
Abstract
Whitefly, Bemisia tabaci is a globally recognized invasive cryptic pest species complex and a primary vector for 90% of begomoviruses. Understanding the species composition and diversity within the B. tabaci cryptic species complex is essential for developing effective pest management strategies. The Asia II 1 genetic group of B. tabaci is notably widespread in India and across Asia, demonstrating significant genetic diversity. Our study investigates the haplotype diversity of Asia II 1 using the mtCOI barcoding gene, analyzing 676 sequences from various Asian countries and 190 sequences from India. We identified 241 distinct haplotypes in Asia II 1 across Asia, with the highest haplotype diversity in China (Hd: 1.000) and the lowest in Vietnam (Hd: 0.667). Nucleotide diversity peaked in Pakistan (pi: 0.0145) and was lowest in Vietnam (pi: 0.0010). In India, we identified 77 haplotypes with a diversity of 0.926 and nucleotide diversity of 0.0076. When grouped by hostplant families, 79 haplotypes were recorded, with the highest diversity in Cucurbitaceae and the lowest in Solanaceae. Our findings suggest that hostplants and geographical location significantly influence genetic group development, offering novel insights into Asia II 1’s genetic structure and evolution. This marks the first comprehensive study of Asia II 1 genetic diversity in Asia and India.
Similar content being viewed by others

Introduction
Evolutionary processes in the ecosystem that contributed significantly to speciation in ecologically and genetically divergent populations have drawn the attention of many1,2 and become one of the most fascinating aspects in evolutionary biology, leading to the study of speciation in phytophagous insect pests3,4. One perception of speciation is more like a continuous phase involving polymorphic populations as they evolve into distinct species called ecological races5. Populations of morphologically conserved lineages that are genetically divergent and perhaps even reproductively isolated are referred to as cryptic species or sibling species because of their previous classification mostly as a single taxon, based on similar morphologies. Cryptic species are more common than previously anticipated and are now known to occur across major metazoan taxa and biogeographic ranges6. With the recent advances in molecular and genetic solutions, the identification and description of cryptic species has increased exponentially over the last two decades7.
Multiple co-distributed cryptic lineage studies incorporating phylogeographic and population genetic perspectives provide an excellent framework for understanding cryptic biodiversity8. Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) is one such cryptic complex for which the taxon was first described in 1889 as Aleyrodes tabaci9. This species complex comprises an unprecedented number of cryptic lineages globally10,11, some of which currently overlap in geographic coverage12. Morphologically indistinguishable lineages of B. tabaci were typically categorised as biotypes, many of which have recently been described as mitochondrial (mtCOI) haplotypes, and exhibit variability in some biological and ecological traits such as the efficiency of plant virus transmission, insecticidal tolerance, dispersal, mating behaviour, and fecundity12,13. The whitefly, B. tabaci cryptic species group comprises morphologically indiscernible lineages of well-known exemplars referred to as biotypes. The destructive sap sucking pest distributed throughout tropical and subtropical latitudes includes the contemporary invasive haplotypes, termed B and Q14. Several well-studied B. tabaci cryptic species exhibit ecological and biological diversity; however, most members are poorly studied or completely uncharacterized.
A considerable effort has already been made to overcome the systematics of this species complex. The previous classification structure of B. tabaci populations into biotypes and host races based on different biological and biochemical markers has been superseded by molecular investigations based on the partial sequence of the mtCOI gene15,16,17. In addition, the genetic distance threshold of 3.5% was defined based on the difference in the distribution of pairwise sequence divergence between the unique mtCOI partial sequences of B. tabaci16. In the subsequent recent analyses, the pattern of putative species clusters were delineated by sequence divergence equal to or higher than 3.5%18.
The distribution of B. tabaci, distribution stretches across the continents, with the Indian subcontinent being postulated as its centre of origin19. It is an agriculturally and economically important insect pest because of its polyphagous nature and super-vectoring of begamoviral diseases20. Presently, B. tabaci is regarded as a complex comprising at least 46 genetic groups21,22 and nearly 13 genetic groups are recorded so far from India, viz., Asia I, Asia I-India, Asia II 1, Asia II 5, Asia II 6, Asia II 7, Asia II 8, Asia II 11, Asia II 13, Middle East Asia Minor (MEAM)-1, MEAM-K, China 3 and China 721,23,24. The eastern and southern parts of India showed maximum diversity compared to northern and north-western India, respectively, with the prevalence of Asia I and Asia II genetic groups. The pattern of dispersion of B. tabaci is driven by a number of factors involving geographical area, host plants, and export-related anthropogenic activities25,27. The Asia II 1 genetic group was found to be more predominant with its widespread distribution across the tropical, subtropical, and temperate zones of India and exhibited the highest haplotype diversity28. Asia II 1 and Asia II 7 are the leading genetic groups occurring in Delhi27 and especially, Asia II 1 is the most abundant and predominantly distributed genetic group in cotton leaf curl disease (CLCuD)-prone north Indian cotton growing states29,30 and in Punjab, the incidence of CLCuD is closely associated with Asia II 1, and the disease incidence was much lower in the areas where Asia II 1 is absent29. Major cotton-growing areas of Pakistan also depicted Asia II 1 as a major genetic group31.
To reveal the prevalence and dominance of constantly evolving B. tabaci genetic group complex, one needs a better understanding of its phylogeographical patterns of genetic variation, species composition, haplotype diversity, migration histories, and demographic records. Dispersion and distribution of Asia II 1 stretches across the country in the recent past and are found prevalent in all the agroclimatic zones of India. This paper aims to reveal the haplotype diversity, phylogenetic status, and genetic differentiation of the Asia II 1 genetic group of B. tabaci across Asia and India, as well as its genetic variation on different host plants in India.
Materials and methods
B. tabaci sample collection
Specimens of B. tabaci were collected from farmer’s fields in various locations as listed in Suppl. File 1; Table S1. Permissions were taken from the farmers for the collection of samples on their land. The adult whiteflies were collected using a hand-held aspirator in 1.5 ml sample collection tubes (Abdos Life Sciences P10204) and stored in 70% ethanol at 200C until further analysis.
Extraction of DNA
Individual whitefly was used for DNA extraction. The whiteflies stored in ethanol were washed twice with sterile water before extraction. Genomic DNA of each whitefly was extracted using the DNASure Mini Tissue Kit (Nucleopore, Genetix NP61305) as per the manufacturer’s protocol and stored at -200C until further use.
mtCOI PCR amplification and sequencing
A partial mtCOI gene fragment of about 820 bp was amplified using the universal primers C1-J-2195 (5’—TTGATTTTTTGGTCATCCAGAAGT-3’) and TL2-N-3014 (5’—TCCAATGCACTAATCTGCCATATTA-3’)32. PCR amplification was carried out in a 25 µl reaction mixture containing 12.5 µl of ready-to-use PCR master mix (Promega M750A), 5.5 µl of nuclease-free water, 1 µl each of forward and reverse primers, and 5 µl of insect DNA. Thermal cycling was performed on a 96-well thermal cycler (Applied Biosystems, Thermo Fisher Scientific) with reaction conditions as follows: initial denaturation for 10 min at 94°C, followed by 35 cycles of 94°C –30 s, 48°C –30 s for annealing, and 72°C –40 s for extension, with a final extension step for 5 min at 720C. Negative control was also kept every time, containing no DNA template to confirm potential contamination especially in the PCR reagents. A 3 µl amplified PCR product was run on 1.2% agarose gel in 1X TAE at 100 V (Jordan Scientific) for 50 min. The gel picture was captured under UV light using the gel documentation unit (ProteinSimple, AlphaImager). The confirmed amplified PCR products were sent to AgriGenome (Kochi, India) for further purification and sequencing6,26.
Genetic group determination
The mtCOI sequences obtained from the own collections were compared with the global Bemisia genetic group datasets21,22. The mtCOI sequences were manually inspected for removal of putative pseudogenes along with ambiguous sites; gap adjustment and trimming of overhangs were carried out using BioEdit v7.2. The aligned sequences were subjected to a basic local alignment search (BLASTn) to confirm species identity23. Newly characterised haplotype sequences from our study were submitted to NCBI, and accession numbers were obtained. The sequences were aligned with the Bemisia genetic group database using the ClustalW program with default parameters in MEGA X (https://www.megasoftware.net/dload_win_gui). The phylogenetic tree was constructed using the maximum likelihood approach, and the sequences clade together with the Asia II 1 genetic group were selected for further studies.
Preparation of datasets
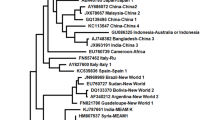
The accessions grouped under Asia II 1 were selected and looked further for their sequence length. The retrieved Asia II 1 sequences of short fragment length below 658 base pairs were discarded. A total of 676 Asia II 1 sequences of Asia Viz., India (n = 190), Pakistan (n = 396), China (n = 30), Nepal (n = 18), Bangladesh (n = 17), Thailand (n = 15), Taiwan (n = 3), and Vietnam (n = 7) were used in the present investigation and are available in Suppl. File 2. The total number of available B. tabaci sequences (mtCOI) from India in GenBank up to 1st September, 2024 are 2906 accessions. Later, these 2906 accessions were analysed generally for their phylogeny. The accessions branching under Asia II 1 were selected again and looked further for their sequence length. The 626 Indian sequences were categorised into Asia II 1, but more than 50% of the retrieved Asia II 1 sequences were junked because of their short fragment length below 658 base pairs (standard cut-off length of sequence with respect to mtCOI). Similarly, a total of 168 good-length sequences of Asia II 1 belonging to India were retrieved and are supplemented with the 22 haplotype sequences generated from our own lab studies (Suppl. File 1; Table S1). The pooled 190 Asia II 1 sequences belonging to India are used in this investigation and are available in Suppl. File 2. The 22 characterized haplotype sequences from our lab studies (Suppl. File 1; Table S1) were supplemented with 168 Asia II I sequences representing India alone were used to generate a phylogenetic tree along with Asia I (GQ281714.1 EU192044.1), Asia II 1 (HM137326, EU192047, GU585369, FJ802389, DQ174519, GU585372), Asia II 5 (AJ748376, AF418666), Asia II 7 (AY686064.1, DQ116660.1), Asia II 8(AJ748358.1, GQ281733.1) and Bemisia species viz., Bemisia afer (GQ139515.1), Bemisia atriplex (GU086362.1), Bemisia subdecipiens (GU220056.1), Trialeurodes vaporariorum (AF418672.2) as an ingroup and outgroup respectively for phylogeny analysis.
The Indian Asia II 1 data set was subdivided into subsample groups like Central India, East India, West India, North India, South India, and Others India (Asia II 1 species of India where collection region/zone has not been mentioned in GenBank) according to geographical region of the country (Supplementary File 3) for analysing comparative genetic variability and haplotype diversity.
Similarly, the Indian Asia II 1 data set from our study was again subdivided into subsample groups according to hostplant families from which B. tabaci is collected, such as Malvaceae, Fabaceae, Solanaceae, Asteraceae, Convolvulaceae, Cucurbitaceae, Euphorbiaceae, and Moraceae, and the accessions that have not been mentioned with hostplant sources in GenBank were denoted as NA (Supplementary File 3) for analysing comparative genetic variability and haplotype diversity.
Phylogenetic structure and haplotype analysis of B. tabaci Asia II 1 sequences
The phylogenetic structure and haplotype analysis of the Asia II 1 genetic group of B. tabaci, prevalent in Asia and India, was investigated. GenBank accessions corresponding to the Asia II 1 genetic group from Asian countries, including India, were retrieved from the NCBI database up until September 1, 2024. These sequences were analysed to confirm phylogenetic relationships using the maximum likelihood method in MEGA X. Genetic distances were calculated using both the P-distance and Kimura 2-parameter (K2P) models in MEGA X34.
Estimation of haplotype diversity
For the haplotype analysis of Asia II 1, the number of haplotypes (H), haplotype diversity (Hd), nucleotide diversity (pi), average number of nucleotide differences (k), total number of mutations (Eta), and G + C content were calculated using the software package DnaSP v6.10.01 (http://www.ub.edu/dnasp/)35. Neutrality tests like Tajima’s D, Fu and Li’s F, and Fu and Li’s D were performed to detect the deviation from the neutral model of evolution using DnaSP v6.10.01. The minimum spanning network of haplotypes was constructed using the PopART version 1.7 package implemented in TCS 1.21 (https://popart.maths.otago.ac.nz/download/)36. To assess differences between and among population groups, analysis of molecular variance (AMOVA) was carried out using the PopART program (version 1.7), which was included in the TCS 1.21.
Results
Genetic structure and haplotype analysis of B. tabaci genetic group Asia II 1 from Asia
A total of 676 curated mtCOI sequences from the B. tabaci Asia II 1 genetic group, originating from Asia, were used for haplotype analysis. This analysis generated 241 distinct haplotypes, of which 195 were singletons. Among the singletons, 117 were found in Pakistan and 48 in India. Other singletons were distributed across China (10), Bangladesh (7), Thailand (6), Nepal (5), and Vietnam (2) (see Supplementary File 3). In addition, 46 haplotypes were shared by at least two sequences. Haplotype H1 was the most dominant, with a frequency of 116 sequences. The second most prevalent was Haplotype H17, shared by 103 sequences, followed by Haplotype H4 with 52 sequences and Haplotype H2 with 45 sequences.
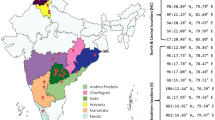
Haplotype H1 exhibited a network connecting 98 sequences from Pakistan, 14 from India, 3 from Bangladesh, and 1 from Nepal. Haplotype H17, with 103 sequences, formed a network comprising 45 sequences from India, 29 from Pakistan, 11 from China, 9 from Thailand, 3 each from Bangladesh and Taiwan, 2 from Vietnam, and 1 from Nepal. The minimum spanning network identified Haplotype H1 as the most widely distributed and ancestral haplotype cluster for the Asia II 1 population of B. tabaci in Asia, followed by Haplotypes H17, H4, and H2 (Figs. 1, Fig. 2A). Based on the unique and shared haplotype numbers in Pakistan and India, the data suggest that Asia II 1 is most prevalent in these two countries, with frequent occurrences of genetic group and haplotype outbreaks.

Geographical distribution of B. tabaci Asia II 1 species across the world and India. The presence of haplotype diversity among these populations across different geographical locations is indicated in multiple colors (The map was generated using online software tool MapChart [https://www.mapchart.net/]).

(A) Minimum spanning network from mtCOI sequences of Asia II 1 haplotypes of B. tabaci belongs to Asian Countries using PopART (Population analysis with reticulate trees) software. 2(B). Minimum spanning network from mtCOI sequences of Asia II 1 haplotypes of B. tabaci India using PopART (Population analysis with reticulate trees) software. 2(C). Minimum spanning network from mtCOI sequences of Asia II 1 haplotypes of B. tabaci belongs to different hostplant families using PopART (Population analysis with reticulate trees) software.
The haplotype diversity of genetic group Asia II 1 concerning Asian countries was found to be higher in Bangladesh (Hd: 0.949), followed by India (Hd: 0.926), Pakistan (Hd: 0.915), Vietnam (Hd: 0.905), Nepal (Hd: 0.856), China (Hd: 0.848), and Thailand (Hd: 0.657). Nucleotide diversity of Asia II 1 sequences from Pakistan was observed to be the highest (pi: 0.03270), followed by India (pi: 0.00763), Bangladesh (pi: 0.00694), Nepal (pi: 0.00585), China (pi: 0.00533), Thailand (pi: 0.00394), and Vietnam (pi: 0.00101) (Table 1). Particularly, the sequences belonging to Taiwan show zero values for nucleotide diversity and haplotype diversity, suggesting that there is no haplotype and nucleotide variation among the sequences and they are 100 percent similar. The number of haplotypes (H), average number of nucleotide differences (k) and total number of mutations (Eta) are represented in Table 1.
The results of AMOVA revealed that, out of the total genetic variation in the mtCOI gene of the Asia II 1 genetic group of B. tabaci populations of Asia, a total 1.93% variation occurred among the populations, whereas the genetic variation within the populations was 98.06% and the FST value observed was 0.01937 (Table 2). The results of a population genetic study utilising neutrality tests in Asian countries revealed negative values for neutrality tests such as Tajima’s D, Fu and Li’s F, and Fu and Li’s D, indicating probable recent population expansion in all of the analysed Asian countries. Tajima’s D, Fu and Li’s F, and Fu and Li’s D neutrality test results were represented in Table 2. Tajima’s D with a negative value or value less than zero implies that the population of interest, i.e., the Asia II 1 genetic group of Asia, is anticipated to experience recent selective sweeps or population expansion following the bottleneck. Negative values of Fu and Li’s F and Fu and Li’s D, on the other hand, show excess allele numbers, most probably driven by recent population growth.
Genetic structure and haplotype analysis of B. tabaci Asia II 1 from India
A total of 22 mtCOI gene sequences obtained from our own collection (Suppl. File 1; Table S1) were blended with 168 sequences of mtCOI from GenBank to achieve a final alignment of 190 sequences with 658 bp in length. The phylogenic tree for all the mtCOI sequences of B. tabaci genetic group Asia II 1 was constructed by involving outgroups and other cryptic species. Looking into the topology of the phylogenetic tree, a higher percentage of sequence similarities were found within the Asia II 1 genetic group and were branched in a single clade where outgroups and other genetic groups branched separately (Fig. 3). Various colours in the phylogenetic tree indicate populations of different geographical regions: Red-North India, Brown-West, and Dark Green-Central India Light green (other parts of India or collection region/zone information is not available), Sky Blue-South, Dark Blue-Asia II 1 Global, Amber-East, Light Blue- genetic groups other than Asia II 1, and Purple denote outgroups.

Phylogenetic tree of mtCOI gene haplotypes of Asia II 1 genetic groups of B. tabaci belongs to India including some outgroups and genetic groups of B. tabaci using maximum likelihood approach in MEGA X. Various colors in the phylogenetic tree indicated different populations of various geographical regions. Red-North India, Brown-West, Dark Green-Central India Light green- Others India (Asia II 1 species of India where collection region/zone has not mentioned), Sky Blue- South, Dark Blue- Asia II 1 Global, Amber-East, Light Blue- genetic groups (Genetic groups other than Asia II 1) and Purple-Outgroups.
A colour-coded pairwise identity matrix for similarity scores of haplotypes of Asia II 1 genetic groups of B. tabaci India merging with some outgroups and genetic groups of B. tabaci using the sequence demarcation tool (SDTv1.2 [http://web.cbio.uct.ac.za/SDT]) is represented in Fig. 4. Each coloured cell represents a percentage identity score between the Asia II 1 sequences (one indicated horizontally to the left and the other vertically at the bottom). A coloured key indicates the correspondence between pairwise identities and the colours displayed in the matrix. SDT yielded identical pairwise identity score distributions for the majority of the Asia II 1 datasets by sharing between 90 and 100% pairwise identity, and distinct differences between the sequences were clearly observable in the coloured matrix for sequence pairs sharing less percent identity.

Color coded pairwise identity matrix for similarity scores of haplotypes of Asia II 1 genetic groups of B. tabaci India merging with some outgroups and genetic groups of B. tabaci using Sequence Demarcation tool Version 1.2 (SDTv1.2).
A total of 77 haplotypes (sequence variants) were identified in the Indian Asia II 1 dataset. There are 23 haplotypes shared by a minimum of 2 to a maximum of 44 sequent variants, and the remaining33 haplotypes were unique. Haplotype H5 is dominant with a frequency of 44 sequences. Supplementary File 3 depicts collection localities and other details about Haplotype 5. There are two other dominant haplotypes, namely Haplotype H8 and Haplotype H1, represented by 20 and 14 sequences, respectively. A total of 13 Asia II 1 accessions out of 22 sequences obtained from own lab collection trips were grouped with Haplotype H5, whereas two of them were grouped with H25, one with H8, and one sequence of own collection from Banswara, Punjab (MN830429.1) was found unique as H36 (Fig. 2B).
Circles in the TCS haplotype network tree for the mtCOI haplotypes of B. tabaci depict the haplotypes identified. The size of each circle is proportional to the frequency of the haplotypes. The lines between each haplotype represented the mutations; each single line counted as a single mutation. Outgroup sequences and cryptic groups were not included in the minimum spanning network due to the level of sequence divergence above 3.5%. The minimum spanning network shows that Haplotype-5 (H5) is the most common as well as the most ancestral haplotype cluster for the population of Asia II 1 B. tabaci in India, followed by H-8 and H-1 (Suppl. File 1; Fig. S2). The dominant cluster H5 shares the majority of the sequences from North India, Central India, and Others (location not specified in NCBI), where cotton covers the major area under cultivation. The highest haplotype diversity was observed in North India (Hd: 0.997), followed by South India (Hd: 0.977), Central India (Hd: 0.927), West India (Hd: 0.889), and East India (Hd: 0.700), where nucleotide diversity was observed highest for South India (pi: 0.0099), followed by West India (pi: 0.00851), North India (pi: 0.00748), East India (pi: 0.00121), and Central India (pi: 0.00797) as per the represented in Table 3; Fig. 1.
The AMOVA revealed that out of the total genetic variation in the mtCOI gene of the Asia II 1 genetic group of B. tabaci populations from different regions of India, a total 12.02% variation occurred among the populations, whereas the genetic variation within the populations was 87.97% and the FST value observed was 0.12028 (Table 2). Tajima’s D neutrality test was significant with positive values for the Central India population and non-significant with negative values for East, North, South, West, and Others (Table 3). Tajima’s D, which is a measure of deviation from neutral evolution where Tajima’s D value of less than or greater than two is usually seen as a strong indicator that a gene is not evolving in a neutral manner. A selective sweep or positive selection is indicated by an abundance of rare alleles in genes with a Tajima’s D value less than − 2. However, an excess of common alleles indicative of balancing selection prevails in genes with a Tajima’s D value greater than 2. Fu and Li’s D and Fu and Li’s F neutrality tests resulted in non-significant negative values for all the population in collected regions (Table 3). A substantial negative Fu and Li’s value indicates an excess of rare mutations or singleton mutations relative to the neutral expectation. This might indicate population expansion or positive selection at linked sites.
Genetic structure and haplotype analysis of B. tabaci Asia II 1 on different hostplant families
When the topology of the phylogenetic tree was explored, a higher percentage of sequence similarities were observed within the Asia II 1 genetic group from different hostplant families and were branched in a single clade, whereas outgroups branched separately. The distinct colours in the phylogenetic tree show distinct hostplant families from which B. tabaci is collected for haplotype analysis from India (Suppl. File 1; Fig. S2).
When the Asia II 1 sequences from India were grouped by respective host plant families, a total of 189 sequences were analyzed, yielding 79 haplotypes (Supplementary Information 3). Haplotype H10 was the most predominant, clustering with 44 sequences, followed by H19 and H34 with 19 and 11 sequences, respectively. Nineteen haplotypes were shared by at least two and up to 44 sequence variants, while the remaining 60 were unique singletons. A minimum spanning network of the host plant families revealed that Haplotype H10 was the most common and widespread cluster within the Asia II 1 genetic group in India, with 17 sequences from the Malvaceae family. Haplotype H34 followed, with 9 Malvaceae sequences, and Haplotype H19, with 6 Malvaceae sequences. Additionally, there were 24 singleton Malvaceae haplotypes (Fig. 2C). This network demonstrated that the Malvaceae family induces greater haplotype variation among the host plant families of B. tabaci, followed by the Fabaceae family. The higher number of haplotypes associated with the Malvaceae family supports the notion that host plants have a significant influence on haplotype and genetic group development.
Although the highest number of haplotypes were observed in Malvaceae, the haplotype diversity was observed to be the highest in Cucurbitaceae (Hd: 0.964), followed by Fabaceae (Hd: 0.919), Malvaceae (Hd: 0.915), and Solanaceae (Hd: 0.881). Nucleotide diversity of Cucurbitaceae is highest (pi: 0.00939), followed by Fabaceae (pi: 0.00812), Malvaceae (pi: 0.00736), and Solanaceae (pi: 0.00525). The number of haplotypes (H), average number of nucleotide differences (k), total number of mutations (Eta), and G + C content are represented in Table 4. The AMOVA for the total genetic variation in the mtCOI gene of the Asia II 1 genetic group of B. tabaci populations of India from various hostplant families shows a total of 13.52% variation among the populations, whereas the genetic variation within the populations observed was 86.47% with the FST value of 0.1352 (Table 2). The values of neutrality tests, viz., Tajima’s D, Fu and Li’s F, and Fu and Li’s D, were represented in Table 4. All the neutrality tests showed negative values, indicating plausible recent population expansion in all of the analysed groups.
Discussion
The genetic diversity and population structure of insect pests inhabiting farmscapes can be influenced by various factors, particularly host plants and local agricultural practices37,38,39. Ecology of B. tabaci relies heavily on dispersal, which not only accounts for host finding and colonisation in constantly changing land cover but also aids in the distribution of desirable genetic traits like insecticide resistance among populations40,41. The identification of new species within B. tabaci was facilitated by sequencing a 657-bp fragment of the mtCOI gene, leading to the introduction of the term "cryptic species complex"16,17. Dinsdale et al.,16 proposed a 3.5% genetic boundary for distinguishing species within the B. tabaci complex. However, Lee et al.,42 suggested that a 4.0% genetic boundary was more appropriate for distinguishing species within the B. tabaci complex than 3.5%. Reclassification based on this 4% genetic divergence revealed the presence of 42 distinct genetic groups. Further global analysis of B. tabaci mtCOI sequences by Kanakala and Ghanim22 reported 44 genetic groups worldwide. Subsequently, Rehman et al.,21 updated the number of genetic groups to 46 based on additional analysis.
The current update of genetic groups includes Africa, Asia I, Asia I-India, Asia II 1 to 13, Spain 1, Asia III, Asia IV, Asia V, Australia, Australia/Indonesia, China 1 to 5, Indian Ocean, Ru, Middle East Asia Minor I and II (MEAM), Mediterranean (MED), MEAM K, New World 1and 2, Japan 1 and 2, Uganda, Italy 1 and Sub-Saharan Africa 1 to 515,17,22,43,44,45. India has reported with 13 genetic groups namely, viz., Asia I, Asia I -India, Asia II 1, Asia II 5, Asia II 6, Asia II 7, Asia II 8, Asia II 11, Asia II 13, Middle East Asia Minor (MEAM)-1, MEAM-K, China 3 and China 721,23.
Among the genetic groups widely distributed across the country, Asia II 1 is predominant, exhibiting national distribution with the highest haplotype diversity. This genetic group is closely associated with cotton leaf curl viruses in specific locations. A key concern with the Asia II 1 group is its potential to expand its distribution, replacing previously established genetic groups in cotton and other agro-ecosystems23,29. This pattern aligns with the findings of Mahmood et al.,46, who reported that Asia II 1 is dominant in Pakistan and the neighboring northern zone of India. Interestingly, Asia II 1 also exhibits dominance in the central zone of India, further highlighting its significant presence in these regions.
The expansion of the Asia II 1 population on cotton in the southern and central regions of Inida may have occurred through the displacement of the Asia I genetic group, as seen in Pakistan47. This situation requires close monitoring to prevent a repeat of what occurred with tomato crops, where the Asia I and Asia II 7 genetic groups were displaced by the more dangerous MEAM-1 genetic group48. Consequently, it is crucial to monitor and assess specific genetic groups of whiteflies for key factors such as fecundity, survival potential, insecticide resistance, and susceptibility to biocontrol agents. This would help predict population growth and the likelihood of genetic group displacement28,29,47,48,49,50,51. Despite the significance of these developments, research on Asian genetic groups particularly B. tabaci Asia II 1 remains limited. This gap underscores the need for further studies to better understand these populations and their potential impact.
The present study reveals significant genetic diversity differences among B. tabaci Asia II 1 sequences from Asian countries, with Pakistan and India showing higher haplotype and nucleotide diversity than others. AMOVA and neutrality tests suggest recent selective sweeps or population expansion within the Asia II 1 group. A recent study identified 31 native and invasive B. tabaci species across 16 Asian countries, with Asia II 1 reported in 10, including India, Pakistan, Bangladesh, Nepal, China, and Japan22,52. Asia II 1 has progressively replaced Asia I in Pakistan’s Punjab and Sindh provinces and now dominates in northern and central India as well46. Additionally, Asia II 1 has found in Southeast Asian nations like Vietnam, Thailand, and Cambodia but is absent from Malaysia46. The study also notes that Asia I has disappeared from Pakistan since 2012, highlighting Asia II 1 dominance46.
An earlier study from India observed that Asia II 1 was predominantly distributed in northern India53. However, results from the current investigation revealed the dominance of Asia II 1 in the northern and central zones of India and the expansion of Asia II 1 to southern India as well. The dominant haplotype, H5, also shares the majority of the sequences from North and Central India. The haplotype diversity was observed to be high in North India, whereas nucleotide diversity was higher in South India. While Asia 1, Asia II 5, and Asia II 8 had a predominant occurrence in Southern India, whereas Asia II 5, Asia I, and Asia II 1 were more prevalent in North-Eastern India21. According to a study conducted on 73 populations that were gathered from different parts of northeastern India, 27 of the 73 populations belonged to Asia II 5, 26 to Asia I, 15 populations belonged to Asia II 1, and 4 were grouped as Asia II 721. Asia II 1 and Asia 1 species of B. tabaci have acquired rapid resistance to organophosphates (Chlorpyrifos, etc.) and pyrethroids (Deltamethrin, etc.) as a result of the extensive application of insecticides54. The insecticide resistance capability of Asia II 1 species might have been the driving force for the expansion of Asia II 1 in India54.
Recent findings from a larger survey in India indicate that Asia II 1 predominates in the northern region of the country, reflecting the circumstances in Pakistan. In other south Asian countries like Bangladesh and Nepal, they have a similar situation like India where Asia 1 and Asia II 5 acts as a dominant species followed by Asia II 1 respectively resulting in the dominance of Asia II 1 in other countries. This seems Asia II 1 is being dominant in other regions of the world46,55,56. The study highlights significant genetic diversity within the B. tabaci Asia II 1 genetic group across various Asian countries, with Pakistan and India emerging as key regions for its spread and prevalence. Haplotype H1, dominant in Pakistan, and H17, prevalent in India, underscore the central role of these two nations in shaping the population dynamics of Asia II 1. Other countries, such as Nepal and Bangladesh, also exhibited considerable genetic variation, while Taiwan showed no diversity, indicating complete sequence similarity. The majority of genetic variation was found within populations rather than between them, suggesting extensive gene flow across the region. Additionally, the negative neutrality test values across all countries point to recent population expansion or selective sweeps in the Asia II 1 group (Suppl. File 1; Fig. S2). These findings underscore the dominance of Asia II 1 in Pakistan and India and highlight the need for continued monitoring to understand its evolutionary trajectory and impact on agriculture throughout Asia. The higher haplotype diversity within the Asia II 1 genetic group might be due several agro-ecological factors including predominant use of different insecticide in different locations and the different restiance mechanism induced in these populations46,57,58,59.
The results from the present investigation also revealed that the Malvaceae family induces more haplotype variation among hostplant families, followed by the Fabaceae family, even though the nucleotide and haplotype diversity was found to be high in Cucurbitaceae. This study also outlines the influence of geographic location on the dominance of haplotypes. Asia II 1 is an indigenous species that can be found throughout Southeast Asia and the Indian subcontinent60. According to a comparative study on host suitability, Asia II 1 and MEAM1 performed well on all hosts in a similar fashion, while Asia II 1 thrived best on cotton plants and had a higher lifespan and fecundity rate on tomato plants61. After sequencing the nuclear genome of Asia II 1, about 1294 genes with high-impact variants were found. The functional analysis showed that some of the genes are involved in the transmission of viruses, such as 4 genes in the transmission of the tomato yellow leaf curl virus (TYLCV), 96 genes in the transmission of the tomato crinivirus (ToCV), and 14 genes in pesticide resistance60. One particular study correlated the incidence of B. tabaci Asia II 1 with the incidence of CLCuD29. A direct assessment of the cotton leaf curl Multan virus (CLCuMuV) transmission efficiency in four cryptic species, viz., two native (Asia 1 and Asia II 1) and two invading (MEAM1 and MED) species, reported that Asia II 1 was the species that transmits CLCuMuV the most efficiently61,62.
Strengths and limitations
The study’s strengths lie in its comprehensive genetic analysis of B. tabaci Asia II 1, providing valuable insights into its haplotype diversity, geographic influence, and host plant interactions, which are crucial for pest management. However, limitations include incomplete regional data, particularly in Southern and Central India, insufficient exploration of host plant interactions, and a narrow focus on the Asia II 1 genetic group without considering its interactions with other genetic groups. Additionally, the study lacks quantitative assessment of environmental factors affecting pest dominance.
Conclusion
The Asia II 1 genetic group of B. tabaci is increasingly dominant across Asia due to its extensive presence, damage potential, and ability to outcompete other species. This rapid expansion, particularly in countries like India and Pakistan, can be linked to dynamic changes in haplotype and nucleotide diversity, high gene flow, adaptability to various host plants, swift insecticide resistance, and competitive advantages. Its nuclear genome, enriched with genes for virus transmission and pesticide resistance, virus-induced behavioral changes, and co-evolution with specific host-virus-vector interactions, further contribute to its spread. Environmental factors like temperature, humidity, local agricultural practices, and mono-cropping of cotton also play a role in its dominance, posing a significant threat to existing genetic groups in agro-ecosystems.
The study identified 241 haplotypes from 676 mtCOI sequences of Asia II 1 from Asia, with Haplotype H1 (116 sequences) being the most prevalent. In India, 77 haplotypes were found from 190 sequences, with Haplotype H5 (44 sequences) dominant, and from 189 host plant sequences in India, 79 haplotypes were identified, with Haplotype H10 (44 accessions) predominant. These haplotypes, especially H1, H5, and H10, provide new insights into Asia II 1’s genetic diversity, suggesting multiple genetic lineages and evolutionary paths crucial for pest management and understanding its evolution.
Data availability
All data generated or analysed in this study are included in this article and in the Supplementary Information files. The details of sequences of B. tabaci AsiaII-1 generated in this study have been deposited with NCBI Nucleotide Database vide accession numbers: KF298449, KF298451, KR149673, KR149674, KR149676, MH909092 to MH909096, MK636815 to MK636820, MN830429 to MN830433, MN830437.
References
Schluter, D. Ecology and the origin of species. Trends Ecol Evol. 16(7), 372–380 (2001).
Turelli, M., Barton, N. H. & Coyne, J. A. Theory and speciation. Trends Ecol Evol. 16(7), 330–343 (2001).
Bush, G. L. & Butlin, R. K. Sympatric speciation in insects. Adaptive Speciat. https://doi.org/10.1017/CBO9781139342179.013 (2004).
Dres, M. & Mallet, J. Host races in plant-feeding insects and their importance in sympatric speciation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 357(1420), 471–492 (2002).
Diehl, S. R. & Bush, G. L. An evolutionary and applied perspective of insect biotypes. Annu. Rev. Entomol. 29(1), 471–504 (1984).
Pfenninger, M. & Schwenk, K. Cryptic animal species are homogeneously distributed among taxa and biogeographical regions. BMC Evol. Biol. 7(1), 121–132 (2007).
Bickford, D. et al. Cryptic species as a window on diversity and conservation. Trend. Ecol. Evol. 22(3), 148–155 (2007).
Beheregaray, L. B. & Caccone, A. Cryptic biodiversity in a changing world. J. Biol. 6(4), 1–5 (2007).
Tay, W. T., Evans, G. A., Boykin, L. M. & De Barro, P. J. Will the real Bemisia tabaci please stand up?. PLoS ONE 7(11), e50550 (2012).
Kunz, D., Tay, W. T., Elfekih, S., Gordon, K. H. J., & De Barro, P. J. Take out the rubbish–Removing NUMTs and pseudogenes from the Bemisia tabaci cryptic species mtCOI database. BioRxiv, 724765 (2019).
Vyskocilova, S., Tay, W. T., van Brunschot, S., Seal, S. & Colvin, J. An integrative approach to discovering cryptic species within the Bemisia tabaci whitefly species complex. Sci. Rep. 8(1), 10886 (2018).
Brown, J. K., Frohlich, D. E. & Rosell, R. C. The sweetpotato or silverleaf whiteflies: biotypes of Bemisia tabaci or a species complex?. Annu. Rev. Entomol. 40(1), 511–534 (1995).
Gill, R.J. & Brown, J.K. Systematics of Bemisia and Bemisia relatives: Can molecular techniques solve the Bemisia tabaci complex conundrum–a taxonomist’s viewpoint. In Bemisia: Bionomics and management of a global pest 5–29 (2009).
Hadjistylli, M., Roderick, G. K. & Brown, J. K. Global population structure of a worldwide pest and virus vector: genetic diversity and population history of the Bemisia tabaci sibling species group. PLoS ONE 11, e0165105 (2016).
Boykin, L. M. et al. Global relationships of Bemisia tabaci (Hemiptera: Aleyrodidae) revealed using Bayesian analysis of mitochondrial COI DNA sequences. Mol. Phylogenetics Evol. 44(3), 1306–1319 (2007).
Dinsdale, A., Cook, L., Riginos, C., Buckley, Y. M. & De Barro, P. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann. Entomol. Soc. Am. 103(2), 196–208 (2010).
De Barro, P. J., Liu, S. S., Boykin, L. M. & Dinsdale, A. B. Bemisia tabaci: a statement of species status. Annu. Rev. Entomol. 56, 1–19 (2011).
Liu, S. S., Colvin, J. & De Barro, P. J. Species concepts as applied to the whitefly Bemisia tabaci systematics: how many species are there?. J. Integr. Agric. 11(2), 176–186 (2012).
Mound, L.A. Biology and identity of whitefly vectors of plant pathogens. (1983).
Perumal, Y., Marimuthu, M., Salim, A. P. & Ponnusamy, B. Host plant mediated population variations of cotton whitefly Bemisia tabaci Gennadius (Aleyrodidae: Homoptera) characterized with random DNA markers. Am. J. Biochem. Biotechnol. 5(1), 40–46 (2009).
Rehman, M., Chakraborty, P., Tanti, B., Mandal, B. & Ghosh, A. Occurrence of a new cryptic species of Bemisia tabaci (Hemiptera: Aleyrodidae): an updated record of cryptic diversity in India. Phytoparasitica 49, 869–882 (2021).
Kanakala, S. & Ghanim, M. Global genetic diversity and geographical distribution of Bemisia tabaci and its bacterial endosymbionts. PLoS ONE 14(3), 139–156 (2019).
Ramesh, K. B., Mahendra, C., Kelageri, S. S., Rajna, S. & Subramanian, S. Distribution and mitotype diversity of Bemisia tabaci. Indian Journal of Entomology 35, 35–39 (2023).
Gouda, M. R., Rajna, S., Gambhir, S. & Subramanian, S. Unravelling genetic diversity of whitefly species on different host plants from New Delhi. J. Environ. Biol. 45, 259–267 (2024).
Ram Kumar, N., Chang, J. C., Narayanan, M. B. & Ramasamy, S. Phylogeographical structure in mitochondrial DNA of whitefly, Bemisia tabaci Gennadius (Hemiptera: Aleyrodidae) in southern India and Southeast Asia. Mt DNA Part A. 28(5), 621–631 (2017).
Hashmi, T. R., Devi, S. R., Meshram, N. M. & Prasad, R. Assessment of bacterial endosymbionts and the host, Bemisia tabaci (Hemiptera: Aleyrodidae), using rRNA and mitochondrial cytochrome oxidase I gene sequences. Commun. Integr. Biol. 11(1), 143–152 (2018).
Hashmi, T. R., Dey, D. & Prasad, R. Distribution frequency of endosymbionts and genetic characterization of Bemisia tabaci (Hemiptera: Aleyrodidae) on Fabaceous host plants in India. Orient. Insects 52(2), 128–142 (2018).
Prasanna, H. C. et al. Cryptic species composition and genetic diversity within Bemisia tabaci complex in soybean in India revealed by mtCOI DNA sequence. J. Integr. Agric. 14(9), 1786–1795 (2015).
Ahmed, M. Z. et al. Genetic identity of the Bemisia tabaci species complex and association with high cotton leaf curl disease (CLCuD) incidence in Pakistan. Pest Manag. Sci. 67(3), 307–317 (2011).
Prabhulinga, T. et al. Mitochondrial COI based genetic diversity and phylogeographic structure of whitefly Bemisia tabaci (Gennadius) on cotton in India. Int. J. Trop. Insect Sci. 40, 1–12 (2020).
Hameed, S., Hameed, S., Sadia, M. & Malik, S. A. Genetic diversity analysis of Bemisia tabaci populations in Pakistan using RAPD markers. Electron. J. Biotechnol. 15(6), 6–6 (2012).
Simon, C. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Gouda, M. R., Gaikwad, K., Prabha, R., Kumar, A. & Subramanian, S. Elucidation of ejaculatory bulb proteins in Bemisia tabaci Asia-1 and Asia II-1 and confirmation of their mating transfer via RNAi. Mol. Biol. Rep. 51, 861 (2024).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Clement, M., Snell, Q., Walke, P., Posada, D. & Crandall, K. TCS: Estimating gene genealogies. In Proc. of the International Parallel and Distributed Processing Symposium, Vol. 2, pp. 7–pp (IEEE Computer Society, 2002).
Mayr, E. Systematics and the Origin of Species from the Viewpoint of a Zoologist (Harvard Univ, 1999).
Ehrlich, P. R. & Raven, P. H. Butterflies and plants: A study in coevolution. Evolution 18, 586–608 (1964).
Park, Y., Nam, H. Y., Baek, S., Lee, S. H. & Lee, J. H. Population genetic structure of Bemisia tabaci MED (Hemiptera: Aleyrodidae) in Korea. PLoS ONE 14(7), 0220327 (2019).
Li, H. R., Pan, H. P., Tao, Y. L., Zhang, Y. J. & Chu, D. Population genetics of an alien whitefly in China: Implications for its dispersal and invasion success. Sci. Rep. 7, 1–8 (2017).
Naranjo, S. E., Castle, S. J., De Barro, P. J. & Liu, S. S. Population dynamics, demography, dispersal and spread of Bemisia tabaci. In Bemisia: Bionomics and management of a global pest 185–226 (2009).
Lee, W., Park, J., Lee, G. S., Lee, S. & Akimoto, S. I. Taxonomic status of the Bemisia tabaci complex (Hemiptera: Aleyrodidae) and reassessment of the number of its constituent species. PLoS ONE 8, e63817 (2013).
Firdaus, S. et al. The Bemisia tabaci species complex: Additions from different parts of the world. Insect Sci. 20(6), 723–733 (2013).
Hu, J. et al. New putative cryptic species detection and genetic network analysis of Bemisia tabaci (Hempitera: Aleyrodidae) in China based on mitochondrial COI sequences. Mitoch DNA Part A 29(3), 474–484 (2018).
Roopa, H. K. et al. Prevalence of a new genetic group, MEAM-K, of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae) in Karnataka, India, as evident from mtCOI sequences. Fla. Entomol. 98, 1062–1071 (2015).
Mahmood, M. A. et al. Dominance of Asia II-1 species of Bemisia tabaci in Pakistan and beyond. Sci. Rep. 12(1), 1528 (2022).
Ashfaq, M. et al. DNA barcoding of Bemisia tabaci complex (Hemiptera: Aleyrodidae) reveals southerly expansion of the dominant whitefly species on cotton in Pakistan. PLoS ONE 9(8), 104485 (2014).
Banks, G. K. et al. First report of the Bemisia tabaci B biotype in India and an associated tomato leaf curl virus disease epidemic. Plant Dis. 85(2), 231–231 (2001).
Legg, J. P., French, R., Rogan, D., Okao-Okuja, G. & Brown, J. K. A distinct Bemisia tabaci (Gennadius)(Hemiptera: Sternorrhyncha: Aleyrodidae) genetic group cluster is associated with the epidemic of severe cassava mosaic virus disease in Uganda. Mol. Ecol. 11(7), 1219–1229 (2002).
McKenzie, C. L. et al. Distribution of Bemisia tabaci (Hemiptera: Aleyrodidae) biotypes in North America after the Q invasion. J. Econ. Entomol. 105(3), 753–766 (2012).
Chowda-Reddy, R. V. et al. Bemisia tabaci phylogenetic groups in India and the relative transmission efficacy of tomato leaf curl Bangalore virus by an indigenous and an exotic population. J. Integr. Agric. 11(2), 235–248 (2012).
Lestari, S. M. et al. Genetic diversity of cryptic species of Bemisia tabaci in Asia. Arch Insect Biochem. Physiol 112(2), 21981 (2023).
Ellango, R. et al. Distribution of Bemisia tabaci genetic groups in India. Environ. Entomol. 44(4), 1258–1264 (2015).
Naveen, N. C. et al. Insecticide resistance status in the whitefly, Bemisia tabaci genetic groups Asia-I, Asia-II-1 and Asia-II-7 on the Indian subcontinent. Sci. Rep. 7, 40634 (2017).
Ahmed, N. et al. Circular DNA enrichment sequencing reveals the viral/satellites genetic diversity associated with the third epidemic of cotton leaf curl disease. Biol. methods protoc. 6(1), 005 (2021).
Farooq, T., Umar, M., She, X., Tang, Y. & He, Z. Molecular phylogenetics and evolutionary analysis of a highly recombinant begomovirus, Cotton leaf curl Multan virus, and associated satellites. Virus Evol. 7, 54 (2021).
Islam, W. et al. A nation-wide genetic survey revealed a complex population structure of Bemisia tabaci in Pakistan. Acta Trop. 183, 119–125 (2018).
Rajna, S., Mahapatro, G., Subramanian, S. & Chander, S. Determination of insecticide resistance in cotton whitefly in North India. Ind. J. Agric. Sci. 94, 404–409 (2024).
Kelageri, S. S. Toxicity variations of insecticides against cotton whitefly Bemisia tabaci (Gennadius) (Doctoral dissertation, Division of Entomology, ICAR-Indian Agricultural Research Institute, New Delhi, 2022).
Hussain, S. et al. Whole genome sequencing of Asia II-1 species of whitefly reveals that genes involved in virus transmission and insecticide resistance have genetic variances between Asia II-1 and MEAM1 species. BMC Genom. 20, 1–13 (2019).
Ahmed, M. Z. et al. Host suitability comparison between the MEAM1 and AsiaII 1 cryptic species of Bemisia tabaci in cotton-growing zones of Pakistan. Pest Manag. Sci. 70(10), 1531–1537 (2014).
Pan, L. L., Cui, X. Y., Chen, Q. F., Wang, X. W. & Liu, S. S. Cotton leaf curl disease: which whitefly is the vector?. Phytopathology 108(10), 1172–1183 (2018).
Acknowledgements
Authors greatly acknowledge the financial assistance and constant support provided by NAHEP-CAAST, Indian Agricultural Research Institute, Pusa Campus, New Delhi. Authors equally thankful to NCBI and all the reference authors who have submitted B. tabaci Asia II-1 sequences in NCBI GenBank. The DST-INSPIRE, is acknowledged by the first author for supporting in terms of Senior Research Fellowship during course of PhD programme.
Funding
The corresponding author gratefully acknowledges the support from NAHEP- CAAST programme of ICAR- IARI, New Delhi toward purchase of chemicals and molecular biological reagents needed for conducting this study.
Author information
Authors and Affiliations
Contributions
SS: conceptualization, visualization, methodology, data curation, supervision, reviewing and editing. KBR: methodology, investigation, software analysis, writing original draft preparation, validation, formal analysis, reviewing and editing. CM, MNRG and RS: Software analysis and reviewing and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study does not include any animals which require any kind of ethical clearance. We confirm that all experiments were performed in accordance with relevant guidelines and regulations of the Institute.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ramesh, K.B., Mahendra, C., Gouda, M.N.R. et al. Genetic structure and haplotype analysis of predominant genetic group of Bemisia tabaci Asia II 1 from Asia and India. Sci Rep 15, 11672 (2025). https://doi.org/10.1038/s41598-025-96483-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96483-9
