
Abstract
Microscopic mechanisms of the crystallization from the melt provide for conversion of short-range order of a liquid state into the long-range order of a solid crystalline state and determine in much the primary microstructure. The ”choice” of proper crystalline structure is not a trivial process that is ruled by thermodynamics. On the other hand from the microscopic point of view it takes time for atoms to be incorporated into ordered structure that leads to formation of some intermediate region between disordered liquid and crystal. In the paper the origin and properties of the region that was called ”the pre–crystallization layer” are investigated within atomistic simulation approach by the classical molecular dynamics. The simulations were carried out for model Fe–Ni–Cr alloy that is crystallized into face centered cubic lattice. It was shown that the properties of the pre–crystallization layer are different from those of liquid and solid. Its structural peculiarities lead to formation of primary defects of just solidified material crystal structure and depend on the orientation of solidification front relative to the crystal lattice.
Similar content being viewed by others

Introduction
Rapid development of additive technologies stimulated the search for ways or modes that would allow to control and handle the properties of a material during manufacture process (see, for example 1). This requires deeper understanding of basic processes responsible for the solidification/crystallization of a liquid. Microscopic mechanisms of a solidification provide for conversion of short-range order of a liquid state into the long-range order of a solid crystalline state and determine in much the primary microstructure (see 2,3,4).
In the paper the origin and properties of the region that was called ”the pre–crystallization layer” are investigated within atomistic simulation approach by the classical molecular dynamics. Here a model austenitic alloy Fe-14Ni-19.5Cr (Ni and Cr concentrations are in weight percent) was studied as an imitation of stainless steel 316L that is widely used in industry including additive manufacturing. The nominal chemical composition of 316L austenitic steel can be found in paper 5. Minor alloying elements besides Ni and Cr in our model composition were equally distributed as Ni and Cr atoms. All classical Molecular Dynamics (MD) simulations were carried out with the Finnis-Sinclair type interatomic potential 6 which provides the crystallization of the overcooled melt into face centered cubic (fcc) lattice with Ni and Cr atoms distributed as disordered substitutional solution. MD simulations were carried out with massively-parallel classical MD code LAMMPS 7,8,9. For sample structure analysis we used orientation analyses by Polyhedral Template Matching (PTM) 10 implemented in scientific data visualization and analysis software for atomistic simulations OVITO 11,12,13 and Adaptive Template Analysis (ATA) 14 which allows precise, high confidence recognition of crystal structures and various defects in virtual samples at elevated temperatures up to the melting point. The Fe-14Ni-19.5Cr system is characterized by very small difference between solidus and liquidus temperatures 5 and so one can speak of the melting temperature. As it was shown in 3 the interatomic potential used here reproduces melting parameters of 316L steel quite well. Similarly to paper 3 to take into account electron heat conductivity the Two Temperature Model was used.
The results obtained in CMD simulations are always influenced by the model used and the methods employed. These factors may introduce unavoidable artifacts that could affect the interpretation of the data. That’s why we paid special attention to the choice of the interatomic model, structure analysis methods, and consistency of simulations results performing statistically independent CMD simulations at various conditions (temperatures below and higher melting temperature, different crystallographic direction of solidification).
The emphasis of the paper is on the study of the phenomenon that we called the pre-crystallization layer (PCL) which separates liquid and solid states during solidification and has a finite thickness. Both the PTM and ATA techniques used for the PCL identification have free adjustable RMSD parameters tuning the sensitivity of the methods. In both methods RMSD has the same meaning. Prior the PCL study we have chosen the same RMSD value 0.13 for them. The choice was made based on analysis of single phase liquid and fcc samples at various conditions to obtain stable results and avoid false positive crystal structures detection in the liquid and vice versa 10,14. The PCL is a generalization to the crystallization front. The layer was reliably identified in the testing of two techniques used for the analysis of crystal structures being formed during crystallization in two-phase systems. The region identified by the ATA as crystal was a bit greater than that identified by the PTM because some atoms were assigned to the crystal by the ATA but to the melt by the PTM. Further research into the properties of the identified layer showed it to be not simply an artifact of the two structure analysis methods but a layer whose physical properties differed from both the crystal and the melt.
Note here that in papers 15,16 atoms belonging to some intermediate layer in the vicinity of the crystallization front were revealed and then analysed from the point of view of the mechanism (ballistic or diffusive) of their building into the crystalline. Here we focus on the structural properties of the PCL itself.
The PCL is also shown to be the source of crystallization defects, whose number/frequency depends on the velocity of the crystallization front and its orientation relative to the lattice of the solidified material (see 3). Twining during 316L steel crystallization is studied. Twining is the main mechanism that governs microstructure formation. It is shown that twin planes in the PCL form mainly in parallel to the crystallization front and the characteristic distance between them (the twin size) depends on the solidification rate and crystallization front orientation relative to the lattice of the growing crystal.
Criteria for the PCL identification and characterization
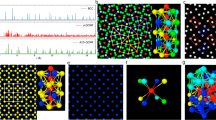
Illustration of stages of the PCL identification in a two-phase sample of the Fe-Ni-Cr alloy is presented in Fig. 1. Here the system measured \(~18.5\times 18.5\times 113 nm\) (\(\sim 3\) million atoms) was considered. We began with structural analysis by the PTM (see Fig. 1b) and the ATA (see Fig. 1c). Then we excluded the atoms identified as fcc crystal by the PTM. Note that a lot of isolated atoms in the crystal region remained not assigned to the crystal by the PTM because the method is sensitive to thermal noise. Then we removed the atoms identified by the ATA as not belonging to any ordered structure (see Fig. 1d). What remained in the sample after that are dynamic clusters in the melt, isolated atoms of thermal noise in the crystal region, and a thin layer of atoms near the solid-liquid interface. Just this layer we characterize as the pre-crystallization layer (see Fig. 1e). In Fig. 2, the PCL resulted from the analysis is shown with and without dynamic clusters in the melt. The types of structures from the ATA are shown by different colors. Thus, the PCL is a region near the solid-liquid boundary identified as the crystal by the ATA and as the melt by the PTM. The PCL is not stationary. Even in the absence of the crystallization propagation (\(T=T_m\)) the PCL constantly fluctuates (as the ripple of waves on water). The amplitude of these fluctuations is determined by the solid-liquid interface free energy 17. This excess surface energy has anisotropic nature depending on boundary orientation relative to the crystal.

Stages in the identification of the PCL: (a) – the sample before processing (atoms are colored by the type – Fe, Ni, Cr); (b) – PTM processing (green – fcc, grey – melt); (c) – ATA processing (cyan – fcc, green – hcp, yellow – bcc, red – icosahedral, blue – melt); (d) – removal of the fcc crystal determined by the PTM and the structure identified as the melt by the ATA; (e) – the PCLs only are shown.

The PCL in the model Fe-14Ni-19.5Cr alloy at the melting temperature: (a) – pre-crystallization layer with crystal-like dynamic clusters in the melt and (b) – finally identified PCL.
Next step is to determine some distinctive properties of the PCL by an alternative structure recognition technique. Here we used the formulation of averaged Bond Orientational Order (BOO) parameters 18 \(Q_i\) which was originally introduced by Steinhardt 19. The BOO parameters are scalar metrics of local atom neighbourhood and defined from 0 to 1 interval. To distinguish different kind of structures local BOO parameters \(Q_4\) and \(Q_6\) are widely used. Figure 3 presents distributions of parameters \(Q_4\) and \(Q_6\) in the PCL for the last perpendicular to crystallographic directions [100], [110], [111] at the melting temperature. The averaging of the distributions was done over 20 snapshots taken every 10 ps. Grey and green show atomic distributions in liquid and fcc phases, respectively. Red color marks the distribution for the PCL. In the \(Q_4-Q_6\) diagram, the PCL is seen to be between the melted material and the fcc. This proves its existence in terms of the structural parameters \(Q_4\) and \(Q_6\).

Structural parameters \(Q_4\)-\(Q_6\) distributions for the PCL (red tones) perpendicular to crystallographic directions [100], [110], [111] at the melting temperature in comparison with \(Q_4\) and \(Q_6\) distributions for the melt (grey tones) and the fcc (green tones).
Table 1 provides data on the distribution of atoms in different structures of the PCL as dependent on the degree of overcooling/overheating and different crystallization directions. The two-phase systems were constructed so as to allow one of the crystallographic directions [100], [110], or [111] to run along the Z axis (perpendicular to the crystallization front). A thin middle region in the crystal was kept at a given temperature. The entire system was kept under ambient pressure with the Nose-Hoover thermostat 20,21. Our simulations were done with the Two-Temperature Model (TTM) by Duffy and Rutherford 22,23. The use of the TTM allows us to take into account electron heat transfer which becomes important for the case of high temperature gradients during fast crystallization from melt. Parameters of the TTM are presented in 3. Results for the identified types of structures were obtained by averaging over time for 20 snapshots taken every 10 ps. It is seen from the Table 1 that the PCL structural composition depends both on temperature and crystallization direction. One can notice that for the [111] direction the number of atoms belonging to the hcp is considerably greater that for other directions. The hcp structure in fcc materials corresponds to stacking faults (edge dislocation cores) and the twin boundaries, and its noticeable fraction may lead to the formation of the defects during crystallization. The abnormally high number of ordered structures other than fcc in the PCL is another criterion of its identification as a peculiar structure. In a number of additional calculations the hcp percentage was determined in the PCLs having different orientations at \(T=0.9T_m\) (see Fig. 4).

The hcp percentage in the PCLs having different orientations at \(T=0.9T_m\).
Mechanism of the PCL formation
To study the mechanism of the PCL formation it is necessary to analyze the melt structure in order to see the distribution and concentration of dynamic clusters in it. For this purpose we used a system of 4 million atoms. To make results more representative, we processed 21 snapshots taken every 10 ps. Performed with the ATA cluster analysis shows the concentration of clusters versus their size for different degrees of overcooling/overheating averaged over 100 ps (see Fig. 5). At all temperatures the highest peak corresponds to the smallest possible close-packed fcc/hcp or icosahedral structures of 13 atoms. The bigger clusters have irregular shapes and are incorporate different structures (see insets in Fig. 5). The clusters are of dynamic character meaning that their lifetime is much less than 1 ps.
Table 2 presents the fractions of atoms belonging to crystal-like structures identified by the ATA in the melt at different temperatures (the first row). For each of the identified structure and each temperature two numbers are presented. The first is the fraction of atoms belonging to the particular structure relative to all atoms in the melt. The second is the percentage of atoms belonging to the particular structure of all ”crystal-like” atoms. As seen from Table 2, the concentration of crystal-like structures reaches \(~4.5\%\) at the melting temperature. Note that at the same temperatures the fractions of ico clusters in the PCL (see Table 1) and in the melt (see Table 2) differ drastically. The ico structure has no potential for long range order formation, but the icosahedral clusters most readily transform into the hcp or fcc when building in the growing crystal and thus adding to the formation of the fcc matrix or extended defects like twin boundaries or stacking faults.

Dynamic clusters size distributions in the melt for different degrees of overcooling/overheating.
Now consider the mechanism that governs the incorporation of atoms (clusters) into the crystal during the crystallization. To illustrate the process quantitatively we select a group of four atoms located in the melt near the crystallization front (marked by color circles with dot in the inset in Figure 6a). As the phase boundary moves, the atoms first get into the PCL (see the inset in Fig. 6b) and then into the crystal (see the inset in Fig. 6c). Figure 6a-c show how the positions of 4 atoms change (relative to their final positions) with time. Figure 6d shows the structural parameter \(Q_6\) of the atoms versus time. The grey region in Fig. 6 corresponds to the period when the atoms are in the melt and can easily diffuse through the liquid. In this case the value of \(Q_6\) is not higher than \(0.25-0.30\). Then the peach region lies - the period when the atoms are in the PCL. Note that their displacements are much smaller than in the melt. The crystalline structure begins to manifest itself, the parameter \(Q_6\) begins to grow and reaches its mean value for the crystal. Note that atomic mobility in the XY plane is higher than along the Z direction (crystal growth direction), i.e. the growing crystal limits their mobility in the crystallization direction. The green region in Fig. 6 corresponds to the period when the PCL has travelled rather far and the atoms have been incorporated into the crystalline structure and now fluctuate with deviations typical of the fcc crystal at a given temperature.

Incorporation of atoms from the melt into the lattice when the crystal/melt boundary moves in the direction [111] (along Z axis) at temperature \(T=0.95T_m\). (a–c) – relative coordinates of four atoms versus time. (d) – bond order parameter \(Q_6\) of the atoms versus time. The grey region corresponds to the period when the atoms are in the melt. The green one shows when the atoms are deep in the fcc lattice. The intermediate peach-colored region shows when the atoms are in the PCL.
The same four atoms trajectories incorporating from the melt into the fcc lattice are shown in the BOO parameters \(Q_4\)-\(Q_6\) diagram in Fig. 7a. Lines with arrows of the same colors as in Fig. 6 are shown in front of \(Q_4\)-\(Q_6\) distributions calculated for pure melt (grey tones) and bulk fcc (green tones) at the same temperature \(T=0.95T_m\). One can see that \(Q_4\)-\(Q_6\) parameters of the atoms stochastically move from the liquid region to the fcc one as the crystallization front passes. Four atoms were chosen randomly for demonstration of the detailed individual atom behavior during the crystal growth process. Figure 7b presents statistical data for all the atoms belonging to the PCL at different moments. Scatter plot in Fig. 7b shows \(Q_4\)-\(Q_6\) parameters distribution for all atoms identified as the PCL for 20 uncorrelated system snapshots. The scatter plot consists of \(\sim 500000\) points. The inset in Figure 7b presents averaged over the snapshots distribution of relative atom positions. We used mean PCL atom coordinates at each time as zero level. Negative relative positions are closer to the crystal while positive are for the liquid. At \(T=0.95T_m\) crystallization front moves with nearly constant velocity \(\sim 13\) m/s 3. Thus, the positions of atoms in the PCL correspond to different times prior and after crystallization front passing by (see upper scale in the inset). The distribution is sliced into colored chunks by the relative position (time). The same colors are used for the points in the scatter plot in the BOO diagram. One can see in both Fig. 7a,b that as time advances the PCL atoms move from liquid-like bond ordering closer to the fcc-like. Full width at half maximum of the distribution in the inset in Fig. 7b we will use as the PCL thickness.

(a) – The structural parameters \(Q_4\) and \(Q_6\) of four atoms (the same as in Fig. 6) incorporating from the melt into the lattice when the crystal/melt boundary moves in the direction [111] at temperature \(T=0.95T_m\). Four atoms trajectories are shown by lines with arrows of the same colors as in Figure 6 in comparison with \(Q_4\)-\(Q_6\) distributions for the melt (grey tones) and the fcc (green tones). (b) – Scatter plot of \(Q_4\)-\(Q_6\) parameters for all atoms belonging to the PCL for 20 system snapshots (\(\sim 500000\) atoms). Colors of the points correspond to positions of atoms relative to mean position of the PCL at a moment. Inset in (b) shows averaged distribution of the PCL atoms, the colors of the distribution chunks are the same as for points in \(Q_4\)-\(Q_6\) diagram. As the crystallization front moves with constant velocity (\(\sim 13\) m/s for \(T=0.95T_m\)) positions relative to the front correspond to different times prior and after crystallization front passage.
Hereafter we tried to evaluate quantitative characteristics of the PCL other than order parameters \(Q_4\) and \(Q_6\). In Fig. 8 the thickness of the PCL as dependent on temperature is presented. Results are obtained by averaging data from 20 snapshots. For all crystallization directions under study, the PCL is seen to be less than 1 nm in thickness (\(\sim 2\) lattice parameters). The point at \(0.85~T_m\) jumps far out of the general trend because of the large number of defects in the crystallizing structure assigned to the the PCL in automated processing. As seen from Fig. 8 the PCL thickness decreases as the degree of overcooling increases due to the higher crystallization rate. As the temperature goes down, the activation barrier for kinetic rearrangements increases. At the same time the driving force \(\Delta G\) for the formation of the equilibrium fcc lattice below melting temperature increases as well. Competition of those two factors leads to faster moving of the crystallization front and trapping more defects at lower temperatures 3.

The PCL thickness versus overcooling/overheating during crystallization/melting in different crystallographic directions.
Atomic volumes of the fcc crystal, melt, and PCL at different temperatures are presented in Fig. 9. Results for the PCL are obtained with the use of the Voronoi tessellation. One can see that the PCL density lays between the melt and crystal densities.

Average volume per atom calculated with Voronoi tessellation for the PCL versus temperature during the crystallization/melting in different crystallographic directions.
Let’s look now thoroughly at the structure of the PCL. Figure 10 gives an illustration of the PCL. The layer is seen to have, in principle, a crystalline-like structure since the horizontal planes with the indices (100) are easily noticed, but fluctuations are much higher than those in the crystal. For the verification, we performed additional calculations with the moving crystallization front at \(0.85T_m\), where the system was tracked over 10 ps (10 thousand steps) and atomic coordinates were saved every 10 time steps. From such a detailed series of snapshots we can infer that atoms in the PCL are not strictly limited in number. They can go into the crystal or go back to the melt even at a rather high degree of overcooling, i.e at high crystallization rate. The truth however is that the “rather high” degrees of overcooling give quite moderate boundary velocities. Take, for example, the crystallization front velocity of 10m/s at \(0.9T_m\) (on the velocities of the crystallization front see 3), meaning that 100 ps is required to move the phase boundary for a distance of the order of the PCL thickness (1 nm).

The PCL with the [100] direction along the Z axis: (a) after PTM procedure; (b) with the PCL colored blue. The zoomed image shows the atomic structure in more detail.

The mechanism of defect formation in the PCL for crystal/melt boundary moving upward in the [111] direction at \(0.85T_m\). Colors of atoms show different structures.
In 3 it was shown that the twinning is proper mainly to the crystallization in [111] direction. Now one may argue it is the higher hcp concentration in the PCL (see Table 1 and Fig. 4) that is very likely to play as a catalyst in the formation of ingrain defects. Figure 11 shows snapshots of the PCL moving upward along the Z axis in [111] crystallographic direction at \(0.85T_m\). Colors of atoms mark different structures. Figure 11 illustrates the mechanism of defect formation from the PCL (as it was shown earlier the fraction of the hcp structure is maximal for this direction). At a time of 80ps, a twin embryo nucleates in the PCL. As the SLI moving on, atoms continue their incorporation into the hcp plane. At this stage, there is a probability for the embryo either to collapse or to grow. As a result, after \(\sim 100~ps\) when the PCL covers a distance greater than its thickness, the defect becomes trapped in the lattice.
Summary
Numerical simulation with classical molecular dynamics approach was carried out to study mechanisms of crystallization of a model Fe-Ni-Cr alloy imitating stainless steel 316L which crystallizes in the fcc structure. The main goal of the paper is to proof the existence of so-called Pre-Crystallization Layer (PCL) of finite thickness separating liquid and crystalline states during crystallization. It was shown that the PCL is distinctive of the liquid and solid states in the degree of ordering (\(Q_4-Q_6\) diagram), structure (relative fractions of hcp-, fcc-, and bcc-like atoms), density, diffusivity etc. The thickness of the PCL is about 1 nm and is wealky dependent on the degree of overcooling and orientation of the crystallization front relative to crystal lattice. At some particular relative orientations (close to [111] direction of the crystallization) the PCL is characterized by the higher fraction of the hcp structure that proved to be favorite to the generation of extended ingrain defects – twin boundaries or stacking faults in just crystallized material. Both types of the defects in the fcc lattice have the hcp structure. As the direction of the crystallization declines from [111] the fraction of the hcp in the PCL rapidly decreases and the probability of twinning decreases as well that is in agreement with our previous results (see 3). Note here that the fraction of the hcp clusters forming in the liquid is three times higher than that of the fcc. This may be the reason for high concentration of the hcp in the PCL where they build in the crystal primarily forming the hcp structure. Then, it transforms into the fcc but the kinetics of the transformation depends on the orientation of the PCL relative to the fcc matrix. It proves that the kinetics is slower for [111] direction of crystallization and the hcp structures live for a longer time which increases the probability of the generation of extended ingrain defects. Described here pre-crystallization layer is not a feature of the considered material only. We believe that any material crystallizing in closed packed structure (fcc or hcp) should have similar peculiarities of boundary between solid and liquid phases.
Data availibility
The datasets analyzed during the current study available from the corresponding author on reasonable request.
References
Ivanov, I. A. et al. Effect of laser-induced ultrasound treatment on material structure in laser surface treatment for selective laser melting applications. Sci. Rep. 11, 23501. https://doi.org/10.1038/s41598-021-02895-8 (2021).
Dremov, V. V., Chirkov, P. V. & Karavaev, A. V. Molecular dynamics study of the effect of extended ingrain defects on grain growth kinetics in nanocrystalline copper. Sci. Rep. 11, 934. https://doi.org/10.1038/s41598-020-79861-3 (2021).
Dremov, V. V. et al. Atomistic simulation of primary microstructure formation in metals during crystallization from the melt. Sci. Rep. 14, 28105. https://doi.org/10.1038/s41598-024-79228-y (2024).
Kichigin, R. M., Chirkov, P. V., Karavaev, A. V. & Dremov, V. V. Atomistic simulation of specificities of microstructure formation. Phys. Met. Metall. 1224–1231, 28105. https://doi.org/10.1134/S0031918X24602178 (2024).
Pichler, P., Simonds, B. J., Sowards, J. W. & Pottlacher, G. Measurements of thermophysical properties of solid and liquid NIST SRM 316L stainless steel. J. Mater. Sci. 55, 4081. https://doi.org/10.1007/s10853-019-04261-6 (2020).
NIST Interatomic Potentials Repository. https://www.ctcms.nist.gov/potentials/entry/2019--Mendelev-M-I--Fe-Ni-Cr/2019--Mendelev-M-I--Fe-Ni-Cr--LAMMPS--ipr1.html.
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19. https://doi.org/10.1006/jcph.1995.1039 (1995).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171. https://doi.org/10.1016/j.cpc.2021.108171 (2022).
LAMMPS Molecular Dynamics Simulator. http://lammps.org/.
Larsen, P. M., Schmidt, S. & Schiotz, J. Robust structural identification via polyhedral template matching. Modell. Simul. Mater. Sci. Eng. 24, 055007. https://doi.org/10.1088/0965-0393/24/5/055007 (2016).
Stukowski, A. & Albe, K. Extracting dislocations and non-dislocation crystal defects from atomistic simulation data. Model. Simul. Mater. Sci. Eng. 18, 015012. https://doi.org/10.1088/0965-0393/18/8/085001 (2010).
Stukowski, A. Computational analysis methods in atomistic modeling of crystals. J. Metals 66, 399–407. https://doi.org/10.1007/s11837-013-0827-5 (2014).
OVITO - Open Visualization Tool - Scientific data visualization and analysis software for atomistic simulation models in materials science and related disciplines. http://www.ovito.org/.
Sapozhnikov, F. A., Ionov, G. V. & Dremov, V. V. An adaptive template method for analyzing crystal structures and defects in molecular dynamics simulations of high-rate deformations. Russ. J. Phys. Chem. B 2, 238–245. https://doi.org/10.1134/S1990793108020115 (2008).
Yan, Z., Xu, B., Wang, F., Li, J. & Kong, L. T. Effects of undercooling on atomic crystallization behaviors and growth mechanisms of pure metals. J. Appl. Phys. 132, 075301. https://doi.org/10.1063/5.0098537 (2022).
Hua, M. A joint diffusion/collision model for crystal growth in pure liquid metals. Nat. Commun. 15, 5749. https://doi.org/10.1038/s41467-024-50182-7 (2024).
Hoyt, J. J., Asta, M. & Karma, A. Method for computing the anisotropy of the solid-liquid interfacial free energy. Phys. Rev. Lett. 86, 5530–5533. https://doi.org/10.1103/PhysRevLett.86.5530 (2001).
Lechner, W. Accurate determination of crystal structures based on averaged local bond order parameters. J. Chem. Phys. 129, 114707. https://doi.org/10.1063/1.2977970 (2008).
Steinhardt, P. J., Nelson, D. R. & Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 28, 784–805. https://doi.org/10.1103/PhysRevB.28.784 (1983).
Nose, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519. https://doi.org/10.1063/1.447334 (1984).
Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697. https://doi.org/10.1103/PhysRevA.31.1695 (1985).
Duffy, D. M. & Rutherford, A. M. Including of the effects of electronic stopping and electron-ion interactions in radiation damage simulations. J. Phys. Condenced Matter 19, 016207. https://doi.org/10.1088/0953-8984/19/1/016207 (2006).
Rutherford, A. M. & Duffy, D. M. The effect of electron-ion interaction on radiation damage simulations. J. Phys. Condenced Matter 19, 496201. https://doi.org/10.1088/0953-8984/19/49/496201 (2007).
Acknowledgements
The work was supported by the Grant number MT-96 under the EOTP Rosatom research program.
Author information
Authors and Affiliations
Contributions
V.V.D. conceived CMD simulations, contributed to the discussion, and wrote the manuscript. P.V.C. and R.M.K. performed CMD simulations, carried out analysis of CMD samples, visualized the results, and contributed to the discussion. A.V.K. performed CMD simulations, analyzed the results, and contributed to the discussion and writing of the manuscript. F.A.S. performed automated structure analysis with ATA software tool. E.B.C., I.A.I., V.S.D., S.V.S. supervised the project and were responsible for conceptualizing the project goals, as well as contributed to the discussion and results analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chirkov, P.V., Dremov, V.V., Kichigin, R.M. et al. Molecular dynamics simulation and analysis of pre-crystallization layer between liquid and solid states. Sci Rep 15, 12705 (2025). https://doi.org/10.1038/s41598-025-97121-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-97121-0
