
Abstract
Glycolysis is a critical metabolic pathway in cancer cells, fulfilling their energy requirements, supporting biosynthesis, maintaining redox balance, and enabling survival in hostile environments. Alpha-enolase (ENO1) has been identified as a key promoter of tumor progression through its involvement in glycolysis. This study aims to elucidate the relationship between ENO1, glycolysis, and gemcitabine sensitivity in pancreatic cancer (PC). The expression levels of ENO1 in PC were analyzed using the GEPIA2 database, Kaplan-Meier survival plots, and immunohistochemistry (IHC). To assess the impact of ENO1 on gemcitabine sensitivity, we manipulated ENO1 expression in PC cell lines through overexpression and silencing techniques. Subsequent analyses included flow cytometry assays, glucose uptake and lactate production measurements, and cytotoxicity assays. The underlying mechanisms by which ENO1 modulates gemcitabine sensitivity were explored using Western blotting (WB). ENO1 was found to be significantly overexpressed in PC tissues, and elevated ENO1 levels were associated with poorer prognosis in PC patients. Overexpression of ENO1 reduced the sensitivity of PC cells to gemcitabine, enhancing cell proliferation, migration, and invasion by altering the cell cycle and inhibiting apoptosis. Conversely, silencing ENO1 decreased glycolysis in PC cells and heightened their sensitivity to gemcitabine. Furthermore, glycolysis inhibition—achieved through ENO1 knockdown, glucose deprivation, or treatment with 2-Deoxy-D-glucose (2-DG)—further enhanced the susceptibility of PC cells to gemcitabine. Mechanistically, ENO1 was found to regulate the expression of gemcitabine resistance-related genes, particularly ribonucleotide reductase catalytic subunit M1 (RRM1), via MYC through the glycolytic pathway, thereby contributing to gemcitabine resistance. This study demonstrates that ENO1 plays a crucial role in PC progression and is closely linked to gemcitabine resistance through its regulation of the glycolytic pathway.
Similar content being viewed by others

Introduction
PC is a highly malignant gastrointestinal tumor with a poor prognosis and is the seventh leading cause of cancer-related mortality1,2. Radical resection remains the only potential cure for this fatal disease; however, most patients have already lost the opportunity for surgical resection by the time of diagnosis. Additionally, patients who undergo surgical resection often experience recurrence and metastasis within two years, resulting in a five-year survival rate of less than 30%3. Thus, improving the effectiveness of medical treatment, particularly chemotherapy, remains a major research priority.
Among the various chemotherapeutic agents, gemcitabine has remained a cornerstone of treatment for advanced PC since 19974. While it has demonstrated some clinical benefits in prolonging progression-free survival (PFS), disease-free survival (DFS), and overall survival (OS), the complete remission (CR) rate and prognosis remain poor5,6. One of the primary important reasons for this disappointing outcome is the presence of intrinsic and acquired chemoresistance to gemcitabine7,8,9. However, the development of resistance to gemcitabine significantly limits its clinical efficacy and contributes to the poor prognosis associated with this disease.
Chemoresistance is closely related to the tumor cell metabolism10,11,12. Glycolysis, a central metabolic pathway, plays a crucial role in energy production and biosynthesis required for the rapid proliferation of cancer cells. In many cancers, including PC, the Warburg effect-a phenomenon where cancer cells preferentially utilize glycolysis even in the presence of oxygen-supports tumor growth and survival under metabolic stress13. Moreover, PC is characterized by poor vascularization, resulting in a sparse vascular distribution that resulting in a hypoxic microenvironment that promotes the Warburg effect and contributes to chemotherapeutic drug resistance8. Given this, targeting the Warburg effect in PC represents a promising strategy to reverse gemcitabine resistance.
ENO1 is a glycolytic enzyme that catalyzes the reversible conversion of 2-phosphoglycerate (2-PGA) to phosphoenolpyruvate (PEP) during glycolysis14. ENO1 overexpression activates the relatively energy-deficient aerobic glycolytic pathway contributing to chemotherapy resistance. ENO1-stimulated glycolysis has been shown to play a critical role in cisplatin resistance in gastric cancer as metabolic reprogramming in cisplatin-resistant cells is characterized by increased glycolysis. Inhibition of glycolysis has been found to significantly reverse this drug resistance15. Additionally, studies have shown that ENO1 expression is elevated in PC tumor tissues, and its upregulation is positively correlated with disease progression and gemcitabine sensitivity16,17. Emerging evidence has shown that targeting ENO1 could sensitize hypoxia induced gemcitabine chemoresistance through modulation of redox homeostasis. However, the precise relationship between ENO1, glycolysis and gemcitabine resistance in PC remains unclear and requires further investigation.
Given the central role of ENO1 in glycolysis and its potential impact on chemoresistance, this study aims to investigate the relationship between ENO1, glycolysis, and gemcitabine resistance in PC. We hypothesize that ENO1 overexpression contributes to gemcitabine resistance by enhancing glycolysis and upregulating RRM1 through MYC, a well-known transcription factor involved in metabolic reprogramming and oncogenesis. Understanding the ENO1-MYC-RRM1 axis may provide new insights into the mechanisms of gemcitabine resistance and identify potential therapeutic targets to improve the efficacy of chemotherapy in PC.
Materials and methods
Cell culture
Human PC cell lines PANC-1, MIA PaCa-2, and SW1990 were purchased from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (China) and cultured according to the standard protocol suggested by the ATCC: DMEM (Gibco, USA) supplemented with 10% foetal bovine serum (CLARK, USA), 100 U/ml penicillin, and 100 µg/ml streptomycin (Gibco, USA). The cells were incubated in a humidified incubator maintained at 37 °C with 5% CO2.
Patient specimens and immunochemistry (IHC)
All PC patient specimens were collected from surgical oncology patients at the Affiliated Hospital of Xuzhou Medical University from March 2014 to June 2019. IHC assays were carried out, and the staining intensity scores of ENO1 were determined as previously described18. According to the instructions of the PV-method IHC kit (Zhongshan biotech, China), the slides were routinely dewaxed and hydrated. Then, the slides were incubated with reagent A (endogenous peroxidase blocker) to block endogenous peroxidase activity. Next, the slides were incubated with primary antibody anti-ENO1 (1:200, 11204-1-AP, Proteintech, China) overnight at 4 °C. Subsequently, the slides were treated with reagent B (Polymer Helper) to enhance the reaction and then incubated with reagent C (polyperoxidase-anti-rabbit IgG) for secondary antibody incubation. DAB (Zhongshan biotech, China) was used to generate brown precipitates. PBS was used instead of the primary antibody as a negative control. The staining results were scored based on the percentage of positive cells and the intensity of staining. (1) Score for the percentage of positive cells: 0 (0–5% positive cells); 1 (6–25%); 2 (26–50%); 3 (51–75%); 4 (76–100%); (2) Score for staining intensity: 0 for no staining, 1 for pale yellow, 2 for brown, 3 for tan. The final score was calculated as the product of the two scores.
Analysis of gene expression
GEPIA2 (http://gepia.cancer-pku.cn/#analysis) and TNMplot (https://tnmplot.com/analysis/) were utilized to evaluate the expression of ENO1, RRM1 and MYC19,20. The KM plotter (https://kmplot.com/analysis/) was applied to explore the relationships between the expressions of ENO1 and RRM1, and OS, as well as DFS21.
Cell lines establishment and cell transfection
Short hairpin RNAs (shRNAs) targeting ENO1 and ENO1 overexpression experiments were conducted as our previous description17. For siRNA transfections, Lipofectamine RNAiMAX transfection reagents (Thermo Fisher Scientific, USA) were utilized in accordance with the manufacturer’s protocol.
Western blot
WB was performed as described previously17. Primary antibodies against ENO1 (11204-1-AP), β-actin (20536-1-AP), human Myc proto-oncogene protein (c-MYC) (10828-1-AP), RRM1 (10526-1-AP) and the secondary antibody, HRP-goat anti-rabbit IgG (H + L) (SA00001-2), were purchased from Proteintech (China).
Flow cytometry assay
For the assessment of cell apoptosis, cells were treated with different concentration gemcitabine for 36 h. The cell apoptosis was measured by Annexin V-FITC Apoptosis Detection Kit (Sigma-Aldrich) according to manufacturer’s instructions. Then, the cells were analyzed using the BD FACScan System (Becton, Dickinson and Company, USA).
For the analysis of the cell cycle, tumor cells were harvested and then fixed with 75% pre‑cold ethanol for 24 h at ‑20 °C. Cells were analyzed using the BD FACScan System after staining with propidium iodide (Sigma-Aldrich).
Cytotoxicity assay
Gemcitabine (Eli Lilly and Company, USA) was diluted in sterile PBS. Cells were then inoculated with gemcitabine at final concentration gradients of 0 nM, 2 nM, 4 nM, 8 nM, 16 nM, 32 nM, 64 nM, 128 nM, and 256 nM and incubated for 24 h. In the glycolysis-inhibited group, cells were cocultured in glucose-restricted medium, and medium with 5 mM 2-DG (Sigma, USA) added. Glucose-free DMEM (Clark, USA) was mixed with DMEM containing 4500 mg/L glucose at volume ratios of 12.5%, 25%, and 100%. Cell viability was determined using the CCK-8 kit.
Glucose uptake and lactate production detection
Cells were seeded into 6-well plates. Once the cells had attached, the medium was replaced with 3 mL of fresh medium. The supernatant medium was collected at 24 h, 48 h, and 72 h of incubation. The glucose and lactate concentrations were measured using a glucose assay kit (Folin-Wu colorimetry) (Regen Bio, China) and a lactate assay kit (Nanjing Jiancheng Biological Institute, China), respectively. The cells were trypsinized and then counted. Glucose uptake and lactate production levels were normalized to µM/106 cells. Fold changes were normalized to the control group.
Statistical analysis
All statistical analyses were completed using SPSS 23.0. Independent samples t-tests were used to compare the differences between the two groups. When *p < 0.05, or **p < 0.01, the difference is considered statistically significant.
Result
Upregulation of ENO1 correlated to poor prognosis in PC
To investigate the expression of ENO1 in PC, we analyzed the expression levels of ENO1 using the GEPIA2 database. ENO1 was significantly overexpressed in PC tissues compared to normal tissues (Fig. 1A). In the GSE16515 and GSE287 cohorts, the mRNA level of ENO1 was notably elevated in PC tissues (Fig. 1B). Kaplan-Meier survival curves were utilized to investigate the association between ENO1 expression and OS as well as DFS. The results revealed that patients with low expression of ENO1 had better survival outcomes (Fig. 1C). Additionally, Kaplan-Meier analysis revealed that in terms of T3, N0, N1, and M0 subgroups, patients with high ENO1 expression had a considerably poorer prognosis compared to those with low ENO1 expression (Fig. 1D–G). Furthermore, we assessed ENO1 expression in tumor and adjacent non-tumor tissues from 34 PC patients at our hospital. ENO1 was predominantly localized in the cytoplasm and was significantly overexpressed in PC tissues relative to normal tissues (Fig. 1H). These findings suggest that ENO1 may serve as an important prognostic marker and potential therapeutic target in PC. Further research is needed to explore the specific mechanisms by which ENO1 contributes to PC progression and evaluate its feasibility as a therapeutic target.

High ENO1 expression in PC is associated with poor prognosis. (A) The expression levels of ENO1 based on GEPIA 2. (B) ENO1 expression levels in PC samples versus normal samples from the GSE16515 and GSE28735 databases. (C) Kaplan-Meier analysis showing OS and DFS rates of PC patients with high versus low ENO1 expression. (D) OS and DFS rates for T3 stage PC patients with high versus low ENO1 expression. (E) OS and DFS rates for PC patients without lymph node metastasis, comparing high and low ENO1 expression. (F) OS and DFS rates for PC patients with lymph node metastasis, comparing high and low ENO1 expression. (G) OS and DFS rates for PC patients without distant metastasis, comparing high and low ENO1 expression. (H) IHC results showing ENO1 expression in non-cancerous versus cancerous pancreatic tissue samples. ** P < 0.01.
ENO1 contribute to resistance of gemcitabine through regulating cell cycle, cell apoptosis and glycolysis
To further investigate the mechanisms of ENO1, we explored overexpression of ENO1 and its impact on gemcitabine resistance in MIA PaCa-2 and PANC-1 cells (Fig. 2A). Gemcitabine has been reported to induce irreversible damage in cancer cells primarily by interfering with DNA synthesis, inhibiting the proliferation of cancer cells, and inducing apoptosis5,7. We then assessed changes in the cell cycle after ENO1 overexpression. Flow cytometry analysis demonstrated that ENO1 overexpression significantly alters the cell cycle distribution in both MIA PaCa-2 and PANC-1 cells. Specifically, there was a notable decrease in the percentage of cells in the G1 phase and an increase in the percentage of cells in the S and G2/M phases in ENO1-overexpressing cells compared to vector control cells (Fig. 2B). To evaluate gemcitabine sensitivity, 50 nM gemcitabine was added to assess the apoptotic level. Annexin V/PI staining reveald that ENO1 overexpression reduced the apoptosis rate induced by gemcitabine in both MIA PaCa-2 and PANC-1 cells, indicating that ENO1 decreases the sensitivity of these cells to gemcitabine (Fig. 2C). ENO1 is a key enzyme in glycolysis, and previous studies have shown that the Warburg effect is a potential target for intervention in drug resistance. This promoted us to measure cellular glycolysis by assessing glucose consumption and lactate production. The results showed that glucose consumption and lactate production were higher in the ENO1 overexpression group compared to the control group at 24 h, 48 h and 72 h, indicating increased glycolysis in MIA PaCa-2 and PANC-1 cells following ENO1 overexpression (Fig. 2D,E). Furthermore, the half-maximal inhibitory concentration (IC50) value for gemcitabine in MIA PaCa-2 and PANC-1 cells was significantly increased after ENO1 overexpression (Fig. 2F). Collectively, these findings demonstrate that ENO1 overexpression in PC cell lines promotes cell cycle progression, reduces apoptosis in response to gemcitabine, and enhances glycolytic activity.

ENO1 contributes to gemcitabine resistance and glycolysis in vitro. (A) WB analysis detecting ENO1 expression levels in MIA PaCa-2 and PANC-1 cells. (B) Cell cycle distribution by flow cytometry analysis in MIA PaCa-2 and PANC-1 cells after ENO1 overexpression. (C) Flow cytometry assay showing the percentage of apoptotic cells treated with or without 50 nM gemcitabine for 24 h. (D and E) Cellular glucose consumption (D) and lactate production (E) after overexpression of ENO1. (F) IC50 values for gemcitabine on MIA PaCa-2 and PANC-1 cells after 24 h of incubation in vitro. ** P < 0.01.
Inhibition of glycolysis promotes gemcitabine chemosensitivity in ENO1 overexpressed PC cells
ENO1 has been reported to be involved in glycolysis and glycolysis plays a crucial role in determining the sensitivity of cancer cells to gemcitabine17,22,23. To investigate whether inhibition of glycolysis could alleviate gemcitabine resistance, we cocultured ENO1-overexpressing cells with glucose-restricted medium as well as the glycolytic inhibitor 2-DG. As shown in Fig. 3A,B, lowering the glucose concentration in the culture medium (to 25% and 12.5%) increased the sensitivity of both MIA PaCa-2 and PANC-1, as well as ENO1-overexpressing cells, to gemcitabine. The IC50 value for gemcitabine with reduced glucose was more pronounced in ENO1-overexpressing cells, suggesting that these cells are more reliant on glycolysis for survival and are more vulnerable when glycolysis is inhibited. Furthermore, both 25% and 12.5% glucose concentrations resulted in higher glucose consumption and lactate production compared to control cells (Fig. 3C,D). The glycolysis inhibitor 2-DG enhanced gemcitabine sensitivity in both MIA PaCa-2 and PANC-1 cells, significantly decreased the IC50 of gemcitabine, particularly in ENO1-overexpressing cells (Fig. 3E). Additionally, treatment with 2-DG reduced glucose consumption in ENO1-overexpressing cells, with a more significant reduction observed at 48 and 72 h (Fig. 3F). This indicates that 2-DG effectively inhibits the glycolytic activity promoted by ENO1 overexpression. No significant change was observed in lactate production (Fig. 3F). These results collectively demonstrate that inhibition of glycolysis can effectively sensitize ENO1-overexpressing cells to gemcitabine.

ENO1 enhances gemcitabine resistance and glycolytic activity in vitro. (A) IC50 values for gemcitabine in MIA PaCa-2 cells with or without ENO1 overexpression under different glucose concentrations (100, 25, and 12.5%). (B) IC50 values for gemcitabine in PANC-1 cells with or without ENO1 overexpression under different glucose concentrations (100, 25, and 12.5%). (C and D) Glucose consumption and lactate production in MIA PaCa-2 (C) and PANC-1 (D) cells with or without ENO1 overexpression at different time points. (E) IC50 values for gemcitabine in MIA PaCa-2 cells with or without ENO1 overexpression after 2-DG treatment. (F) Glucose consumption and lactate production in MIA PaCa-2 and PANC-1 cells with or without ENO1 overexpression after 2-DG treatment. * P < 0.05, ** P < 0.01.
Knockdown of ENO1 enhances gemcitabine sensitivity and reduces glycolysis
The above results suggest that PC enhances cellular glycolysis levels through ENO1 overexpression, thereby promoting gemcitabine resistance. To further investigate this, shRNA targeting ENO1 was performed in SW1990 cells, which have high ENO1 expression, to establish a stable cell line with ENO1 knockdown (Fig. 4A). Flow cytometry results revealed an increase in the G1 phase population and a decrease in the S and G2/M phases in both shRNA#1 and shRNA#2 cells compared to the control (Fig. 4B). ENO1 knockdown increased the sensitivity of SW1990 cells to gemcitabine, leading to higher apoptosis rates in both shRNA#1 and shRNA#2 cells (Fig. 4C). Moreover, the IC50 values for gemcitabine, glucose consumption, and lactate production were significantly decreased in both shRNA#1 and shRNA#2 cells when glucose content in the culture medium was reduced (Fig. 4D). Additionally, upon ENO1 depletion, treatment with 2-DG further decreased the IC50 values for gemcitabine, glucose consumption, and lactate production (Fig. 4E–G). These findings suggest that targeting ENO1 to reduce glycolysis may be an effective strategy to enhance the sensitivity of PC cells to gemcitabine and improve treatment outcomes.

ENO1 knockdown enhances gemcitabine sensitivity and reduces glycolytic activity in vitro. (A) WB analysis detecting the expression levels of ENO1 in SW1990 cells after knockdown with shRNA#1 and shRNA#2. (B) Cell cycle distribution analyzed by flow cytometry in SW1990 cells following ENO1 knockdown. (C) Flow cytometry assay assessing apoptosis in SW1990 cells treated with 10 nM gemcitabine for 24 h after ENO1 knockdown. (D) IC50 values for gemcitabine in SW1990 cells after knockdown with shRNA#1 and shRNA#2. (E) IC50 values for gemcitabine, glucose consumption and lactate production in SW1990 cells at different time points under 25% glucose conditions following ENO1 knockdown. (F) IC50 values for gemcitabine, glucose consumption and lactate production in SW1990 cells at different time points under 12.5% glucose conditions following ENO1 knockdown. (G) IC50 values for gemcitabine, glucose consumption and lactate production in SW1990 cells at different time points after 2-DG treatment following ENO1 knockdown. * P < 0.05, ** P < 0.01.
ENO1 increases RRM1 expression via regulating the expression of MYC
To further investigate the mechanism by which ENO1 affects gemcitabine resistance in PC through the glycolytic pathway, we examined the expression of gemcitabine-related genes. We found that expression levels of RRM1 were significantly increased in MIA PaCa-2 and PANC-1 cells following ENO1 overexpression (Fig. 5A), whereas knockdown of ENO1 results in reduced expression of RRM1 (Fig. 5A). The expression of RRM1 was significantly upregulated in the PC tissues compared to normal tissues, based on data from the GEPIA2 and TNMplot databases (Fig. 5B). Kaplan-Meier curves were applied to investigate the association between RRM1 expression and OS as well as DFS. The results revealed that patients with high RRM1 expression had shorter survival time (Fig. 5C). No significant change was observed in the N1 subgroup (Fig. 5D). Furthermore, patients with high RRM1 expression had a shorter OS compared to those with low RRM1 expression, but no significant change was observed in DFS (Fig. 5E). Pearson correlation analysis revealed a positive correlation between RRM1 and ENO1 expression levels in PC tissues (Fig. 5F). Previous studies have shown that ENO1 mediates cell cycle progress via MYC, and one of the major causes of cellular senescence due to MYC deficiency is the downregulation of RRM-dependent deoxyribonucleotide(dNTP) pools24,25,26,27. A positive correlation was also observed between MYC and RRM1 expression levels (Fig. 5G), as well as between ENO1 and MYC expression levels (Fig. 5H) in PC tissues. These findings underscore the importance of the ENO1-MYC-RRM1 axis in the progression of PC and its potential as a target for therapeutic intervention to overcome gemcitabine resistance.

RRM1 expression correlates with poor prognosis and is regulated by ENO1 through MYC in PC. (A) WB analysis assessing expression levels of RRM1 and MYC in MIA PaCa-2 and PANC-1 cells following ENO1 overexpression, as well as ENO1 knockdown in SW1990 cells. (B) The expression levels of RRM1 in PC tissues analyzed using the GEPIA2 and TNMplot database. (C) Kaplan-Meier analysis showing OS and DFS rates of PC patients with high versus low RRM1 expression. (D) OS and DFS rates for PC patients with lymph node metastasis, comparing high and low RRM1 expression. (E) OS and DFS rates for PC patients without distant metastasis, comparing high and low RRM1 expression. (F) Pearson correlation analysis between RRM1 and ENO1 expression levels based on the GEPIA2 database. (G) The expression levels of MYC in PC tissues analyzed based on the GEPIA2 database. (H) Pearson correlation analysis between MYC and ENO1 expression levels based on the GEPIA2 database. (I) Pearson correlation analysis between MYC and RRM1 expression levels based on the GEPIA2 database.
MYC and RRM1 are critical mediators of ENO1-driven gemcitabine resistance and glycolytic activity in PC cells
To assess the necessity of MYC and RRM1 for ENO1 function, we employed siRNAs to deplete MYC or RRM1 in ENO1-overexpressing MIA PaCa-2-ENO1 and PANC-1-ENO1 cells. Our findings revealed that silencing MYC (siMYC) reduces RRM1 expression in both cell lines, confirming that MYC is upstream of RRM1 in the regulatory pathway. Silencing RRM1 (siRRM1) did not affect MYC levels, indicating that RRM1 acts downstream of MYC (Fig. 6A). Subsequent flow cytometry analysis showed that silencing MYC or RRM1 led to an increase in the G1 phase population and a corresponding decrease in the S and G2/M phases, indicating that both MYC and RRM1 are involved in promoting cell cycle progression in ENO1-overexpressing cells (Fig. 6B). MYC or RRM1 silencing also resulted in increased susceptibility of cells to gemcitabine, as evidenced by a higher apoptotic cell population (Fig. 6C) and lower IC50 values for gemcitabine (Fig. 6D). Additionally, depletion of MYC or RRM1 decreased glycolytic activity in ENO1-overexpressing cells, as evidenced by reduced glucose consumption and lactate production (Fig. 6E,F). Taken together, our results suggest that the ENO1-MYC-RRM1 axis plays a critical role in promoting gemcitabine resistance and glycolysis in PC, highlighting potential therapeutic targets for overcoming chemoresistance.

Knockdown of MYC or RRM1 partially reverses the effects of ENO1 overexpression on cell sensitivity to gemcitabine. siRNAs targeting MYC or RRM1 were transfected into MIA PaCa-2-ENO1 and PANC-1-ENO1 cells. (A) WB analysis of RRM1 and MYC. (B) Cell cycle distribution was analyzed by flow cytometry. (C) Flow cytometry assay to assess apoptosis after treatment with 50 nM gemcitabine for 24 h. (D) IC50 values for gemcitabine. (E and F) Glucose consumption and lactate production measured in MIA PaCa-2-ENO1 (E) and PANC-1-ENO1 (F) cells at indicated time points.
Discussion
PC is notoriously resistant to chemotherapeutic agents, including gemcitabine, which remains a cornerstone in the treatment of this aggressive cancer28,29. Resistance to gemcitabine is a significant challenge in the clinical management of PC, often leading to treatment failure and poor patient outcomes30,31. Previous studies have implicated several mechanisms in gemcitabine resistance, including enhanced DNA repair, altered drug metabolism, and changes in apoptotic pathways5,32. However, the role of metabolic reprogramming, particularly glycolysis, in mediating this resistance is less well understood.
In this study, we provide compelling evidence that ENO1 plays a crucial role in promoting gemcitabine resistance in PC by regulating key cellular processes such as the cell cycle, apoptosis, and glycolysis. Our data reveal that ENO1 is significantly upregulated in PC tissues compared to normal tissues, and high ENO1 expression is strongly correlated with poor prognosis. Specifically, patients with elevated levels of ENO1 exhibit shorter OS and DFS, emphasizing the potential of ENO1 as both a prognostic biomarker and a therapeutic target in PC. We also confirmed the expression of ENO1 by IHC in 34 clinical PC tissues, with results showing significantly higher ENO1 expression in PC tissues compared to paracancerous tissues, which was consistent with previous results16,18,33.
ENO1, a key glycolytic enzyme, has been shown to facilitate tumor progression by enhancing glycolysis-a metabolic hallmark of cancer cells that provides energy and biosynthetic precursors necessary for rapid proliferation15,34,35. Our previous study suggested that ENO1 promotes cell growth, tumor progression and gemcitabine resistance by increasing the expression of Yes-associated protein 1 (YAP1)17. In this study, we build on previous research by demonstrating that overexpression of ENO1 in PC cells leads to altered cell cycle dynamics, with a decrease in the G1 phase and an increase in the S and G2/M phases, thereby promoting cell proliferation and gemcitabine resistance. Additionally, ENO1 overexpression reduces apoptosis in response to gemcitabine treatment, indicating a direct role of ENO1 in decreasing the chemosensitivity of PC cells.
Glycolysis plays a crucial role in determining the sensitivity of cancer cells to gemcitabine36,37. By supporting energy production, biosynthesis, and stress management, glycolysis can contribute to chemoresistance38,39. Crucially, we found that ENO1-driven gemcitabine resistance is closely linked to enhanced glycolytic activity, as evidenced by increased glucose consumption and lactate production in ENO1-overexpressing cells. Consistent with these results, we observed a significant increase in gemcitabine sensitivity following knockdown of ENO1 in SW1990 cells, suggesting that the effect of ENO1 on glycolysis is paralleled with gemcitabine sensitivity. Based on the above results, we hypothesize that ENO1-mediated glycolysis is a key factor influencing PC gemcitabine chemosensitivity. Interestingly, inhibition of glycolysis by glucose-restricted medium or the glycolysis inhibitor 2-DG produces similar effects as ENO1 silencing, further supporting the critical role of glycolysis in mediating gemcitabine resistance.
To further explore the molecular mechanisms underlying ENO1-mediated gemcitabine resistance, we examined key enzymes involved in gemcitabine transport and metabolism. RRM1, which plays a crucial role in DNA synthesis and repair, was identified as a downstream target of ENO1 and associated with resistance to gemcitabine40,41,42. RRM1 contributes to the increased production of dNTPs, the essential building blocks for DNA synthesis and repair42. Gemcitabine’s mechanism of action involves inhibiting ribonucleotide reductase and incorporating into DNA to halt replication5. Elevated RRM1 levels can counteract this effect by maintaining dNTP pools, enabling cancer cells to repair DNA damage and resist gemcitabine-induced cell death43. MYC is a potent oncogene that regulates the expression of a wide range of genes involved in cell growth, metabolism, and apoptosis24,44,45. In this study, we demonstrated that ENO1 overexpression leads to upregulation of RRM1, a key enzyme involved in DNA synthesis and repair that has been previously implicated in gemcitabine resistance. Importantly, we showed that the regulation of RRM1 by ENO1 is mediated through MYC, a well-known oncogenic transcription factor. Silencing MYC or RRM1 in ENO1-overexpressing cells not only decreased glycolytic activity but also enhanced gemcitabine sensitivity, highlighting the therapeutic potential of targeting this axis.
Our research is a preliminary exploratory study, and there are many limitations. Firstly, there are differences between in vitro and in vivo environments, but animal experiments were not conducted to further verify the effect of ENO1 overexpression in solids tumors on malignant behaviors such as tumorigenesis, metastasis, glycolysis and gemcitabine resistance in PC cells. Secondly, the IHC samples were compared between tumor tissue and paracancerous tissue. To investigate the relationship between ENO1 expression and gemcitabine resistance, a comparison of expression levels in tumor tissues from chemotherapy-sensitive and chemotherapy-resistant patients—or between the periods of gemcitabine chemotherapy sensitivity and resistance relapse in the same patients—would be necessary, which requires further in-depth investigation in the future.
In conclusion, our study elucidates the critical role of ENO1 in promoting gemcitabine resistance in PC through upregulation of RRM1 via c-Myc and glycolysis (Fig. 7). These findings suggest that targeting ENO1 and its downstream effectors may represent a promising strategy to overcome chemoresistance and improve therapeutic outcomes for patients with PC. Further research is warranted to validate these findings in clinical settings and to explore combination therapies that target glycolysis and the ENO1-MYC-RRM1 axis.

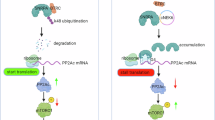
A proposed pathway illustrating ENO1-mediated gemcitabine resistance in PC Cells. This diagram illustrates a pathway where ENO1, through the regulation of MYC and RRM1, promotes cell cycle progression, inhibits apoptosis, and enhances glycolysis. These effects collectively contribute to the development of gemcitabine resistance in PC cells.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ENO1:
-
Alpha-enolase
- PC:
-
Pancreatic cancer
- IHC:
-
Immunohistochemistry
- WB:
-
Western blot
- 2-DG:
-
2-Deoxy-D-glucose
- RRM1:
-
Ribonucleotide reductase regulatory subunit M1
- PFS:
-
Progression-free survival
- DFS:
-
Disease-free survival
- OS:
-
Overall survival
- CR:
-
Complete remission
- 2-PGA:
-
2-phosphoglycerate
- PEP:
-
Phosphoenolpyruvate
- FBS:
-
Fetal bovine serum
- shRNA:
-
Short hairpin RNAs
- c-MYC:
-
Human Myc proto-oncogene protein
- IC50:
-
The half-maximal inhibitory concentration
- dNTP:
-
Deoxyribonucleotide
- YAP1:
-
Yes - associated protein 1
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49 (2024).
Huang, J. et al. Worldwide burden of, risk factors for, and trends in pancreatic cancer. Gastroenterology 160, 744–754 (2021).
Mula, M. & Sander, J. W. Current and emerging drug therapies for the treatment of depression in adults with epilepsy. Expert Opin. Pharmaco. 20, 41–45 (2019).
Burris, H. R. et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 15, 2403–2413 (1997).
de Sousa, C. L. & Monteiro, G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 741, 8–16 (2014).
Feng, M. et al. LAT2 regulates glutamine-dependent mTOR activation to promote glycolysis and chemoresistance in pancreatic cancer. J. Exp. Clin. Cancer Res. 37, 274 (2018).
Binenbaum, Y., Na’Ara, S. & Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Update 23, 55–68 (2015).
Qin, C. et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol. Cancer 19, 50 (2020).
Zhou, C. et al. LncRNA PVT1 promotes gemcitabine resistance of pancreatic cancer via activating Wnt/beta-catenin and autophagy pathway through modulating the miR-619-5p/Pygo2 and miR-619-5p/ATG14 axes. Mol. Cancer 19, 118 (2020).
Laker, R. C. et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8, 548 (2017).
Chen, L. Y. et al. CBX3 promotes proliferation and regulates glycolysis via suppressing FBP1 in pancreatic cancer. Biochem. Biophys. Res. Co 500, 691–697 (2018).
Ryan, D. G. & O’Neill, L. Krebs cycle rewired for macrophage and dendritic cell effector functions. Febs Lett. 591, 2992–3006 (2017).
Fendt, S. M. 100 Years of the Warburg effect: A cancer metabolism endeavor. Cell 187, 3824–3828 (2024).
Wang, X. X. et al. TWIST1 transcriptionally regulates glycolytic genes to promote the Warburg metabolism in pancreatic cancer. Exp. Cell Res. 386, 111713 (2020).
Qian, X. et al. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget 8, 47691–47708 (2017).
Sun, L. et al. Over-Expression of alpha-enolase as a prognostic biomarker in patients with pancreatic cancer. Int. J. Med. Sci. 14, 655–661 (2017).
Ma, H. et al. ENO1 contributes to the gemcitabine resistance of pancreatic cancer through the YAP1 signaling pathway. Mol. Carcinogen. 63, 1221–1234 (2024).
Yin, H., Wang, L. & Liu, H. L. ENO1 overexpression in pancreatic cancer patients and its clinical and diagnostic significance. Gastroent. Res. Pract. 2018, 3842198 (2018).
Tang, Z. et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102 (2017).
Bartha, A. & Gyorffy, B. TNMplot.com: A web tool for the comparison of gene expression in normal, tumor and metastatic tissues. Int. J. Mol. Sci. 22, (2021).
Gyorffy, B. Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innovation (Camb) 5, 100625 (2024).
Wang, Z. et al. Lactate promotes the growth of patient-derived organoids from hepatopancreatobiliary cancers via ENO1/HIF1alpha pathway and does not affect their drug sensitivities. Cell Death Discov. 8, 214 (2022).
Wang, L., Bi, R., Yin, H., Liu, H. & Li, L. ENO1 silencing impaires hypoxia-induced gemcitabine chemoresistance associated with redox modulation in pancreatic cancer cells. Am. J. Transl. Res. 11, 4470–4480 (2019).
Zhang, K. et al. alpha-enolase inhibits apoptosis and promotes cell invasion and proliferation of skin cutaneous melanoma. Mol. Biol. Rep. 49, 8241–8250 (2022).
Tang, H., Pan, L., Tang, L. & Liu, J. Alpha-enolase 1 knockdown facilitates the proliferation and invasion of villous trophoblasts by upregulating COX-2. Mol. Genet. Genom. Med. 11, e2220 (2023).
Hu, C. et al. CUDC-907, a novel dual PI3K and HDAC inhibitor, in prostate cancer: antitumour activity and molecular mechanism of action. J. Cell Mol. Med. 24, 7239–7253 (2020).
Mannava, S. et al. Ribonucleotide reductase and thymidylate synthase or exogenous deoxyribonucleosides reduce DNA damage and senescence caused by C-MYC depletion. Aging (Albany NY) 4, 917–922 (2012).
Coveler, A. L. et al. A phase 1b/2 randomized clinical trial of oleclumab with or without durvalumab plus chemotherapy in patients with metastatic pancreatic ductal adenocarcinoma. Clin. Cancer Res. (2024).
Philip, P. A. et al. Nab-paclitaxel plus gemcitabine in patients with locally advanced pancreatic cancer (LAPACT): a multicentre, open-label phase 2 study. Lancet Gastroenterol. 5, 285–294 (2020).
Suker, M. et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol. 17, 801–810 (2016).
Petrelli, F. et al. FOLFIRINOX-based neoadjuvant therapy in borderline resectable or unresectable pancreatic cancer: a meta-analytical review of published studies. Pancreas 44, 515–521 (2015).
Larson, A. C., Doty, K. R. & Solheim, J. C. The double life of a chemotherapy drug: Immunomodulatory functions of gemcitabine in cancer. Cancer Med. US 13, e7287 (2024).
Sun, H. et al. ENO1 expression and Erk phosphorylation in PDAC and their effects on tumor cell apoptosis in a hypoxic microenvironment. Cancer Biol. Med. 19, 1598–1616 (2022).
Fu, Q. F. et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J. Hematol. Oncol. 8, 22 (2015).
Ni, J., Huang, Y., Li, C., Yin, Q. & Ying, J. Beyond ENO1, emerging roles and targeting strategies of other enolases in cancers. Mol. Ther. Oncolytics 31, 100750 (2023).
Hu, X. et al. Shikonin reverses cancer-associated fibroblast-induced gemcitabine resistance in pancreatic cancer cells by suppressing monocarboxylate transporter 4-mediated reverse Warburg effect. Phytomedicine 123, 155214 (2024).
Wang, D., Zhang, P., Liu, Z., Xing’, Y. & Xiao, Y. NXPH4 promotes gemcitabine resistance in bladder cancer by enhancing reactive oxygen species and glycolysis activation through modulating NDUFA4L2. Cancers 14, (2022).
Penny, H. L. et al. Targeting glycolysis in macrophages confers protection against pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 22, (2021).
Xi, Y. et al. hENT1 reverses chemoresistance by regulating glycolysis in pancreatic cancer. Cancer Lett. 479, 112–122 (2020).
Ueno, H., Kiyosawa, K. & Kaniwa, N. Pharmacogenomics of gemcitabine: can genetic studies lead to tailor-made therapy? Br. J. Cancer 97, 145–151 (2007).
Xie, H. et al. Predictive and prognostic roles of ribonucleotide reductase M1 in resectable pancreatic adenocarcinoma. Cancer Am. Cancer Soc. 119, 173–181 (2013).
Nakano, Y. et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br. J. Cancer 96, 457–463 (2007).
Hasselluhn, M. C. et al. An NFATc1/SMAD3/cJUN complex restricted to SMAD4-deficient pancreatic cancer guides rational therapies. Gastroenterology 166, 298–312 (2024).
Yang, J. et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol. Life Sci. 77, 305–321 (2020).
Biancur, D. E. & Kimmelman, A. C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. BBA Rev. Cancer 1870, 67–75 (2018).
Funding
This study was supported by the Department of Science & Technology, Xuzhou, Jiangsu, China (Grant number: KC20054).
Author information
Authors and Affiliations
Contributions
These Authors contributed equally: YL and HM. The project design was completed by JW and YL. The operation experiment was carried out by YL, HM, XX, YD and LL. Data analysis for YL. YL and HM finished writing the manuscript. Data review and article review by JW and WZ. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was conducted in compliance with the declaration of Helsinki. Informed consent was obtained from all subjects. The ethics approval statements for human subjects were provided by the Ethnic Committee of the Affiliated Hospital of Xuzhou Medical University (NO. XYFY2015-JS001-01).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, Y., Ma, H., Xiong, X. et al. Deletion of ENO1 sensitizes pancreatic cancer cells to gemcitabine via MYC/RRM1-mediated glycolysis. Sci Rep 15, 9941 (2025). https://doi.org/10.1038/s41598-025-94319-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94319-0
