
Abstract
Bifidobacterium breve is a well-recognized probiotic species. B. breve JKL2022, a strain isolated from the feces of healthy infants that exhibits superior conjugated linoleic acid (CLA)-converting activity, was functionally characterized for probiotic safety and applicability through genomic and in vitro analyses. The JKL2022 genome comprises a 2,313,948 bp sequence assembled into a single contig, encoding a total of 1,998 genes. In silico predictive analyses confirmed the absence of virulence factors and acquired resistance genes while verifying its intrinsic antimicrobial resistance profile. Several CAZymes were identified, consistent with the strain’s fermentation profile. Additionally, the gene encoding the key enzyme for CLA conversion was identified as a 993-bp lai gene, underscoring the species-level differences in microbial CLA metabolism. The functionality, stress tolerance, and safety of JKL2022 were further confirmed through experimental assessments. JKL2022 exhibited tolerance to acid and bile salts, auto-aggregation, and cell surface hydrophobicity, indicating its potential to survive gastrointestinal transit. Furthermore, JKL2022 exhibited α-glucosidase inhibitory activity and tested negative for starch hydrolysis, hemolysis, and gelatinase activity. The inherent probiotic properties of Bifidobacterium, combined with the strain-specific CLA conversion using growing cells and postbiotic preparations, contribute to the potential health benefits of B. breve JKL2022, as verified in this study.
Similar content being viewed by others
Introduction
Bifidobacterium breve is a beneficial bacterial species that colonizes the human gastrointestinal tract and is associated with a variety of health-promoting effects. Members of this species are Gram-positive, anaerobic symbionts of the human intestine1,2. B. breve strains have a long history of probiotic use and are included in the list of Qualified Presumption of Safety (QPS) biological agents3 and are generally recognized as safe (GRAS) by the U.S. Food and Drug Administration (FDA)4. Recent research has focused on supplementing B. breve strains capable of producing conjugated linoleic acid (CLA) in various animal models, including pigs, chickens, and mice, to harness the health benefits of CLA, which include anti-atherosclerotic, antidiabetic, anti-obesity, and immunomodulatory properties5,6.
Among natural dietary sources of fatty acids, dairy products (milk, butter, and cheese) and lean meats (beef, lamb, and mutton) contain the highest levels of CLA, reaching up to 3.0–8.0 mg/g fat, whereas fish and vegetables contain negligible CLA levels (< 0.1 mg/g fat). The relatively low CLA content in these food products poses a challenge in achieving the recommended daily CLA intake of 3–6 g for potential health benefits7,8. Several strategies have been explored to enhance the CLA content in dairy and meat products, including dietary modifications (e.g., forage type and feeding stage) and processing techniques (e.g., fermentation and storage)9,10,11. In particular, grass-feeding and oil supplementation (e.g., sunflower, soybean, or flaxseed oil) have been shown to increase CLA levels in dairy milk. For example, dietary supplementation with soybean, rubber seed, or flaxseed oil increased the fatty acid content in cow’s milk, with cis-9, trans-11 CLA levels increasing by 501.85% and trans-10, cis-12 CLA levels increasing by 125%12,13. Additionally, food-grade, CLA-producing lactic acid bacteria (LAB) have been extensively studied for their potential application in enhancing the CLA content of fermented dairy products14,15. Notably, CLA-producing bacteria generate highly specific isomers rather than a mixture of different isomers, which offers advantages in terms of isomer-dependent health effects15,16.
In ruminants, biohydrogenation is the primary process through which linoleic acid (LA) derived from plant materials is converted into stearic acid, with CLA as an intermediate metabolite. This process is facilitated by the metabolic activity of ruminal microorganisms (e.g., Butyrivibrio fibrisolvens) and an endogenous enzyme system in the mammary tissue (Δ9-desaturase). By contrast, food-grade LAB and CLA-producing bacteria employ distinct enzymatic systems. For instance, Cutibacterium acnes (formerly Propionibacterium acnes) utilizes the polyunsaturated fatty acid isomerase (PAI) to convert LA exclusively into CLA (trans-10, cis-12 CLA)17, whereas other Cutibacterium species produce a mixture of isomers (cis-9, trans-11 CLA: 75–95%; trans-10, cis-12 CLA: 15–25%)18. Members of Lactobacillus spp. use a multi-enzyme system involving CLA hydrolase (myosin cross-reactive antigen), CLA decarboxylase (acetoacetate decarboxylase), and CLA dehydrogenase (short-chain dehydrogenase/oxidoreductase), resulting in varying CLA isomer compositions19,20. For example, Lactiplantibacillus plantarum JCM1551 produces only cis-9, trans-11 CLA21, Lpb. plantarum PL62 produces 47% cis-9, trans-11 CLA and 53% trans-10, cis-12 CLA22, and Limosilactobacillus reuteri ATCC55739 produces 59% cis-9, trans-11 CLA and 41% trans-10, cis-12 CLA23. Similarly, Bifidobacterium species, particularly B. breve, can convert LA into CLA via a membrane-spanning linoleic acid isomerase (LAI), producing cis-9, trans-11 CLA as the predominant isomer24,25.
In this study, we characterized the whole genome of the CLA-producing B. breve JKL2022 (KACC81214BP), a strain isolated from the feces of healthy infants26, using predictive in silico analyses and in vitro characterization to evaluate its probiotic functionality and safety. The inherent probiotic properties of B. breve JKL2022, coupled with its remarkable CLA-converting activity, hold promise for probiotic applications. Current research efforts focus on the development of functional probiotics for both human and animal use, as well as functional compounds derived from JKL2022 to enhance the CLA content of various animal-derived food products.
Materials and methods
Bacterial strains and culture conditions
B. breve JKL2022 (KACC81214BP) and other Bifidobacterium strains (B. breve JCM7017, JCM7019, JCM1273, JCM1192, and B. animalis subsp. lactis BB12) were routinely cultured in de Man, Rogosa, and Sharpe (MRS) broth (BD Difco, USA) supplemented with 0.05% (wt/vol) L-cysteine hydrochloride (cys-MRS) (Sigma, USA) at 37 °C under anaerobic conditions (GasPak™ EZ anaerobic chamber, BD Difco). All strains were preserved in 10% skim milk with glycerol (3:1, vol: vol) at -80 °C.
Whole genome analysis
The genomic DNA of B. breve JKL2022 was extracted using the QIAamp® PowerFecal® DNA extraction kit (QIAGEN, USA) following the manufacturer’s protocol. Whole genome sequencing was performed at CJ Bioscience (Korea) using the Pacific Biosciences (PacBio, USA) Sequel II platform. De novo assembly of sequence reads was conducted using the PacBio SMRT analysis program v. 2.3.0. Functional genome annotation was performed using the Rapid Annotation using Subsystem Technology (RAST) pipeline with default parameters27 and KEGG annotation via EzBiocloud. The genome was further annotated using BlastKOALA with the refprok.pep KEGG database28 for pathway classification and dbCAN3 CGC-Finder29 for CAZyme gene cluster identification using default parameters. Transfer RNAs (tRNAs) were identified using tRNAscan-SE v. 1.3.1, and ribosomal and non-coding RNAs (rRNAs) were identified using INFERNAL v. 1.1.3 with the Rfam 12.0 database. Putative genes encoding beneficial enzymes and stress-related proteins were identified through NCBI BLAST searches.
Virulence factors and antimicrobial resistance genes
Identification of virulence factors and antimicrobial resistance genes (ARGs) is essential when characterizing probiotic candidates to ensure safety and efficacy. Genes encoding virulence factors were identified using VirulenceFinder v. 2.0 (Center for Genomic Epidemiology, CGE, https://cge.food.dtu.dk/services/VirulenceFinder/) using the genome sequence of JKL2022. Putative enzymes associated with the production of harmful metabolites (e.g., arylsulfatase, ß-glucuronidase, nitroreductase, azoreductase, D-lactate dehydrogenase, and amino acid decarboxylase) were identified in the JKL2022 genome using the EzBioCloud BLAST function. Moreover, ARGs were detected using the Resistance Gene Identifier (RGI) from the Comprehensive Antibiotic Resistance Database (CARD, https://card.mcmaster.ca/analyze/rgi), and the ResFinder v. 4.4.2 (parameters: 90.0% ID threshold, 60.0% minimum length) and ResFinderFG v. 2.0 (parameters: 98.0% ID threshold, 60.0% minimum length) (http://genepi.food.dtu.dk/). The detected ARGs were correlated with the antimicrobial susceptibility profile of strain JKL2022 based on the in vitro antibiotic susceptibility assay.
Prophage and mobile genetic elements
Phages integrated into the JKL2022 genome (prophages) were identified using sequence similarity analysis with the PhageBoost tool30. Mobile genetic elements (MGEs) associated with antimicrobial resistance genes and virulence factors were detected using MobileElementFinder v. 1.0.3 (https://cge.food.dtu.dk/services/MobileElementFinder/) with the MGE database v. 1.0.231. Genes located within prophage and MGE regions were identified based on the annotated genome of JKL2022 and correlated with the identified ARGs.
Prediction of genetic markers
Potential genetic markers were identified to enable rapid identification and differentiation of strain JKL2022 from other B. breve strains. Housekeeping genes with at least one base difference from other Bifidobacterium strains were selected through sequence analysis.
Probiotic characteristics
Acid and bile salt tolerance
The resistance of B. breve JKL2022 to acidic conditions and bile salt exposure was evaluated as follows. JKL2022 was exposed to artificial gastric juice (AGJ) composed of 0.2% pepsin (Roche Diagnostics, USA) and 0.35% NaCl, with the pH adjusted to 2.0 and 3.0 using 3 N HCl. Briefly, cells from an overnight culture (15–18 h) were harvested by centrifugation (8,000 × g, 4 °C, 10 min), washed twice with 1× PBS, and resuspended in an equal volume of PBS to achieve a 1× cell concentration (approximately 1 × 109 CFU/mL). Subsequently, 1% (v/v) of the washed cells was inoculated into 5 mL AGJ (pH 2.0 and 3.0) and incubated at 37 °C. Viability was assessed by spread plating on cys-MRS agar after 0, 15, 30, 60, and 120 min of exposure followed by anaerobic incubation at 37 °C. Viable cell counts were reported as log(CFU mL− 1).
Survival in the presence of bile was determined by inoculating 1% (v/v) of washed cells into 5 mL MRS broth supplemented with 0.5% (w/v) porcine bile extract (PBE; Sigma Aldrich, USA). Cultures were incubated anaerobically at 37 °C, and viable cell counts were determined at 6 h intervals over a 24 h period. The survival rate at each time point was calculated using the formula:
where CFU mL− 1 at time t represents the average number of colony-forming units after exposure, and CFU mL− 1 at time t0 represents the average number of colony-forming units of the initial inoculum. Experiments were performed in triplicate, with three replicates per experiment.
Auto-aggregation capacity
The auto-aggregation capacity of JKL2022 was assessed following a previously established protocol. An overnight culture of JKL2022 was harvested, washed with sterile distilled water, and resuspended to an optical density (OD) of 0.3 at 660 nm using an INNO microplate spectrophotometer (LTEK Co., Ltd.). A 1 mL aliquot of the cell suspension was transferred to a 1.75 mL microcentrifuge tube and incubated at ambient temperature (25 °C) for 60 min, followed by centrifugation at 980 × g for 2 min. The supernatant was transferred to a 96-well plate, with each well containing 250 µL. Absorbance at 660 nm was measured, and the degree of auto-aggregation was calculated using the formula:
where ODinitial represents the optical density of the cell suspension at 660 nm before incubation, and ODfinal represents the optical density after incubation.
Cell surface hydrophobicity assay
Bacterial adhesion to hydrocarbons was evaluated using a previously established method32. Briefly, an overnight culture of JKL2022 was harvested, washed, and resuspended in 1× PBS to an OD of 0.7 at 600 nm. A 3 mL aliquot of the JKL2022 cell suspension was mixed with 1 mL of xylene, dichloromethane, or hexane, followed by incubation at 37 °C for 10 min. The mixture was vortexed and incubated for 1 h at 37 °C to allow phase separation. The aqueous phase was collected, and absorbance at 600 nm was measured. Surface hydrophobicity was calculated using the formula:
where ODinitial represents the optical density at 600 nm before incubation and ODfinal represents the optical density of the aqueous layer after incubation.
Functional characterization
Carbohydrate fermentation profile
The carbohydrate fermentation profiles of B. breve JKL2022, JCM7017, and JCM7019 were determined using the API 50CH kit (bioMérieux, USA) according to the manufacturer’s instructions, with minor modifications. Briefly, cell suspensions were prepared in 0.85% NaCl equivalent to 2 McFarland standard (approximately 6.0 × 108 CFU/mL). A 2% (v/v) inoculum of the cell suspension was introduced into API CHB medium supplemented with 0.05% L-cysteine. The cupules were then filled with the inoculated suspension (~ 150 µL) and incubated anaerobically at 37 °C. A color change from purple to yellow (or from purple to black for esculin) was indicative of carbohydrate fermentation. The results were denoted as follows: (-) negative fermentation, (+) positive fermentation, and (w) weak fermentation.
Additionally, the carbohydrate-active enzymes of the strains were analyzed using the data from the CAZy database (https://www.cazy.org/bB.html). The number of genes associated with glycoside hydrolases, glycosyl transferases, carbohydrate esterases, and carbohydrate-binding module families was assessed. The genome of B. breve UCC2003 was used as a reference.
Starch hydrolysis test
The ability of the strains to hydrolyze starch was evaluated by streaking overnight cultures of Bifidobacterium spp. onto cys-MRS agar supplemented with 1% (w/v) soluble starch (Duksan, Korea). The plates were incubated anaerobically at 37 °C for 24–48 h, after which they were flooded with iodine solution. The presence of a clearing zone around the bacterial growth indicated a positive result for starch hydrolysis.
α-Glucosidase inhibitory activity
The α-glucosidase inhibitory activity of the B. breve strains was assessed following previously established methods33. Briefly, 25 µL of JKL2022 cell-free supernatant (CFS), obtained from overnight cultures of JKL2022, was added to a reaction mixture containing 150 µL of 1× PBS and 75 µL of 0.02 M p-nitrophenyl-α-D-glucopyranoside (PNPG) solution (Sigma). The mixture was incubated at 37 °C for 10 min. The enzymatic reaction was initiated by adding 50 µL of α-glucosidase (0.17 U/mL, Sigma), followed by further incubation at 37 °C for 10 min. The reaction was terminated by adding 1 mL of 0.1 M Na2CO3 (Sigma). The amount of p-nitrophenol released was measured spectrophotometrically at 405 nm. The inhibition rate was calculated using the formula:
where A represents the absorbance of the reaction mix containing only α-glucosidase (positive control), B represents the absorbance of the reaction mix without α-glucosidase and CFS (blank control), C represents the absorbance of the reaction mix containing both α-glucosidase and CFS (treatment group), and D represents the absorbance of the reaction mix containing only CFS (negative control).
Conjugated linoleic acid production
CLA production by Bifidobacterium spp. was assessed in vitro by culturing the strains in cys-MRS broth supplemented with 1% linoleic acid (LA, 50 mg/mL, Sigma), incubated at 37 °C for 24 h. CLA concentration was quantified following a previously established protocol24. Briefly, 400 µL of culture was transferred to a sterile 1.75 mL microfuge tube and mixed with 800 µL of isopropanol and 600 µL of hexane. The mixture was vortexed briefly and centrifuged at 980 × g for 5 min to facilitate phase separation. The upper phase was collected and diluted in methanol (100:900 µL), and its absorbance at 233 nm was measured using an INNO spectrophotometer.
Safety assessment
Antibiotic susceptibility
The antibiotic susceptibility of B. breve JKL2022 was evaluated using a modified broth microdilution assay34. An overnight culture of B. breve JKL2022 grown in cys-MRS was harvested by centrifugation (8,000 × g, 4 °C, 10 min), washed with 0.85% NaCl, and resuspended in 2 mL of 0.85% NaCl equivalent to the 0.5 McFarland standard (approximately 1.5 × 108 CFU/mL).
The antibiotics listed in Table 3 were prepared at an initial concentration of 2,048 µg/mL in modified cation-adjusted Mueller-Hinton broth (CAMHB; Merck, Germany). Sterile 96-well plates were prepared by adding 100 µL of sterile modified CAMHB to each well. Then, 100 µL of each antibiotic solution (2,048 µg/mL) was added to the first lane, followed by serial two-fold dilutions across the plate until lane 10 (final concentration, 2 µg/mL). A 10 µL aliquot of JKL2022 cell suspension was inoculated into wells in lanes 1–11. Lane 11 contained B. breve JKL2022 without antibiotics, serving as a negative control, and lane 12 contained only modified CAMHB, serving as a blank control. The plates were incubated anaerobically at 37 °C, and cell growth was assessed at 24 and 48 h by measuring absorbance at 600 nm using an INNO spectrophotometer. Antibiotic resistance was interpreted based on the reported intrinsic resistance profile of Bifidobacterium derived from molecular characterization35 and the prevalence of ARGs among human gut-derived bacteria36.
Hemolytic activity
The hemolytic activity of B. breve strains was assessed by streaking overnight cultures onto tryptic soy agar (TSA) or cys-MRS plates supplemented with 5% (v/v) sheep blood, followed by anaerobic incubation at 37 °C for 24–48 h. Hemolytic activity was classified based on colony morphology: the presence of a clear zone around the colonies indicated ß-hemolysis, a greenish discoloration indicated α-hemolysis, and the absence of a clearing zone indicated γ-hemolysis33.
Gelatinase activity
The gelatinase activity of the strains was evaluated in vitro using nutrient gelatin stab cultures. Overnight cultures of Bifidobacterium spp. were inoculated by stabbing into nutrient gelatin and incubated anaerobically at 37 °C for 24–48 h. Liquefaction of gelatin observed following 1 h of refrigeration was indicative of gelatinase activity37.
Statistical analysis
All in vitro experiments were performed in triplicate, and the data are expressed as the means ± standard deviations, analyzed using the GraphPad Prism (v. 9.5.1). The surface adhesion capacity of bifidobacteria was analyzed using the ordinary one-way analysis of variance (ANOVA) (α, 0.05) with Tukey’s multiple comparison post-hoc test. The differences in α-glucosidase inhibition were measured using the Brown-Forsythe and Welch ANOVA test (α, 0.05) with Dunnett’s T3 multiple comparison post-hoc test.
Results and discussion
The genomic and phenotypic characteristics of B. breve JKL2022 related to its probiotic functionality were evaluated through various in silico and in vitro analyses. Consistent with the established probiotic traits of the genus Bifidobacterium, JKL2022 exhibited key functional attributes supporting its probiotic potential. Genomic analyses confirmed the presence of genes encoding beneficial enzymes and the absence of virulence factors, reinforcing both the functional and safety aspects of the strain. Moreover, in vitro assessments aligned with the genomic profile, demonstrating a clear correlation between the genotype and phenotype of the strain. Notably, the superior CLA conversion activity of JKL2022 represents a unique functional attribute, further enhancing its probiotic potential.
Whole genome analysis
The genome of B. breve JKL2022 consists of a single 2,313,948 bp contig with a guanine and cytosine (G + C) content of 58.7% (Fig. 1A). The genome encodes 1,936 protein-coding genes, 53 tRNA genes, and 9 rRNA genes. The EggNog/COG functional categorization revealed a high abundance of genes involved in carbohydrate transport and metabolism (212), replication, recombination, and repair (172), amino acid transport and metabolism (161), translation, ribosomal structure, and biogenesis (134), transcription (113), and cell wall and membrane envelope biogenesis (89), as shown in Fig. 1B and C. Annotation using BlastKOALA resulted in 1,044 annotated entries out of the 1,930 entries (54.1%), classified into metabolism, genetic information processing, environmental information processing, cellular processes, organismal systems, and human diseases (Fig. 2). Among the metabolism-related genes, the most abundant were those associated with global and overview maps (797), carbohydrate metabolism (145), and amino acid metabolism (145).

Genome properties of JKL2022. (A) Circular genome map of Bifidobacterium breve JKL2022. Circles represent the following characteristics from the outermost circle to the center: (1) contig information, (2) coding sequences on forward strand, (3) coding sequences on reverse strand, (4) transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), (5) GC skew, and (6) GC ratio. G, guanine; C, cytosine; CDS, coding sequences. (B) Functional annotation based on Kyoto Encyclopedia of Genes and Genomes and (C) Cluster of Orthologous Groups.

Genotype analysis of Bifidobacterium breve JKL2022 based on (A) KEGG Mapper Reconstruction using the BlastKOALA, (B) predicted CAZyme gene clusters using the dbCAN3 program, and (C) distribution of carbohydrate metabolism genes.
Table 1 presents a summary of genes associated with probiotic functionalities identified in the JKL2022 genome. Notably, the CLA-converting activity of B. breve JKL2022 was attributed to a 993-bp lai gene (JKL2022_00014), which encodes a putative linoleic acid isomerase with a membrane-spanning configuration24. Additional genes encoding beneficial functions include carbohydrate-active enzymes (CAZymes) belonging to glycoside hydrolase (1,3-ß-galactosyl-N-acetylhexosamine phosphorylase), glycosyl transferase (α-D-xyloside xylohydrolase, maltose α-D-glucosyltransferase, and sucrose phosphorylase), carbohydrate esterase (N-acetylglucosamine-6-phosphate deacetylase), and carbohydrate-binding module families, as identified using the Carbohydrate-Active Enzymes database (http://www.cazy.org/)38. The abundance of CAZymes suggests that B. breve JKL2022 possesses a diverse carbohydrate metabolism capability, facilitating adaptation to local carbohydrate availability and contributing to the degradation of carbohydrates in the host gut39.
Furthermore, JKL2022 harbors a gene encoding a choloylcholine hydrolase (JKL2022_00976), also known as a bile salt hydrolase. Bile salt hydrolases play a critical role in probiotic function by facilitating bile salt detoxification, leading to the formation of deconjugated bile acids and enhancing the strain’s survivability during gastrointestinal transit. Several stress-related genes were also identified, including heat shock protein (HsP) HsP.16.4 (JKL2022_00089), putative HspR (JKL2022_00146), GroEL (JKL2022_01010), chaperone proteins dnaK1 (JKL2022_00143), GrpE (JKL2022_00144), DnaJ (JKL2022_00145), and the large-conductance mechanosensitive channel (JKL2022_01468). The presence of these genes suggests that JKL2022 can tolerate temperature, pH, and osmotic stress—critical factors in probiotic viability during production and host consumption.
Additionally, the genome of JKL2022 was screened for potential genetic markers that may facilitate strain identification in mixed Bifidobacterium populations. Table 2 lists candidate genetic markers with at least one base-pair difference, many of which correspond to housekeeping genes. These markers could serve as the basis for developing a rapid PCR-based screening method for JKL2022, enabling strain-specific detection and identification independent of sequencing analysis.
Virulence factors
In addition to possessing genes associated with probiotic functionality, it is equally important that a probiotic strain does not harbor virulence factors, as their presence could pose risks to the host and potentially induce disease. The absence of virulence factors enhances the probiotic safety profile by eliminating the possibility of horizontal gene transfer (HGT) to other bacteria and reducing the risk of pathogenicity. Common virulence factors in lactic acid bacteria include adherence factors, toxins, surface-associated proteins, and proteins involved in collagen and fibronectin binding. Predictive analyses using the VirulenceFinder 2.040 confirmed that JKL2022 lacks genes associated with virulence. Furthermore, the PathogenFinder prediction tool (https://cge.food.dtu.dk/services/PathogenFinder/) classified JKL2022 as non-pathogenic, with a probability score of 0.241.
Antimicrobial resistance genes
Antimicrobial resistance typically arises through three fundamental mechanisms: (1) enzymatic degradation of antimicrobial compounds, (2) modification of antimicrobial target molecules or proteins, and (3) alterations in membrane permeability to antibiotics41. Prediction of ARGs using the CARD-RGI42 identified the presence of the rifamycin-resistant ß-subunit of RNA polymerase (rpoB) (92.48% ID). ResFinderFG v. 2.0 identified the dfr (98.91% ID) and van_ligase (99.92% ID) genes, associated with resistance to trimethoprim and D-cycloserine, respectively. In contrast, no acquired ARGs were detected using ResFinder v.4.4.2.
All three ARGs identified in the JKL2022 genome share a common resistance mechanism, target modification or replacement, attributed to mutations in their nucleotide sequences. The dfr gene (JKL2022_01586) encodes dihydrofolate reductase, the target of trimethoprim. dfrF was originally identified in Streptococcus pyogenes; however, B. breve and B. longum have been reported to harbor sequence variants of this resistance determinant43. JKL2022_01586 exhibited ten nucleotide differences compared with the dfr sequence. The van_ligase factor was identified as D-alanine-D-alanine ligase (JKL2022_00307), the target of D-cycloserine44. Notably, JKL2022_00307 showed only a single nucleotide difference from the reference sequence, suggesting a high likelihood of susceptibility to D-cycloserine. rpoB (JKL2022_01402) encodes the RNA polymerase-binding protein RbpA, a known target of rifamycins. Bifidobacteria exhibit intrinsic resistance to rifamycins due to mutations within the rifampicin-binding pocket and adjacent regions (rpoB-mutants)45.
However, the presence of these genetic determinants alone does not confirm a resistant phenotype and necessitates experimental validation. The AMR profile of B. breve JKL2022 was experimentally assessed using the broth microdilution method, revealing concordance between the predicted genotype and the intrinsic resistance to aminoglycosides (Table 3). In 2005, Zhou et al. reported the antimicrobial susceptibility profile of probiotic Bifidobacterium strains, highlighting the intrinsic and non-transmissible nature of the genetic factors conferring antimicrobial resistance46. The low-level intrinsic resistance of bifidobacteria (e.g., vancomycin, aminoglycosides, and metronidazole) due to their natural cell physiology is considered safe and presents minimal risks for contributing to multi-drug resistance, as they are not acquired via HGT, are non-transferable, and do not pose a public health risk46,47. On the other hand, the presence of mobile ARGs would render the strain unsafe for probiotic use due to the risk of AMR dissemination.
Prophage and mobile genetic elements
Chromosomal genes can be transferred between bacterial cells through various mechanisms, including mobile genetic elements (e.g., plasmids) and phage-mediated lateral transduction48. This approach enables bacterial cells to acquire exogenous genetic material beyond the DNA inherited from the parent cell. A single prophage was identified in the genome of JKL2022, located between 506,666 and 522,012 bp. This prophage region harbors 12 genes, including the insertion sequence IS408 (JKL2022_00428), an uncharacterized membrane protein YobI (JKL2022_00430), putative transposases (JKL2022_00431, JKL2022_00433), and several hypothetical proteins. The presence of MGEs associated with antimicrobial resistance was further assessed using the MGE web tool, which predicted a single MGE at positions 35,423–36,691 bp (complement). Additionally, multiple putative transposases associated with the insertion sequence element IS1353 (JKL2022_01155, JKL2022_01431, JKL2022_01852, JKL2022_01863, and JKL2022_01095) were identified in the genome. Notably, none of these MGEs are located in proximity to the previously identified ARGs, indicating a low risk of ARG transmission via HGT.
Stress tolerance
For probiotics to exert beneficial effects on the host, they must survive passage through the gastrointestinal tract (GIT) and establish a stable niche. Previous studies have reported that bifidobacteria exhibit resistance to lysozyme concentrations of up to 200 mg/L, enabling survival in the oral cavity49. Following this, they are exposed to the acidic conditions of the stomach and bile salts in the small intestine. Tolerance to these conditions is a critical trait for probiotic development, as it enhances bacterial survival and persistence. Additionally, auto-aggregation and cell surface hydrophobicity may influence bacterial adhesion to epithelial cells, a key factor in successful niche establishment. These characteristics are sought for in probiotic candidates to ensure proper gut colonization50. In vitro analyses demonstrated that B. breve JKL2022 exhibits acid and bile salt tolerance, further supporting its probiotic potential.
Acid and bile salt tolerance
Acid and bile salt tolerance are critical criteria for probiotic selection, as acid resistance is associated with the ability to maintain intracellular pH homeostasis. The acid tolerance of Bifidobacterium spp. was assessed in AGJ at pH 2.0–3.0. At pH 2.0, all B. breve strains were rapidly inactivated, with no viable cells detected after 15 min of exposure. At pH 3.0, the B. breve strains exhibited moderate acid tolerance, with the JCM strains demonstrating a survival rate of 41.10–45.13% after 120 min (Fig. 3B). Notably, B. breve JKL2022 exhibited superior acid tolerance, with a survival rate of 74 ± 5.54% after 120 min. In contrast, the probiotic B. animalis subsp. lactis remained largely unaffected by the acidic environment, retaining 98.55 ± 0.05% viability after 120 min. JKL2022 harbors the gene encoding the acid tolerance regulatory protein ActR (JKL2022_01381), a member of the two-component ActS/ActR system involved in pH sensing and response to acid stress51. ActS is a sensor kinase, which senses changes in external pH environments and phosphorylates ActR under acidic stress. This process leads to activation or repression of downstream genes involved in acid tolerance, including proton transport and metabolism. Furthermore, B. breve and B. longum have been reported to elicit a stationary phase adaptive acid tolerance response, although not as robust as that of other LAB52. Bifidobacterium species also possess other two-component systems, such as the PhoR-PhoP system, where PhoR acts as the sensor kinase and PhoP is the response regulator, allowing Bifidobacterium to respond to environmental stress (i.e., phosphate starvation)53. These reported mechanisms allow bifidobacterial strains to survive host conditions, particularly during gastrointestinal transit.

In vitro assessment of (A) α-glucosidase inhibition and stress tolerance to (B) acid and (C) bile salt of Bifidobacterium breve JKL2022. Statistically significant difference in α-glucosidase inhibition is denoted by * based on α, 0.05.
Bile salt tolerance was further assessed by evaluating bile salt hydrolase (BSH) activity in vitro. All B. breve strains exhibited BSH activity, with JKL2022 displaying intermediate BSH activity relative to the JCM strains. Among the five strains, JCM1273 exhibited the highest BSH activity, followed by JCM119, JCM7019, JKL2022, and JCM7017. In addition, JKL2022 demonstrated moderate bile salt resistance, maintaining a survival rate > 85% after 24 h of exposure to 0.5% bile salt, comparable to B. animalis BB-12 (Fig. 3C). Bile salt hydrolysis, a key factor in bile resistance, is attributed to the presence of a functional bile salt hydrolase54,55. JKL2022 harbors a single copy of the gene encoding choloylglycine hydrolase (JKL2022_01482), a member of the C59 peptidase family, which catalyzes the degradation of bile salts in mammalian gut56. The process of breaking down conjugated bile salts into free bile acids and amino acids reduces the toxicity of bile salts and potentially alters the bile acid pool57. Ruiz et al. emphasized that the expression of the BSH gene resulted in bile salt stress resistance58. In addition, the F0F1 ATPase, functioning for ATP generation, has been described as the molecular link connecting both acid and bile stress responses in B. animalis59. The presence of these genetic determinants indicates the potential mechanism employed by B. breve JKL2022 for acid and bile salt tolerance, consistent with those previously reported in other probiotic LAB.
Auto-aggregation capacity
All strains exhibited moderate to high auto-aggregation capacity (Fig. 4A). Strains JCM7019, JCM1192, JCM1273, and BB12 demonstrated similarly high auto-aggregation levels (59.38–64.55%, p > 0.05), while JKL2022 and JCM7017 exhibited moderate auto-aggregation (40.90–42.88%, p > 0.05). Strong auto-aggregation is a desirable characteristic in probiotic strains, as it enhances the likelihood of successful gut colonization by promoting adherence to intestinal epithelial cells. Additionally, bacterial aggregation facilitates the formation of a protective barrier that may prevent pathogen adhesion, thereby reducing the ability of pathogenic bacteria to establish a niche60,61.

Surface adhesion capacity of Bifidobacterium spp. cells based on (A) auto-aggregation and cell surface hydrophobicity to hydrocarbons: (B) dichloromethane, (C) hexane, and (D) xylene. a-c Different letters denote significantly different results.
Cell surface hydrophobicity
Cell surface hydrophobicity is an important trait of probiotics that reflects their affinity for hydrophobic environments and plays a crucial role in epithelial cell adhesion61. The hydrophobicity of the bacterial cell surface, which is associated with adhesion capacity, was assessed using hexane, xylene, and dichloromethane (Fig. 4B and D). The tested strains exhibited 20–50% adhesion to hydrocarbons, classifying them as moderately hydrophobic32. The adhesion ability of the bacterial cells to the host could result in transient colonization, promoting immunomodulatory effects and stimulating gut barrier and metabolism62.
Functional characterization
Carbohydrate fermentation profile
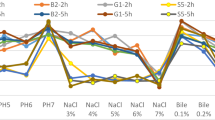
The carbohydrate utilization profile of B. breve JKL2022 was assessed and compared with the reference strains JCM7017 and JCM7019 using the API 50CH kit (Fig. 5). Notably, all three strains exhibited a common sugar fermentation pattern (glucose, lactose, maltose, and raffinose), consistent with literature. However, JKL2022 demonstrated the ability to ferment trehalose and, to a lesser extent, inulin, suggesting its potential applicability in synbiotic formulations.

Carbohydrate profiling of Bifidobacterium breve JKL2022 based on: (A) API 50 CH fermentation profile (scale bar, degree of fermentation); (B) in silico prediction of glycoside hydrolase family members based on Cazy classification (scale bar, number of hits); and (C) in silico prediction of glycosyl transferase (GT), carbohydrate esterases (CE), and carbohydrate-binding module families (scale bar, number of hits).
The fermentation profile of JKL2022 correlated with the presence of carbohydrate-active enzymes. Genes involved in sugar metabolism are organized into gene clusters containing one or more associated glycoside hydrolases and associated regulatory transport proteins. Bottacini et al. (2014) reported the clustering of genes corresponding to the carbohydrate fermentation profile of B. breve strains. For instance, ribose metabolism is associated with the gene cluster 45, which includes rbsD and rbsK, encoding a putative ribose transport system and ribokinase, respectively. Similarly, strain-specific melezitose metabolism corresponds to cluster 10, which contains an α-galactosidase-encoding gene63.
The organization of carbohydrate-active enzymes presented in Fig. 5B and C. indicates the abundance of enzymes belonging to Glycoside Hydrolase Family 13 (GH13) and Glycosyl Transferase Family 2 (GT2)64. Family GH13 is the major glycoside hydrolase family acting on substrates containing α-glucoside linkages (e.g., maltose and trehalose) and, to a lesser extent, monosaccharides (e.g., glucose, fructose, and galactose), ß-linked sugars (e.g., cellobiose, sucrose, and lactose), and sugar alcohols (e.g., mannitol and sorbitol)65. On the other hand, family GT2 includes enzymes that synthesize glycosidic bonds by transferring sugar moieties from donor molecules (i.e., UDP-glucose) to acceptor molecules, resulting in the formation of glycosidic linkages66. Some key GT2 enzymes include glucosyltransferases such as glycerol phosphate polymerase (JKL2022_01530), monoglucosyldiacylglycerol synthase (JKL2022_01528), chondroitin synthase (JKL2022_01845), undecaprenyl-phosphate 4-deoxy-4-formamido-L-arabinose transferase (JKL2022_01865), and putative glycosyltransferase EpsH (JKL2022_00410). GH13 and GT2 enzymes play key roles in the degradation and utilization of dietary and host-derived carbohydrates and prebiotics and exopolysaccharide, cell wall, and cell membrane biosynthesis, respectively67. Thus, they enhance nutrient utilization and support gut colonization by enhancing stress tolerance and adhesion, making them important markers of probiotic potential.
Starch hydrolysis test
None of the tested Bifidobacterium strains exhibited clear zones around their growth, indicating a negative result for starch hydrolysis. Additionally, these strains failed to grow in media containing starch as the sole carbohydrate source. The enzyme α-amylase catalyzes the breakdown of starch into glucose, a process linked to glycemic control, as rapid starch hydrolysis can cause a spike in blood glucose levels68. Several studies indicated the role of inhibiting microbial carbohydrate hydrolyzing enzymes in reducing hyperglycemia69,70. Specifically, the inhibition of α-amylase and α-glucosidase has been the most effectively used mechanism for anti-diabetic drugs70. In this context, the lack of α-amylase activity of JKL2022 indicates that the strain does not contribute to starch-induced increases in blood glucose levels. In conjunction with α-glucosidase inhibition, these observations point to possible compatibility with diabetes management treatments.
α-Glucosidase inhibitory activity
Probiotics have been implicated in regulating blood glucose levels through several mechanisms, including inhibition of α-glucosidase activity, regulation of insulin-related signaling molecules, regulation of gut microflora, and appetite modulation, among others71. In addition, Li et al.72 reported that probiotics play a key role in the regulation of several factors (e.g., glycated hemoglobin A1c (HbA1c), quantitative insulin sensitivity check index (QUICKI), total cholesterol (TC), triglyceride (TG), and low-density lipoprotein cholesterol (LDL-C)) in prediabetes patients. The α-glucosidase inhibitory activity of certain probiotic strains is beneficial in managing type II diabetes. α-Glucosidase is an enzyme that hydrolyzes starches and disaccharides into glucose, facilitating energy production. Inhibiting α-glucosidase in the intestine can slow down glucose absorption in the bloodstream, thereby regulating postprandial blood sugar levels73. All tested strains demonstrated strong anti-α-glucosidase activity (Fig. 3A). Among them, strain JCM1273 exhibited the highest inhibitory activity (87.68 ± 5.34%), followed by JCM1192 (86.68 ± 6.28%), JCM7017 (91.95 ± 7.97%), JCM7019 (73.81 ± 5.94%), BB-12 (72.14 ± 11.95%), and JKL2022 (67.54 ± 6.33%). In 2023, Kumari et al. reported the antidiabetic activity of potential probiotics belonging to Limosilactobacillus, Levilactobacillus, and Lacticaseibacillus genera, inhibiting α-glucosidase and α-amylase activity by up to 85.88% and 75.89%, respectively74. The observed inhibition of α-amylase activity, along with the absence of starch hydrolysis, suggests that JKL2022 may have potential applications in mitigating hyperglycemic conditions33.
Conjugated linoleic acid production
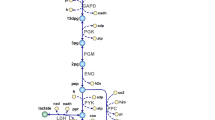
CLA is associated with several health benefits, including anti-obesity, anti-inflammatory, and anti-cancer effects75. In the presence of suitable substrates, such as free LA or α-linolenic acid (LNA), bifidobacteria can convert these compounds into conjugated forms as part of their detoxification mechanisms76. During this process, the bifidobacterial LAI converts toxic LA into CLA24.
Among the tested strains, JKL2022 (0.336 ± 0.025 mg/mL) and JCM7017 (0.295 ± 0.034 mg/mL) exhibited the highest CLA conversion rates, followed by JCM7019 (0.192 ± 0.023 mg/mL), JCM1192 (0.185 ± 0.015 mg/mL), and JCM1273 (0.064 ± 0.010 mg/mL). Other B. breve strains also exhibit strong CLA conversion, reaching up to 57.20 ~ 90.0% conversion77. However, these strains were only reported to exhibit CLA conversion during growth and lack evidence of CLA conversion using washed cells or crude protein extracts. Additionally, experimental evidence confirmed that only JKL2022 can mediate the CLA conversion using washed or partially lysed cells and crude cell-free extracts (Supplementary Data), suggesting that this trait may be strain-specific.
The LAI enzyme of B. breve JKL2022 was previously characterized and heterologously expressed in E. coli BL2124 and confirmed as the key enzyme for bifidobacterial CLA conversion. Figure 6 shows the amino acid sequence alignment of JKL2022 LAI with other bifidobacterial species, showing high sequence homology and the presence of nine transmembrane helices. Furthermore, sequence analysis confirmed that JKL2022 LAI is distinct from the PAI enzyme of Cutibacterium or the MCRA of lactobacilli. Figure 7 illustrates the distinct CLA conversion mechanisms employed by various bacterial genera, including Cutibacterium, Lactobacillus, and Bifidobacterium, emphasizing species-specific enzymatic systems.

Amino acid sequence alignment of bifidobacterial linoleic acid isomerase. BB – Bifidobacterium breve (JCM7017, CP006712.1; NCFB2258, CP006714.1); BL – Bifidobacterium longum (CP176538.1); BP – Bifidobacterium pseudocatenulatum (CP079231.1); BD – Bifidobacterium dentium (AP012326.1); Syn – synthetic construct of LAI (OM158463.1). Predicted protein topology is indicated as outside (O), transmembrane helix (H), or inside (I). Alignment score is based on multiple sequence alignment of nucleotide and amino acid sequences (ClustalW) using B. breve JKL2022 as reference.

Proposed microbial-CLA synthesis for Cutibacterium (PAI), Lactobacillus (CLA-HY, CLA-DH, CLA-DC), and Bifidobacterium (LAI). The tertiary structure of the key enzymes is shown; intermediate metabolites are shown in blue and CLA isomers are shown in yellow.
In our previous study, B. breve JKL2022 was successfully applied as an adjunct culture in cream cheese and yogurt, and exhibited moderate CLA conversion when cultured in modified skimmed-milk media under aerobic and anaerobic conditions14. In this regard, the CLA conversion of JKL2022 using washed cells is promising in enhancing the CLA content of milk and dairy products. Overall, the CLA conversion of strain JKL2022 is expected to compound the inherent beneficial health effects associated with B. breve species.
Safety assessment
Antibiotic susceptibility
The antimicrobial resistance profile of B. breve JKL2022 was assessed using the broth microdilution method (Table 3). B. breve JKL2022 was susceptible to most antibiotics outside the known resistance profile of B. breve, which includes vancomycin, ampicillin, erythromycin, tetracycline, chloramphenicol, and ciprofloxacin. Notably, JKL2022 exhibited susceptibility to rifampicin despite harboring the rifamycin-resistant ß-subunit of RNA polymerase (rpoB). Consistently, JKL2022 displayed a resistant phenotype to clindamycin, gentamicin, kanamycin, streptomycin, mupirocin, metronidazole, and polymyxin B, consistent with its intrinsic resistance and Gram-positive classification.
The antimicrobial resistance phenotype of JKL2022 aligns with AMR genes detected in its genome. The AMR profile of JKL2022 is consistent with previously reported resistance patterns in B. breve strains. Resistance to aminoglycosides is attributed to the absence of specific transport systems required for antibiotic entry into the cell78. Similarly, resistance to polymyxin B, an antibiotic used to treat meningitis, pneumonia, and urinary tract infections, is intrinsic to most bifidobacteria due to their cell membrane structure. Moreover, resistance to polymyxin B often involves modification of the phosphate groups of lipid A with 4-amino-4-deoxy-L-arabinose; in this case, mutations in the UDP 4-deoxy-4-formamido-L-arabinose transferase (JKL2022_01865) may contribute to resistance79. Lastly, as a Gram-positive anaerobe, JKL2022 lacks the specific enzymes required for metronidazole activation, rendering it inherently resistant80.
Hemolytic activity
The ability of the bacterial strains to lyse red blood cells is a critical safety parameter, as hemolytic activity can have severe health implications81. All Bifidobacterium strains exhibited γ-hemolysis (no hemolytic activity) on blood agar, confirming their safety. Additionally, JKL2022 lacks hemolysin-encoding genes, further supporting its non-hemolytic nature.
Gelatinase activity
Gelatinase is recognized as a virulence factor in lactic acid bacteria. Genomic analysis confirmed that none of the tested strains, including JKL2022, harbored genes encoding gelatinase. Consistently, in vitro assessments showed no gelatinase activity. Lactic acid bacteria strains that do not produce gelatinase are considered safer candidates for probiotic development82.
Conclusions
The comprehensive genomic analysis of B. breve JKL2022 identified key probiotic traits, including genes encoding various carbohydrate-active enzymes, the absence of critical virulence factors, and a limited repertoire of antibiotic resistance genes. With respect to probiotic safety, JKL2022 is predicted to be non-pathogenic to human hosts. Consistently, in vitro assessments corroborated the genotypic characteristics of the strain, demonstrating acid and bile salt tolerance, cell surface hydrophobicity, broad carbohydrate metabolism, and the absence of harmful traits, including hemolytic activity, gelatinase activity, and α-amylase activity. Notably, JKL2022 exhibited superior CLA conversion compared to JCM reference strains, suggesting its potential to enhance dietary CLA levels. These findings underscore the probiotic potential of B. breve JKL2022, highlighting the strain’s inherent functional properties and its strain-specific capacity for CLA conversion, which may contribute to additional health benefits.
Data availability
The sequence obtained in this Whole Genome Shotgun project has been deposited in DDBJ/ENA/GenBank under the accession number CP103292. The BioProject accession number is PRJNA871061 and the Biosample accession number is SAMN30403323. Upon a reasonable request, the datasets of this study can be requested from the corresponding author.
References
Turroni, F., Milani, C., Ventura, M. & van Sinderen, D. The human gut microbiota during the initial stages of life: Insights from bifidobacteria. Curr. Opin. Biotechnol. 73, 81–87. https://doi.org/10.1016/j.copbio.2021.07.012 (2022).
Arboleya, S., Watkins, C., Stanton, C. & Ross, R. P. Gut bifidobacteria populations in human health and aging. Front. Microbiol. 7, 1204. https://doi.org/10.3389/fmicb.2016.01204 (2016).
BIOHAZ. Scientific opinion on the maintenance of the list of QPS biological agents intentionally added to food and feed (2013 update). EFSA J. https://doi.org/10.2903/j.efsa.2013.3449 (2013).
Matera, M. & Bifidobacteria Lactobacilli… when, how and why to use them. Global Pediatr. https://doi.org/10.1016/j.gpeds.2024.100139 (2024).
Evangelista, A. G., Correa, J. A. F., Pinto, A. C. M. S., Goncalves, F. D. R. & Luciano, F. B. Recent advances in the use of bacterial probiotics in animal production. Anim. Health Res. Rev. https://doi.org/10.1017/S1466252323000063 (2023).
Chen, Y. et al. Dose-response efficacy and mechanisms of orally administered CLA-producing Bifidobacterium breve CCFM683 on DSS-induced colitis in mice. J. Funct. Foods. https://doi.org/10.1016/j.jff.2020.104245 (2020).
Benjamin, S., Prakasan, P., Sreedharan, S., Wright, A. & Spener, F. Pros and cons of CLA consumption: An insight from clinical evidences. Nutr. Metab. 12, 1–20. https://doi.org/10.1186/1743-7075-12-4 (2015).
Badawy, S. et al. Conjugated linoleic acid (CLA) as a functional food: Is it beneficial or not? Food Res. Int. 172, 1–26. https://doi.org/10.1016/j.foodres.2023.113158 (2023).
Dhiman, T. R., Nam, S. H. & Ure, A. L. Factors affecting conjugated linoleic acid content in milk and meat. Crit. Rev. Food Sci. Nutr. 45, 463–482. https://doi.org/10.1080/10408390591034463 (2005).
Schmid, A., Collomb, M., Sieber, R. & Bee, G. Conjugated linoleic acid in meat and meat products: A review. Meat. Sci. 73, 29–41. https://doi.org/10.1016/j.meatsci.2005.10.010 (2006).
Jang, Y., Elnar, A. G., Hur, S. J. & Kim, G. B. Factors influencing conjugated linoleic acid content of dairy products: Challenges and strategies. Crit. Rev. Food Sci. Nutr.. https://doi.org/10.1080/10408398.2024.2376111 (2024).
Bu, D. P., Wang, J. Q., Dhiman, T. R. & Liu, S. J. Effectiveness of oils rich in linoleic and linolenic acids to enhance conjugated linoleic acid in milk from dairy cows. J. Dairy Sci. 90, 998–1007. https://doi.org/10.3168/jds.S0022-0302(07)71585-0 (2007).
Pi, Y. et al. Effectiveness of rubber seed oil and flaxseed oil to enhance the alpha-linolenic acid content in milk from dairy cows. J. Dairy Sci. 99, 5719–5730. https://doi.org/10.3168/jds.2015-9307 (2016).
Jang, Y., Elnar, A. G., Kang, M. H. & Kim, G. B. Application of conjugated linoleic acid-producing strain, Bifidobacterium breve JKL2022, in the development of probiotic dairy products. Food Sci. Anim. Resour.. https://doi.org/10.5851/kosfa.2024.e91 (2024).
Gorissen, L., Leroy, F., De Vuyst, L., De Smet, S. & Raes, K. Bacterial production of conjugated linoleic and linolenic acid in foods: A technological challenge. Crit. Rev. Food Sci. Nutr. 55, 1561–1574. https://doi.org/10.1080/10408398.2012.706243 (2015).
Roche, H. M. et al. Isomer-dependent metabolic effects of conjugated linoleic acid. Diabetes 51, 2037–2044. https://doi.org/10.2337/diabetes.51.7.2037 (2002).
Rosberg-Cody, E., Johnson, M. C., Fitzgerald, G. F., Ross, P. R. & Stanton, C. Heterologous expression of linoleic acid isomerase from Propionibacterium acnes and anti-proliferative activity of recombinant trans-10, cis-12 conjugated linoleic acid. Microbiology. 153, 2483–2490. https://doi.org/10.1099/mic.0.2006/001966-0 (2007).
Wu, C. et al. Advances in research on microbial conjugated linoleic acid bioconversion. Prog. Lipid. Res. 93, 1–15. https://doi.org/10.1016/j.plipres.2023.101257 (2024).
Kishino, S. et al. Novel multi-component enzyme machinery in lactic acid bacteria catalyzing C=C double bond migration useful for conjugated fatty acid synthesis. Biochem. Biophys. Res. Commun. 416, 188–193. https://doi.org/10.1016/j.bbrc.2011.11.022 (2011).
Salsinha, A. S., Pimentel, L. L., Fontes, A. L., Gomes, A. M. & Rodriguez-Alcala, L. M. Microbial production of conjugated linoleic acid and conjugated linolenic acid relies on a multienzymatic system. Microbiol. Mol. Biol. Rev. 82, 1–21. https://doi.org/10.1128/MMBR (2018).
Kishino, S., Ogawa, J., Omura, Y., Matsumura, K. & Shimizu, S. Conjugated linoleic acid production from linoleic acid by lactic acid bacteria. J. Am. Oil Chem. Soc. 79, 15–163. https://doi.org/10.1007/s11746-002-0451-4 (2002).
Lee, K., Paek, K., Lee, H. Y., Park, J. H. & Lee, Y. Antiobesity effect of trans-10,cis-12-conjugated linoleic acid-producing Lactobacillus plantarum PL62 on diet-induced obese mice. J. Appl. Microbiol. 103, 1140–1146. https://doi.org/10.1111/j.1365-2672.2007.03336.x (2007).
Lee, S. O. et al. Bioconversion of linoleic acid into conjugated linoleic acid during fermentation and by washed cells of Lactobacillus reuteri. Biotechnol. Lett. 25, 935–938. https://doi.org/10.1023/A:1024084203052 (2003).
Elnar, A. G., Jang, Y. J. & Kim, G. B. Heterologous expression and polyphasic analysis of CLA-converting linoleic acid isomerase from Bifidobacterium breve JKL2022. J. Agric. Food Chem. 73, 1425–1440. https://doi.org/10.1021/acs.jafc.4c05746 (2024).
Mei, Y. et al. Computational analysis and heterologous expression of BBI-like proteins from food-grade Bifidobacterium species reveal possibly a key factor in conjugated linoleic acid bioconversion. J. Agric. Food Chem. 71, 8093–8103. https://doi.org/10.1021/acs.jafc.3c00857 (2023).
Jeong, M. Y., Kim, G. B., Elnar, A. G. & Lee, B. H. Novel Bifidobacterium breve JKL2022 strain and method for producing conjugated linoleic acid thereof. South. Korea Patent WO2024005573A1 (2022).
Aziz, R. K. et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 9, 75. https://doi.org/10.1186/1471-2164-9-75 (2008).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. https://doi.org/10.1016/j.jmb.2015.11.006 (2016).
Zheng, J. et al. dbCAN3: Automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51, W115–W121. https://doi.org/10.1093/nar/gkad328 (2023).
Siren, K. et al. Rapid discovery of novel prophages using biological feature engineering and machine learning. NAR Genom Bioinform. 3, lqaa109. https://doi.org/10.1093/nargab/lqaa109 (2021).
Johansson, M. H. K. et al. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: Mobileelementfinder. J. Antimicrob. Chemother. 76, 101–109. https://doi.org/10.1093/jac/dkaa390 (2021).
Farid, W. et al. Gastrointestinal transit tolerance, cell surface hydrophobicity, and functional attributes of Lactobacillus acidophilus strains isolated from Indigenous Dahi. Food Sci. Nutr. 9, 5092–5102. https://doi.org/10.1002/fsn3.2468 (2021).
Won, G. et al. In vitro antidiabetic, antioxidant activity, and probiotic activities of Lactiplantibacillus plantarum and Lacticaseibacillus paracasei strains. Curr. Microbiol. 78, 3181–3191. https://doi.org/10.1007/s00284-021-02588-5 (2021).
Klare, I. et al. Evaluation of new broth media for microdilution antibiotic susceptibility testing of Lactobacilli, Pediococci, Lactococci, and Bifidobacteria. Appl. Environ. Microbiol. 71, 8982–8986. https://doi.org/10.1128/AEM.71.12.8982-8986.2005 (2005).
Ammor, M. S. et al. Molecular characterization of intrinsic and acquired antibiotic resistance in lactic acid bacteria and bifidobacteria. J. Mol. Microbiol. Biotechnol. 14, 6–15. https://doi.org/10.1159/000106077 (2008).
Duranti, S. et al. Prevalence of antibiotic resistance genes among human gut-derived bifidobacteria. Appl. Environ. Microbiol. 83 https://doi.org/10.1128/AEM.02894-16 (2017).
Ben-Miled, H., Benoit-Biancamano, M. O., Ben-Mahrez, K. & Rejiba, S. Alpha-amylase and alphaglucosidase inhibitory properties, beta-galactosidase activity, and probiotic potential of lactic acid bacteria and bifidobacteria from Apis mellifera intermissa and its products. World J. Microbiol. Biotechnol. 39, 205. https://doi.org/10.1007/s11274-023-03648-7 (2023).
Drula, E. et al. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 50, D571–D577. https://doi.org/10.1093/nar/gkab1045 (2022).
Rodriguez, C. I. & Martiny, J. B. H. Evolutionary relationships among bifidobacteria and their hosts and environments. BMC Genom. 21, 26. https://doi.org/10.1186/s12864-019-6435-1 (2020).
Joensen, K. G. et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. https://doi.org/10.1128/JCM.03617-13 (2014).
Darby, E. M. et al. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 21, 280–295. https://doi.org/10.1038/s41579-022-00820-y (2023).
P Alcock, B. et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 51 (D690-D699). https://doi.org/10.1093/nar/gkac920 (2023).
Bergmann, R., van der Linden, M. & Chhatwal, G. S. Nitsche-Schmitz, D. P. Factors that cause Trimethoprim resistance in Streptococcus pyogenes. Antimicrob. Agents Chemother. 58, 2281–2288. https://doi.org/10.1128/AAC.02282-13 (2014).
Lambert, M. P. & Neuhaus, F. C. Mechanism of d-cycloserine action: Alanine racemase from Escherichia coli W. J. Bacteriol. 110, 978–987. https://doi.org/10.1128/jb.110.3.978-987.1972 (1972).
Lokesh, D., Parkesh, R. & Kammara, R. Bifidobacterium adolescentis is intrinsically resistant to antitubercular drugs. Sci. Rep. 8, 11897. https://doi.org/10.1038/s41598-018-30429-2 (2018).
Zhou, J. S., Pillidge, C. J., Gopal, P. K. & Gill, H. S. Antibiotic susceptibility profiles of new probiotic Lactobacillus and Bifidobacterium strains. Int. J. Food Microbiol. 98, 211–217. https://doi.org/10.1016/j.ijfoodmicro.2004.05.011 (2005).
Nunziata, L., Brasca, M., Morandi, S. & Silvetti, T. Antibiotic resistance in wild and commercial non-enterococcal lactic acid Bacteria and bifidobacteria strains of dairy origin: An update. Food Microbiol. 104, 103999. https://doi.org/10.1016/j.fm.2022.103999 (2022).
Hall, J. P. J. Is the bacterial chromosome a mobile genetic element? Nat. Commun. 12, 6400. https://doi.org/10.1038/s41467-021-26758-y (2021).
Kusharyati, D. F. et al. Bifidobacterium from infant stool: The diversity and potential screening. Biodiversitas J. Biol. Divers. 21 https://doi.org/10.13057/biodiv/d210623 (2020).
Binda, S. et al. Criteria to qualify microorganisms as probiotic in foods and dietary supplements. Front. Microbiol. 11, 1662. https://doi.org/10.3389/fmicb.2020.01662 (2020).
Tang, G. et al. Two-component regulatory system ActS/ActR is required for Sinorhizobium meliloti adaptation to oxidative stress. Microbiol. Res. 198, 1–7. https://doi.org/10.1016/j.micres.2017.01.005 (2017).
Waddington, L., Cyr, T., Hefford, M., Hansen, L. T. & Kalmokoff, M. Understanding the acid tolerance response of bifidobacteria. J. Appl. Microbiol. 108, 1408–1420. https://doi.org/10.1111/j.1365-2672.2009.04540.x (2010).
Alvarez-Martin, P. et al. A conserved two-component signal transduction system controls the response to phosphate starvation in Bifidobacterium breve UCC2003. Appl. Environ. Microbiol. 78, 5258–5269. https://doi.org/10.1128/AEM.00804-12 (2012).
da Silva, T. F. et al. Unlocking the potential of probiotics: A comprehensive review on research, production, and regulation of probiotics. Probiot. Antimicrob. Proteins. 16, 1687–1723. https://doi.org/10.1007/s12602-024-10247-x (2024).
Yang, Y. et al. Bile salt hydrolase can improve Lactobacillus plantarum survival in gastrointestinal tract by enhancing their adhesion ability. FEMS Microbiol. Lett. 366, fnz100. https://doi.org/10.1093/femsle/fnz100 (2019).
Kim, G. B. & Lee, B. H. Biochemical and molecular insights into bile salt hydrolase in the gastrointestinal microflora—A review. Asian-Australas J. Anim. Sci. 18, 1505–1512. https://doi.org/10.5713/ajas.2005.1505 (2005).
Kang, M. H., Elnar, A. G. & Kim, G. B. Review on the function, substrate affinity, and potential application of bile salt hydrolase originated from probiotic Srains of Lactobacillus, Bifidobacterium, and Enterococcus. Food Sci. Anim. Resour. 45, 353–374. https://doi.org/10.5851/kosfa.2025.e1 (2025).
Ruiz, L., Margolles, A. & Sanchez, B. Bile resistance mechanisms in Lactobacillus and Bifidobacterium. Front. Microbiol. 4, 396. https://doi.org/10.3389/fmicb.2013.00396 (2013).
Sanchez, B. et al. Adaptation and response of Bifidobacterium animalis subsp. Lactis to bile: A proteomic and physiological approach. Appl. Environ. Microbiol. 73, 6757–6767. https://doi.org/10.1128/AEM.00637-07 (2007).
Zawistowska-Rojek, A., Kosmider, A., Stepien, K. & Tyski, S. Adhesion and aggregation properties of Lactobacillaceae strains as protection ways against enteropathogenic bacteria. Arch. Microbiol. 204, 285. https://doi.org/10.1007/s00203-022-02889-8 (2022).
Rahman, M. M., Kim, W. S., Kumura, H. & Shimazaki, K. -i. Autoaggregation and surface hydrophobicity of bifidobacteria. World J. Microbiol. Biotechnol. 24, 1593–1598. https://doi.org/10.1007/s11274-007-9650-x (2008).
Monteagudo-Mera, A., Rastall, R. A., Gibson, G. R., Charalampopoulos, D. & Chatzifragkou, A. Adhesion mechanisms mediated by probiotics and prebiotics and their potential impact on human health. Appl. Microbiol. Biotechnol. 103, 6463–6472. https://doi.org/10.1007/s00253-019-09978-7 (2019).
Bottacini, F. et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genom. 15, 170–187. https://doi.org/10.1186/1471-2164-15-170 (2014).
Cantarel, B. L. et al. The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–238. https://doi.org/10.1093/nar/gkn663 (2009).
Stam, M. R., Danchin, E. G., Rancurel, C., Coutinho, P. M. & Henrissat, B. Dividing the large glycoside hydrolase family 13 into subfamilies: Towards improved functional annotations of alpha-amylase-related proteins. Protein Eng. Des. Sel. 19, 555–562. https://doi.org/10.1093/protein/gzl044 (2006).
Sinnott, M. L. Catalytic mechanism of enzymic glycosyl transfer. Chem. Rev. 90, 1171–1202. https://doi.org/10.1021/cr00105a006 (1990).
Maske, B. L. et al. A review on enzyme-producing lactobacilli associated with the human digestive process: From metabolism to application. Enzyme Microb. Technol. 149, 109836. https://doi.org/10.1016/j.enzmictec.2021.109836 (2021).
Huligere, S. S. et al. Investigating the antidiabetic efficacy of dairy-derived Lacticaseibacillus paracasei probiotic strains: Modulating alpha-amylase and alpha-glucosidase enzyme functions. Front. Microbiol. 14, 1288487. https://doi.org/10.3389/fmicb.2023.1288487 (2023).
Cattivelli, A., Zannini, M., Conte, A. & Tagliazucchi, D. Inhibition of starch hydrolysis during in vitro co-digestion of pasta with phenolic compound-rich vegetable foods. Food Bioscience. 61 https://doi.org/10.1016/j.fbio.2024.104586 (2024).
Martiz, R. M. et al. Inhibition of carbohydrate hydrolyzing enzymes by a potential probiotic Levilactobacillus brevis RAMULAB49 isolated from fermented Ananas comosus. Front. Microbiol. 14, 1190105. https://doi.org/10.3389/fmicb.2023.1190105 (2023).
Shen, X. et al. The role and mechanism of probiotics supplementation in blood glucose regulation: A review. Foods. https://doi.org/10.3390/foods13172719 (2024).
Li, Y., Wu, Y., Wu, L., Qin, L. & Liu, T. The effects of probiotic administration on patients with prediabetes: A meta-analysis and systematic review. J. Transl Med. 20, 498. https://doi.org/10.1186/s12967-022-03695-y (2022).
Wang, J. B. et al. Screening of probiotics with efficient α-glucosidase inhibitory ability and study on the structure and function of its extracellular polysaccharide. Food Biosci. https://doi.org/10.1016/j.fbio.2021.101452 (2022).
Kumari, V. B. C. et al. Antidiabetic activity of potential probiotics Limosilactobacillus spp., Levilactobacillus spp., and Lacticaseibacillus spp. Isolated from fermented sugarcane juice: A comprehensive in vitro and in Silico study. Nutrients. https://doi.org/10.3390/nu15081882 (2023).
Koba, K. & Yanagita, T. Health benefits of conjugated linoleic acid (CLA). Obes. Res. Clin. Pract. 8, e525–532. https://doi.org/10.1016/j.orcp.2013.10.001 (2014).
Fontes, A. L. et al. Study of the association between genotypic potential and linoleic acid tolerance with microbial production of conjugated linoleic acid. J. Agric. Food Chem. 3, 2199–2207. https://doi.org/10.1021/acsfoodscitech.3c00399 (2023).
Coakley, M. et al. Conjugated linoleic acid biosynthesis by human-derived Bifidobacterium species. J. Appl. Microbiol. 94, 138–145. https://doi.org/10.1046/j.1365-2672.2003.01814.x (2003).
Moubareck, C., Gavini, F., Vaugien, L., Butel, M. J. & Doucet-Populaire, F. Antimicrobial susceptibility of bifidobacteria. J. Antimicrob. Chemother. 55, 38–44. https://doi.org/10.1093/jac/dkh495 (2005).
Breazeale, S. D., Ribeiro, A. A., McClerren, A. L. & Raetz, C. R. A formyltransferase required for polymyxin resistance in Escherichia coli and the modification of lipid A with 4-Amino-4-deoxy-L-arabinose. Identification and function oF UDP-4-deoxy-4-formamido-L-arabinose. J. Biol. Chem. 280, 14154–14167. https://doi.org/10.1074/jbc.M414265200 (2005).
Butta, H., Sardana, R., Vaishya, R., Singh, K. N. & Mendiratta, L. Bifidobacterium: An emerging clinically significant metronidazole-resistant anaerobe of mixed pyogenic infections. Cureus 9, e1134. https://doi.org/10.7759/cureus.1134 (2017).
Pieniz, S., Andreazza, R., Anghinoni, T., Camargo, F. & Brandelli, A. Probiotic potential, antimicrobial and antioxidant activities of Enterococcus durans strain LAB18s. Food Control. 37, 251–256. https://doi.org/10.1016/j.foodcont.2013.09.055 (2014).
Rodrigues, N. P. A., Garcia, E. F. & de Souza, E. L. Selection of lactic acid bacteria with promising probiotic aptitudes from fruit and ability to survive in different food matrices. Braz. J. Microbiol. 52, 2257–2269. https://doi.org/10.1007/s42770-021-00543-x (2021).
Geddes, K. & Philpott, D. J. A new role for intestinal alkaline phosphatase in gut barrier maintenance. Gastroenterology 135, 8–12. https://doi.org/10.1053/j.gastro.2008.06.006 (2008).
Kim, S. H., Song, J. H., Kim, J. & Kang, D. K. Characterisation of a lysophospholipase from Lactobacillus mucosae. Biotechnol. Lett. 42, 1735–1741. https://doi.org/10.1007/s10529-020-02895-0 (2020).
Tanaka, K. et al. Probiotic-derived polyphosphate improves the intestinal barrier function through the caveolin-dependent endocytic pathway. Biochem. Biophys. Res. Commun. 467, 541–548. https://doi.org/10.1016/j.bbrc.2015.09.159 (2015).
Goel, A. & Halami, P. M. Safety assessment of probiotic Lactiplantibacillus plantarum MCC5231 and its persistence in Gastrointestinal tract. Microb. Pathog. 194, 106824. https://doi.org/10.1016/j.micpath.2024.106824 (2024).
Liu, X., Champagne, C. P., Lee, B. H., Boye, J. I. & Casgrain, M. Thermostability of probiotics and their alpha-galactosidases and the potential for bean products. Biotechnol. Res. Int. 2014, 472723. https://doi.org/10.1155/2014/472723 (2014).
Michlmayr, H. & Kneifel, W. beta-Glucosidase activities of lactic acid bacteria: Mechanisms, impact on fermented food and human health. FEMS Microbiol. Lett. 352, 1–10. https://doi.org/10.1111/1574-6968.12348 (2014).
Su, A. C. Y. et al. Lactococcus lactis HkyuLL 10 suppresses colorectal tumourigenesis and restores gut microbiota through its generated alpha-mannosidase. Gut 73, 1478–1488. https://doi.org/10.1136/gutjnl-2023-330835 (2024).
Escamilla-Lozano, Y. et al. Synthesis of fucosyl-oligosaccharides using alpha-l-fucosidase from Lactobacillus rhamnosus GG. Molecules 24 https://doi.org/10.3390/molecules24132402 (2019).
Sela, D. A. et al. Bifidobacterium longum subsp. Infantis ATCC 15697 alpha-fucosidases are active on fucosylated human milk oligosaccharides. Appl. Environ. Microbiol. 78, 795–803. https://doi.org/10.1128/AEM.06762-11 (2012).
Nishiyama, K. et al. Bifidobacterium bifidum extracellular Sialidase enhances adhesion to the mucosal surface and supports carbohydrate assimilation. mBio. https://doi.org/10.1128/mBio.00928-17 (2017).
Acknowledgements
We would like to give thanks to the BT Research Center of Chung-Ang University (Anseong, Republic of Korea) for letting us use their facilities.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation (NRF) funded by the Ministry of Science and ICT (2021R1A2C1093838).
Author information
Authors and Affiliations
Contributions
AGE: Writing – original draft, review & editing, Conceptualization, Methodology, Investigation, Data curation. BGE: Writing – review & editing. G-BK: Writing – review & editing, Conceptualization, Methodology, Supervision, Funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Elnar, A.G., Eum, B. & Kim, GB. Genomic characterization and probiotic assessment of Bifidobacterium breve JKL2022 with strain-specific CLA-converting properties. Sci Rep 15, 15419 (2025). https://doi.org/10.1038/s41598-025-98770-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-98770-x
