. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 24(6), 1260 (2018).
Liu, Y. et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial cite-seq. Nat. Biotechnol. 41(10), 1405 (2023).
Baysoy, A. et al. Spatially resolved in vivo crispr screen sequencing via perturb-dbit. bioRxiv 61, 4556 (2024).
Zhang, D. et al. Spatial dynamics of mammalian brain development and neuroinflammation by multimodal tri-omics mapping. bioRxiv 137, 1270 (2024).
Ryan, M. et al. Tcgaspliceseq a compendium of alternative mrna splicing in cancer. Nucleic Acids Res. 44(D1), D1018 (2016).
Piva, F. et al. Spliceaid 2: A database of human splicing factors expression data and RNA target motifs. Hum. Mutat. 33(1), 81 (2012).
Seiler, M. et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 23(1), 282 (2018).
Ahajjam, S. & Badir, H. Identification of influential spreaders in complex networks using hybridrank algorithm. Sci. Rep. 8(1), 11932 (2018).
Luan, H. et al. Exploration and validation of metastasis-associated genes for skin cutaneous melanoma. Sci. Rep. 12(1), 13002 (2022).
Cai, Z. et al. Fanci serve as a prognostic biomarker correlated with immune infiltrates in skin cutaneous melanoma. Front. Immunol. 14, 1295831 (2023).
Liu, C. J. et al. Gsca: An integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief Bioinform. 24(1), 558 (2023).
Yuan, H. et al. Cancersea: A cancer single-cell state atlas. Nucleic Acids Res. 47(D1), D900 (2019).
Acknowledgements
We would like to thank the TCGA, GEO databases for the availability of the data.
Funding
This research was funded by The National Natural Science Foundation of China, grant number 82103055, 32070395, and 82302642, and The key Science and Technology Research Project of Henan Province of China, grant number 242102311166.
Author information
Authors and Affiliations
Contributions
Conceptualization, WN.G. and R.Z.; methodology, ST.Z.; software, WN.G.; validation, ST.Z., YN.W. and JR.Z.; formal analysis, WN.G.; investigation, YF.Z.; resources, ST.Z.; data curation, R.Z.; writing—original draft preparation, WN.G. and ST.Z.; writing—review and editing, R.Z., LC.Z.; visualization, ST.Z. and YN.W.; supervision, WN.G.; project administration, WN.G.; funding acquisition, WN.G., R. Z., and LC.Z.. All authors have reviewed and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, S., Zhu, R., Wang, Y. et al. Comprehensive systems biology analysis reveals splicing factor contributions to cutaneous melanoma progression. Sci Rep 15, 9486 (2025). https://doi.org/10.1038/s41598-025-93695-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93695-x
