
Abstract
Difficult-to-treat resistant Pseudomonas aeruginosa (DTR-PA) is an MDR subset resistant to all first-line antipseudomonal agents. It is particularly concerning in respiratory infections like bronchiectasis, leading to poor outcomes, limited treatment options, and higher healthcare costs. This study aimed to investigate the antimicrobial resistance profiles and sequence types (STs) of Pseudomonas aeruginosa isolates, providing insight into their resistance patterns and the factors contributing to resistance. In 2021, 38 multidrug-resistant P. aeruginosa isolates were collected from bronchiectasis patients from a single medical center in Ningbo, China. The isolates were obtained from various clinical samples, including sputum, secretion, urine, and blood. Minimum inhibitory concentration testing revealed that 97.4% of the isolates were sensitive to amikacin and tobramycin, with none showing resistance to polymyxin B. Resistance rates to imipenem and meropenem were 84.2% and 57.9%, respectively, with 44.7% of isolates classified as DTR-PA. Multilocus sequence typing identified ST277 (18.4%), ST1076 (13.2%), and ST3012 (13.2%) as the predominant DTR-PA sequence types. The presence of blaPAO, aph(3′)-IIb, and catB7, in all isolates and blaOXA-50 (16 isolates) and crpP genes (24 isolates) in coexisitance in 11 of 16 isolates, suggested a strong association with the DTR phenotype. Phylogenetic analysis grouped DTR-PA isolates into distinct evolutionary lineages (II and III), underscoring their genetic relatedness and potential for clonal spread. Our findings suggest that co-harboring blaOXA-50 and crpP contributes to the development of DTR-PA, highlighting the need for continuous monitoring of these resistance determinants. While the study provides important insights into antimicrobial resistance in DTR-PA, further research is needed to explore resistance development across different infection sites and clinical settings.
Similar content being viewed by others

Introduction
Pseudomonas aeruginosa is a member of the ESKAPE pathogens, a group of bacteria known for their multidrug resistance and significant role in hospital-acquired infections1. This pathogen is also relevant in a One Health context, as it is commonly found in humans, animals, and environmental reservoirs2. P. aeruginosa is a common pathogen linked to community and hospital-acquired infections, particularly in bronchiectasis. Several studies have shown that P. aeruginosa can adversely affect the progression and clinical outcomes of bronchiectasis3. Likewise, evidence confirming the impact of spontaneous clearing or treatment of P. aeruginosa in bronchiectasis remains limited. Even though P. aeruginosa is frequently isolated from pneumonia worldwide, including regions such as Asia–Pacific, Europe, Latin America, and North America. It has been reported that P. aeruginosa is most commonly associated with pneumonia in hospitalized patients (44.6%), followed by bloodstream infections (27.9%) and surgical site infections (19.1%)4 This global prevalence highlights its critical role as a leading pathogen in respiratory infections, emphasizing the need for continued surveillance and effective infection control strategies5. In parallel, the SENTRY global surveillance program has documented a concerning rise in multidrug-resistant (MDR) P. aeruginosa strains, with MDR rates among isolates reaching up to 26.3%6. Notably, a small proportion of isolates (less than 0.1%) were also reported as pan-drug resistant, making P. aeruginosa the most resistant species observed during the surveillance7. Due to resistance and subsequent treatment challenges, the notion of difficult-to-treat resistance (DTR) has emerged to represent the efficacy and safety of clinically available antibiotic alternatives. The modified mortality risk was greater for patients with difficult-to-treat-resistant P. aeruginosa (DTR-PA) compared to those with carbapenem-resistant P. aeruginosa8. Furthermore, various epidemic P. aeruginosa clones, including ST175, ST235, and ST111, characterized by multidrug-resistant profiles, are associated with poorer prognoses9. Consequently, DTR-PA more thoroughly captures the clinical significance of antibiotic resistance concerning mortality risk10. DTR-PA treatment presents challenges due to various mechanisms of antimicrobial resistance, including efflux pumps, biofilm formation, antibiotic-inactivating enzymes, target mutations, and a range of genes acquired through horizontal gene transfer, which complicate clinical interventions11,12. This study aimed to investigate the molecular epidemiology of antibiotic resistance DTR-PA-induced bronchiectasis.
Results
Determination of MIC
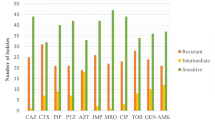
Among the 38 P. aeruginosa isolates tested, 37 (97.4%) were sensitive to amikacin and tobramycin, while none showed resistance to polymyxin B. However, high resistance rates were observed for imipenem (84.2%) and meropenem (57.9%). Seventeen isolates were classified as DTR-PA. The minimum inhibitory concentrations (MICs) of 12 antibiotics against P. aeruginosa are summarized in Table 1.
MLST and antimicrobial resistance genes (ARGs)
The most common sequence types (STs) identified among the 38 P. aeruginosa isolates were ST277 (7/38, 18.4%), ST1076 (5/38, 13.2%), and ST3012 (5/38, 13.2%) (Fig. 1). All isolates belonging to ST1076, ST1623, and ST3012 were classified as DTR-PA. Genetic analysis revealed that blaPAO, aph(3′)-IIb, and catB7 genes were present in all isolates. Additionally, 24 isolates carried the crpP gene, while 16 harbored blaOXA-50. Notably, the coexistence of blaOXA-50 and crpP (11/16 isolates) was strongly associated with developing DTR-PA.

The phylogenetic tree representing 38 P. aeruginosa isolates. The blue background signifies DTR-PA. The red blocks indicate the presence of positive blaOXA-50 or crpP.
Phylogeny and genetic environment
Core genome phylogenetic analysis revealed that the isolates clustered into three distinct evolutionary lineages (Fig. 1). Isolates within lineage I did not include any difficult-to-treat resistant P. aeruginosa (DTR-PA). In contrast, ST1076 isolates clustered into lineage II, while ST1623 and ST3012 isolates grouped into lineage III, all classified as DTR-PA. Furthermore, genetic environment analysis of blaOXA-50 indicated that the presence of inserted elements downstream of blaOXA-50 likely contributed to the non-DTR-PA phenotype (Fig. 2).

Genetic environment of blaOXA-50 in 16 P. aeruginosa isolates.
Discussion
The emergence of DTR-PA in bronchiectasis and other chronic respiratory conditions represents a significant clinical challenge. Patients with bronchiectasis frequently experience recurrent infections, and colonization with DTR-PA is associated with poorer clinical outcomes, including increased exacerbations, reduced lung function, and higher mortality rates13,14. The treatment of DTR-PA infections is particularly challenging due to its resistance to multiple antibiotic classes, necessitating the exploration of alternative therapies such as inhaled antibiotics, bacteriophage therapy, and antimicrobial peptides15. Combination therapies, including β-lactam/β-lactamase inhibitors, polymyxins with aminoglycosides, and dual carbapenem therapy, have shown promise in treating multidrug-resistant P. aeruginosa16. Given the resistance rates observed in our study, personalized treatment strategies incorporating susceptibility-guided combination regimens should be considered to optimize clinical outcomes.
Our findings align with global trends, where increasing carbapenem resistance in P. aeruginosa has been widely documented. For instance, Shortridge et al.5 reported a rise in multidrug-resistant strains across multiple countries, highlighting the role of horizontal gene transfer in the global dissemination of resistance determinants.
In our study, P. aeruginosa isolates exhibited elevated resistance rates, with 84.2% resistant to imipenem and 57.9% to meropenem, consistent with global data. These results are corroborated by studies such as that of Olalekan et al.17, which reported carbapenem resistance in over 40% of P. aeruginosa isolates, including 39% resistance to imipenem and 44% to meropenem, often linked to the presence of carbapenemase-producing strains and the role of horizontal gene transfer in spreading resistance genes.
MLST typing revealed that ST1076, ST1623, and ST3012 were the predominant sequence types among DTR-PA isolates. ST1076 has been reported in hospital-associated infections worldwide18, while ST1623 and ST3012 P. aeruginosa harbouring blaOXA-50 and crpP were identified as DTR-PA. The coexistence of blaOXA-50 and crpP genes was strongly associated with DTR-PA, with 44.7% (17/38) of the isolates identified as DTR-PA , potentially due to the predominance of sputum-derived isolates in our study10. The blaOXA-50 gene, an Ambler Class D β-lactamase, encodes a constitutively expressed oxacillinase that contributes to β-lactam resistance19. Similarly, Shortridge et al.5 reported that P. aeruginosa from pneumonia cases exhibited a slightly higher multidrug resistance (MDR) rate (27.7%) compared to bloodstream infections (23.7%). All the isolates in our study carried blaPAO, aph(3′)-IIb, and catB7, genes commonly associated with carbapenem-resistant P. aeruginosa 20.
blaPAO encodes an intrinsic β-lactamase that hydrolyzes penicillins and cephalosporins19, while aph(3′)-IIb encodes an aminoglycoside phosphotransferase conferring resistance to aminoglycosides such as kanamycin and gentamicin20. Additionally, catB7 acetylates chloramphenicol21, rendering it ineffective. The presence of these genes underscores the multifaceted resistance mechanisms in DTR-PA strains.
Among OXA variants, OXA-51-like subfamily is highly prevalent in P. aeruginosa22. Although OXA-50 has weak carbapenem hydrolyzing activity, its presence, alongside other resistance mechanisms, can enhance carbapenem resistance23. Our findings suggest that the coexistence of blaOXA-50 and crpP contributes to the development of DTR-PA, in contrast to the presence of blaOXA-50 alone. Furthermore, genetic elements inserted downstream of blaOXA-50 were associated with non-DTR-PA phenotypes.. While the prevalence of crpP-like gene has been reported at 25.4% in P. aeruginosa21, its association with fluoroquinolone resistance remains inconsistent24. Thus, the co-harboring of blaOXA-50 and crpP may be critical in driving DTR-PA. Core genome phylogenetic analysis revealed that DTR-PA isolates grouped into lineages II and III. Similarly, ST1076 isolates clustered into lineage II, while ST1623 and ST3012 isolates grouped into lineage III, all classified as DTR-PA. Isolates in lineage II predominantly carried blaOXA-50 and crpP, which have been linked to high-level resistance in clinical P. aeruginosa strains22. Lineage III isolates showed a higher prevalence of ST3012, an emerging sequence type associated with healthcare-associated infections.
However, our study has several limitations. First, the limited number of participants prevents drawing region-wide conclusions. Second, we were unable to evaluate the virulence of the isolates. Furthermore, most strains were derived from sputum samples, limiting the generalizability of our findings. Future studies involving DTR-PA from different infection sites and resistance mechanisms are warranted.
Conclusion
Our study’s primary mechanism underlying DTR-PA was the coexistence of blaOXA-50 and crpP in isolates from bronchiectasis patients. Therefore, continuous monitoring of P. aeruginosa for these resistance genes is crucial. Further research on antibiotic resistance mechanisms in DTR-PA is essential to enhance clinical management and improve patient care.
Methods
Bacterial strains
Thirty-eight P. aeruginosa strains were isolated from patients with bronchiectasis in the respiratory care department of a medical center in Ningbo, China, in 2021. Of these, 33 strains were obtained from sputum samples, three from secretion samples, one from a urine sample, and one from a blood sample.
Antibiotic susceptibility test
The minimum inhibitory concentrations (MICs) of 11 antibiotics against P. aeruginosa were determined using the agar dilution method, while the MIC for polymyxin B was measured using the broth dilution method. According to the tiered drug guidelines recommended by Clinical and Laboratory Standards Institute (CLSI), the tested antibiotics included piperacillin, tazobactam, ceftazidime, cefpirome, aztreonam, imipenem, meropenem, gentamicin, amikacin, tobramycin, ciprofloxacin, and fosfomycin, all obtained from Dalian Meilun Biotech (Dalian, China). Polymyxin B and glucose-6-phosphate (G-6-P) were purchased from Sigma-Aldrich (St. Louis, MO, USA). MIC breakpoints were determined according to CLSI guidelines (https://clsi.org/standards/products/microbiology/documents/m100/). The criteria for classifying strains as DTR-PA were resistance to ceftazidime (≥ 32 mg/L), aztreonam (≥ 32 mg/L), piperacillin/tazobactam (≥ 128/4 mg/L), carbapenems (imipenem or meropenem ≥ 8 mg/L), or ciprofloxacin (≥ 2 mg/L)10.
Genome sequencing and data analysis
Whole-genome sequencing (WGS) of DTR-PA isolates was performed using the Illumina NovaSeq 6000 platform (Novogene Bioinformatics Technology Co., Ltd, Beijing, China), We used paired-end sequencing (PE150) with at least 200X depth for accurate genomic data. Quality control followed standard Illumina protocols, with Fastp (v0.23.1) cleaning raw reads of adapters, ambiguous nucleotides (> 10%), and low-quality bases (Phred score < 5 in > 50% of bases). Resistance genes were identified using the ResFinder v3.0 web server (http://www.genomicepidemiology.org) with a minimum identity of 90% and coverage of 60%. Plasmid typing was conducted via the Plasmid typing database (https://pubmlst.org/bigsdb?db=pubmlst_plasmid_seqdef&page=sequenceQuery). MLST analysis utilized seven housekeeping genes: acsA, aroE, guaA, mutL, nuoD, ppsA, and trpE, with profiles compared to the PubMLST database. Core genome phylogenetic analysis was conducted using Roary (https://sanger-pathogens.github.io/Roary/) based on default parameters, a pan-genome analysis tool, which clustered predicted protein sequences into orthologous gene families using 95% blastp identity and 90% coverage. Roary identified core genes present in all genomes, and their nucleotide sequences were aligned with MAFFT. These alignments were concatenated into a supermatrix for phylogenetic inference, and the core genome phylogenetic tree was reconstructed using FastTree, and the resulting phylogenetic tree was visualized and exported as an SVG file using the Interactive Tree of Life (iTOL) platform25, and further customized, including background colors and additional annotations, using Adobe Illustrator.
Data availability
The sequencing data for DTR-PA had been deposited at GenBank under the accession of PRJNA890049.
References
Miller, W. R. & Arias, C. A. ESKAPE pathogens: Antimicrobial resistance, epidemiology, clinical impact and therapeutics. Nat Rev Microbiol 22(10), 598–616 (2024).
Rizk, A. M. et al. Phenotypic and genotypic characterization of resistance and virulence in Pseudomonas aeruginosa isolated from poultry farms in Egypt using whole genome sequencing. Vet Microbiol 292, 110063 (2024).
Chai, Y. H. & Xu, J. F. How does Pseudomonas aeruginosa affect the progression of bronchiectasis?. Clin Microbiol Infect 26(3), 313–318 (2020).
Reyes, J. et al. Global epidemiology and clinical outcomes of carbapenem-resistant Pseudomonas aeruginosa and associated carbapenemases (POP): A prospective cohort study. Lancet Microbe 4(3), e159–e170 (2023).
Shortridge, D. et al. Geographic and temporal patterns of antimicrobial resistance in Pseudomonas aeruginosa over 20 years from the SENTRY antimicrobial surveillance program, 1997–2016. Open Forum Infect Dis 6(Suppl 1), S63–S68 (2019).
Shortridge, D., Streit, J. M., Mendes, R. & Castanheira, M. In vitro activity of Cefiderocol against U.S. and European gram-negative clinical isolates collected in 2020 as part of the SENTRY antimicrobial surveillance program. Microbiol Spectr 10(2), e0271221 (2022).
Diekema, D. J. et al. The microbiology of bloodstream infection: 20-year trends from the SENTRY antimicrobial surveillance program. Antimicrob Agents Chemother 63(7), 10–1128 (2019).
Machuca, I. et al. Real-world experience of imipenem-relebactam treatment as salvage therapy in difficult-to-treat Pseudomonas aeruginosa infections (IMRECOR study). Infect Dis Ther 14(1), 283–292 (2025).
Lebreton, F. et al. A panel of diverse Pseudomonas aeruginosa clinical isolates for research and development. JAC Antimicrob Resist 3(4), dlab179 (2021).
Kadri, S. S. et al. Difficult-to-treat resistance in gram-negative bacteremia at 173 US HOSPITALS: Retrospective cohort analysis of prevalence, predictors, and outcome of resistance to all first-line agents. Clin Infect Dis 67(12), 1803–1814 (2018).
Horcajada, J. P. et al. Epidemiology and Treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin Microbiol Rev 32(4), 10–1128 (2019).
Botelho, J., Grosso, F. & Peixe, L. Antibiotic resistance in Pseudomonas aeruginosa—Mechanisms, epidemiology and evolution. Drug Resist Updat 44, 100640 (2019).
Zaragoza, R. et al. Update of the treatment of nosocomial pneumonia in the ICU. Crit Care 24(1), 383 (2020).
Flume, P. A. et al. Clinical practice guidelines for pulmonary therapies C: Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med 180(9), 802–808 (2009).
Lynch, J. P. 3rd. & Zhanel, G. G. Pseudomonas aeruginosa pneumonia: Evolution of antimicrobial resistance and implications for therapy. Semin Respir Crit Care Med 43(2), 191–218 (2022).
Bassetti, M., Peghin, M., Vena, A. & Giacobbe, D. R. Treatment of infections due to MDR gram-negative bacteria. Front Med (Lausanne) 6, 74 (2019).
Olalekan, A. et al. High incidence of carbapenemase-producing Pseudomonas aeruginosa clinical isolates from Lagos, Nigeria. JAC Antimicrob Resist 5(2), dlad038 (2023).
Magalhaes, B. et al. Combining standard molecular typing and whole genome sequencing to investigate Pseudomonas aeruginosa epidemiology in intensive care units. Front Public Health 8, 3 (2020).
Girlich, D., Naas, T. & Nordmann, P. Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob Agents Chemother 48(6), 2043–2048 (2004).
Torres, R. T., Cunha, M. V., Ferreira, H., Fonseca, C. & Palmeira, J. D. A high-risk carbapenem-resistant Pseudomonas aeruginosa clone detected in red deer (Cervus elaphus) from Portugal. Sci Total Environ 829, 154699 (2022).
Xu, Y. et al. The prevalence and functional characteristics of CrpP-like in Pseudomonas aeruginosa isolates from China. Eur J Clin Microbiol Infect Dis 40(12), 2651–2656 (2021).
Pandey, D., Singhal, N. & Kumar, M. Investigating the OXA variants of ESKAPE pathogens. Antibiotics (Basel) 10(12), 1539 (2021).
Petrova, A., Feodorova, Y., Miteva-Katrandzhieva, T., Petrov, M. & Murdjeva, M. First detected OXA-50 carbapenem-resistant clinical isolates Pseudomonas aeruginosa from Bulgaria and interplay between the expression of main efflux pumps, OprD and intrinsic AmpC. J Med Microbiol 68(12), 1723–1731 (2019).
Hernandez-Garcia, M. et al. Presence of chromosomal crpP-like genes is not always associated with ciprofloxacin resistance in Pseudomonas aeruginosa clinical isolates recovered in ICU patients from Portugal and Spain. Microorganisms 9(2), 388 (2021).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res 47(W1), W256–W259 (2019).
Funding
This study was supported by funding from the National Key R&D Program of China (2023YFC2308400); and National Natural Science Foundation of China (82072314).
Author information
Authors and Affiliations
Contributions
Lulu Jin: Conceptualization, Methodology, Amina shahzadi: Writing- Original draft preparation, Data curation. Jing Guo: Software, Data curation, project management. Haowei Ye: Software, Data curation and investigation. Hao Xu: Software and investigation. Xinling Pan: Reviewing and Editing, Software and Project management. Danfeng Lou: Investigation, Reviewing and Editing, Software and Project management.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jin, L., Ye, H., Xu, H. et al. Genomic epidemiology and characterization of difficult-to-treat resistant Pseudomonas aeruginosa isolates co-harboring blaOXA-50 and crpP causing bronchiectasis. Sci Rep 15, 12932 (2025). https://doi.org/10.1038/s41598-025-95950-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-95950-7
Keywords
This article is cited by
-
Antimicrobial resistance patterns and carbapenemase gene distribution in pediatric Pseudomonas aeruginosa isolates: molecular and epidemiological insights from an Iranian referral center
BMC Microbiology (2026)
-
Parallel microevolution of multidrug resistance and hypermucoviscosity in two carbapenem-resistant Pseudomonas aeruginosa clinical isolates with strikingly divergent virulence outcomes
BMC Microbiology (2026)
