
Abstract
Streptococcus mutans, a primary cariogenic bacterium, plays a central role in dental caries, one of the most widespread chronic diseases globally. Glucosyltransferases (GTFs) are key virulence factors in this process, as they synthesize extracellular polysaccharides that contribute to biofilm formation and pathogenicity. Targeting GTFs has emerged as a promising strategy for preventing dental caries, with previous studies demonstrating its potential efficacy. This study builds on our prior work by providing detailed molecular insights into the binding modes of previously identified GTF inhibitors. Using computational tools, including density functional theory, molecular docking, and molecular dynamics simulations, we examined the binding interactions and structural stability of selected inhibitors. All investigated candidates demonstrated superior binding behavior compared to the reference ligand, acarbose, as indicated by multiple structural parameters. Structural dynamics analysis revealed significant stability in the binding interactions of Complex III and V, with average deviations of 2.06 ± 0.38 and 2.07 ± 0.30 Å, respectively. Similarly, a trend in structural compactness was observed, with gyration values of 32.98 ± 0.23 and 33.01 ± 0.24 Å, respectively. Principal component analysis indicated that the constructed pattern approaches zero with the achievement of a global energy minimum, particularly for Complex III and V. Furthermore, MM/PBSA free energy calculations identified Compound V as the most favorable binder, with a binding free energy of −24.20 kcal/mol. Our findings provide valuable molecular-level insights into the inhibitory mechanisms of GTF-targeting compounds, strengthening their potential as anti-cariogenic agents. By elucidating key binding interactions, this study contributes to the ongoing search for improved scaffolds that may hinder biofilm-mediated infections and advance therapeutic strategies against dental caries.
Similar content being viewed by others

Introduction
Dental caries, a biofilm-mediated bacterial disease is a global oral health concern affecting the majority of people worldwide1,2. It is a dynamic multifactorial disease that encompasses both physical and chemical processes of dental surface3,4. The main factors underlying this disease include poor oral hygiene, fermentable carbohydrates, and pathogenic traits of cariogenic microorganisms predominantly Streptococcus mutans. S. mutans is involved in metabolizing dietary sugars via a glycolytic pathway, resulting in the production of acids such as propanoic acid and butanoic acids. This acidic environment disturbs the physiological pH and facilitates demineralization of the dental structure, resulting in loss of tooth5,6. The virulence traits of pathogen Streptococcus mutans enhances its survival at low pH allowing it to thrive in acidic environment7. Simultaneously, the secretions of glycosyltransferases, an extracellular enzyme, play a dominant role in the formation of high molecular weight glucans. The three types of Glucosyltransferases, GtfB (GTF-I), GtfC (GTFSI), and GtfD (GTF-S) produced by the Streptococcus mutans metabolize sucrose to produce complex glucans with distinct linkages. These complex glucans facilitate the attachment of other microorganisms which results in the formation of biofilm8,9.
Biofilms provide immense benefit to microorganisms by providing them with a protected environment, evading host immune response, and offering enhanced microbial resistance. In order to counteract S. mutans infection and biofilm development, a number of treatments have been attempted. Plaque removal mechanisms, broad-spectrum antibiotics, and antimicrobial substances such as chlorhexidine, fluoride salts, and quaternary ammonium compounds are some of the measures used. Chlorhexidine, a widely utilized antiseptic, suppresses plaque formation but has restrictions such as staining and taste alteration. Fluoride salts rigidify enamel as well as inhibit bacterial metabolism, while the application of amoxicillin and vancomycin antibiotics is rare for controlling severe infection. Nevertheless, antibiotic resistance as well as the non-selective effect of these medications presents a real challenge. Natural products, on the other hand, also possess strong antibiofilm activities against S. mutans. The active components in plant lectins, cranberry, raw leaves extracts of Morus alba, and the constituents within barley coffee10,11. Flavonoids and polyphenols in particular showcased remarkable anti-biofilm attributes in many of the previous investigations. Several small molecules, including apigenin12,13, anthraquinones14, α-angostin15, chitosan16, tt-farnesol17,18, 7-epiclusianone19,20, honokiol21 and myricetin22,23, have been characterized in this regard. Despite their effectiveness in controlling nuisance and medically challenging infections, there remains an issue of selectivity. Further, the emergence of resistance among the bacterial community has challenged the current therapeutics and remedies designed to combat biofilm-mediated infections24,25. Recent progress in this field has led to the development of targeted antimicrobials aiming to specifically eliminate the bacterial load of the principal etiological agent while preventing the loss of beneficial microbiota of the oral cavity26. Since the GTFs play distinct and coinciding roles in the dental biofilm formation, and as evidenced, targeting GTFs activity consequently inhibits the formation of EPS. Impairing the virulence without jeopardizing the existence of S. mutans and other oral cavity species could serve as a precise and appealing strategy for the treatment of dental caries compared to traditional treatments. Thus, the precision targeting approach is beneficial in countering the development of cariogenic biofilm and bacterial pathogenesis, while safeguarding the natural microbiota.
Inhibiting GTF involves distinct molecular mechanisms, targeting the enzyme either at genes or protein level27. Understanding these events empowers the efficient utilization of the inhibitors, aiding in the development of potent and selective therapeutics. Nijampatnam and colleagues have an enduring dedication to creating selective anti-biofilm against S. mutans virulence28. The exploration resulted in the identification of selective GTFC small molecule inhibitors29,30,31. In our previous attempt small molecules against S. mutans GTFs were identified utilizing a combination of in silico approaches followed by the validation in cellular assays32. However, the precise mechanism of inhibition for these candidates remains unclear. Advances in silico methods have revolutionized drug discovery, providing understanding concerning mechanistic insights at the atomic scale. The accuracy and effectiveness of computational tools, notably the molecular dynamic (MD) simulation, offer numerous advantages in elucidating molecular actions at the atomic level33. This is exemplified by the research conducted by Baidya and colleagues, who by employing a range of computational tools, provided a mechanistic basis of action for selected proton pump inhibitors antagonizing the function of acetyltransferase34. Recently, Chen and collaborators provided detailed insights into the relative disruptive capacities of the five most common anthocyanidins and provided an understanding of the underlying mechanisms employing the dynamic simulations35.
In the present study, a computational framework was established to investigate the binding modes and temporal stability of selected candidates with reference to the standard inhibitor acarbose, exploring the static and dynamic binding modes.Next, ligand-induced structural alterations within the target protein at both the macroscopic and microscopic levels were monitored. It was followed by the computation of associated variance in generated structural ensembles by Principal Component Analysis (PCA). The observed findings provide a molecular-level comprehension revealing possible explanations for the observed functional discrepancies amongst the candidates under examination. These findings are proposed to offer valuable information for the design and optimization of new compounds to impede biofilm-mediated infections.
Results and discussion
Dental caries is a biofilm-mediated bacterial infection which involves GTFs, which are responsible for the formation of biofilm. Inhibiting GTF activity prevents polysaccharide synthesis and consequently the development of dental caries. The current research is an extension of our earlier work32 to present molecular level details on the binding modes of identified inhbitors as potential antibiofilm inhbitors against Streptococcus mutans biofilm.
Density functional theory; from wave functions to densities
Geometry optimizations
Compounds (I–VI) were optimized at B3LYP/6-31G (d, p) level of DFT. Optimized geometries are shown in (Fig. S1). Frequency analysis was performed after geometry optimization to confirm the structures as true minimum. Minimization energies and dipole moments of compounds are shown in (Table 1). Compound III was more stable with the minimization energy − 1307334.77 kcal/mol followed by Compound IV with the energy − 1290783.46 kcal/mol.
Frontier molecular orbital analysis
Optimized structures were followed for the quantum chemical calculations to describe the intrinsic attributes of the hit compounds such as the gap between the energies associated with the highest occupied molecular orbital (EHOMO) and lowest unoccupied molecular orbital (ELUMO), enabling the determination of the global parameters. The properties include chemical softness (s), chemical hardness (η), chemical potential (µ), electronegativity (χ), nucleophilicity (ε), and electrophilicity index (ω). The inhibitory efficiency of the hit compounds has been investigated by calculating the local and global parameters. The distinguishing factors underlying the observed activity were assessed to understand the potential abilities of these inhibitors. Collectively, these studies offered valuable insight into the inhibition mechanisms of these hits towards GTF in S mutans. The high softness and low hardness value of a molecule indicate that it is a potent inhibitor.
In other words, molecules with larger softness values are more reactive and tend to form bonds easily, whereas those with smaller hardness values need to be supplied with more energy in order to perform a chemical reaction. The reactivity indices obtained globally for the hit compounds, as presented in (Table 2), indicate that Compounds I and IV possess the smallest hardness values and largest softness values, which represent their high reactivity. These properties also imply that they are strong inhibitors. Compounds II, III, V, and VI have lower softness values, implying lower reactivity.
The counter plot of the HOMO-LUMO produced shows the spatial distribution of red and green colors, the former representing the positive phase of the molecular orbitals and the latter the negative phase, as shown in (Fig. 1). The spatial extent of the HOMO and LUMO orbitals of a ligand plays a major role in specifying the type of interaction it might have with a prospective receptor. The ligand’s HOMO orbital interacts with the receptor’s LUMO, and vice versa. As a result, raising the energy of the ligand’s HOMO level decreases the energy gap between the HOMO and LUMO levels of the ligand and the receptor, thus increasing the binding probability. Lowering the energy of the ligand’s LUMO level also increases the binding probability36.

Visualization of HOMO-LUMO counter plots for the test compounds, providing insights into their electronic structures and potential reactivity.
Molecular Docking simulations
Following DFT optimization of the compounds, molecular docking analysis of the Compounds (I-VI) was performed to analyze their conformations and binding modes. Before conducting molecular docking analysis, a docking protocol benchmarking was executed. This involved comparing the deviation between the placement of the cognate ligand and the corresponding simulated pose. The relocked pose presented in Fig. S2 highlights the remarkable precision of MOE software in reproducing a similar pose. The docking analysis demonstrated strong binding affinities for all selected potential hits with a docking scores varying from −7.27 to −6.33 kcal/mol. The docking scores for Compounds I-VI are −6.45, −6.33, −6.46, −7.27, −6.90, and −6.47 for respectively, compared to the −12.09 Acarbose, used as a standard inhibitor herein, delays the absorption of carbohydrates37. The interaction pattern for the reference compound is shown in (Fig. S3). The scores docking show good correlation with biological activity reported previously32, thus highlighting the strong affinities to inhibit biofilm formation.
All these compounds exhibit 80% biofilm inhibition except for Compound I which display 76% biofilm inhibition. In addition, Compound I, with a MIC value of 15.62 µg/mL and a docking score of -6.45, exhibits better antimicrobial activity compared to Compounds 2–6, which have higher MIC values (125–250 µg/mL) despite similar docking scores. This indicates that while binding affinity may influence activity. Furthermore, the ADMET analysis also indicate these compounds exhibited favorable pharmacokinetic properties. These identified compounds stand out as promising antibiofilm inhibitors against S.mutans biofilm formation as compounds are non-toxic and safe for oral consumption.
The interaction patterns exhibited by the protein-ligand complexes are illustrated in (Fig. 2). The residues Asp588, Asp593, Asp475, Arg477, Asp480, Asn481 Asn862, Asp909, Glu515, Gln592, Gln960 and Trp517 were involved in hydrogen bonding. Hydrophobic contacts were also observed with Trp517, Leu482, and Leu434. The synergistic interplay of both the hydrophobic and hydrophilic interactions imparted overall stability to the of the complexes.

Mapping the 3D interaction landscape of (a) Compound I, (b) Compound II, (c) Compound III, (d) Compound IV, (e) Compound V, and (f) Compound VI within the protein’s binding site.
Molecular dynamics simulations
Molecular dynamics stands as an indispensable tool in unraveling the molecular mechanisms of macromolecules offering valuable insights into their structural and binding patterns. Through MD simulations, a diverse array of conformational patterns, experienced by a protein can be monitored.
To explore the dynamics triggered by the binding of the selected actives, the complexes were simulated in the presence of solvent. The resultant trajectories were then accessed for the stability profile both at global as well as local levels, compactness, and conformational landscape of these complexes, assisting in comprehending the intricate molecular mechanisms guiding the behavior of these small molecules in a dynamic state with respect to time.
Quantifying structural variability: root mean square deviation
The root mean square deviation (RMSD) is a quantitative measure used to evaluate the deviation between the native conformation of the protein and its final conformation attained during molecular dynamics. This macroscopic examination thus offers valuable insights into stability and dynamics behavior, where the smaller values correspond to a higher level of stability. The RMSD pattern for the selected candidates presented in (Fig. 3) highlights the stable binding modes though negligible deviations were recorded in some instances. All the systems have demonstrated an approximate projected deviation value of around 3 Å. In contrast, the values recorded for the Acarbose bound GTFSI revealed the value of 2.19 ± 0.28 Å. When calculating the average deviation, it was noted that Compound VI was significantly stable, bearing a substantial resemblance to the wild type acarbose, as evidenced by its average RMSD value of 2.07 ± 0.30 Å. These observations underscore the remarkable potential of Compound VI as an effective anti-biofilm agent against S. mutans. In close proximity, Compounds III and V demonstrated average RMSD values of 2.06 ± 0.38 and 2.07 ± 0.30 Å, respectively, indicating minimal deviations from the conformation observed for acarbose bound structure. Upon analysis of the RMSD of Cα atoms of GTFSI complexed with Compound I, an average value of 2.81 ± 0.60 Å was observed. However, a subtle variation from acarbose was noted between 55 and the last 100 ns of simulation. Compound IV on the other hand showcased the deviations oscillating around 2.85 ± 0.47 Å, characterized by slight fluctuations spanning from 40 to 70 ns, followed by stabilization towards the terminal time frame. Conversely, the pronounced divergence pattern with the highest deviation values of 3.0 ± 0.766 Å was observed for GTFSI bound to Compound II. The RMSD analysis underscores stable binding modes with minor deviations, notably Compound VI was identified as a promising compound in rendering stability to the target protein thus a potent inhibitor of antibiofilm.

Illustrating the time dependent RMSD of selected Compound I (red), Compound II (pink), Compound III (cyan) Compound IV (green), Compound V (orange), and Compound VI (violet) within the protein’s binding site providing insights into their structural stability. The black line represents the pattern observed for Acarbose bound to the target protein.
Revealing protein folding dynamics: radius of gyration
The radius of gyrations is valuable in assessing protein dynamics during molecular simulations, revealing insight regarding the protein’s folding upon ligand binding. The higher the value of radius of gyration indicates the decreased structural rigidity while lower values signify increased stability and compact state. As depicted in Fig. 4, all systems exhibited gyration scores between 32.96 ± 0.22 to 33 ± 0.23 Å, showcasing discernible fluctuations from the cognate ligand, acarbose. Similarly, the overlapping folding pattern was recorded for GTFSI bound to Compound IV, V, and VI with the average gyration values of 33.12 ± 0.26, 33.01 ± 0.24, and 32.96 ± 0.22 Å respectively. In comparison to the acarbose, a more folded and compact structural ensemble was obtained for the target protein when bound to Compound I and III displaying average scores of 32.49 ± 0.28 and 32.98 ± 0.23 Å. However, a distinctively less compact state was revealed by the binding of Compound II, reflected in the average Rg value of 33.48 ± 0.36 Å.
Summarizing the results of gyration, the results of radius of gyration show that all potential antibiofilm inhibitors impart structural rigidity to the protein, indicating that all systems were well converged.

The gyration patterns of selected compounds—Compound I (red), Compound II (pink), Compound III (cyan), Compound IV (green), Compound V (orange), and Compound VI (violet)—within the protein’s binding site during the simulation, shedding light on the folding dynamics. The black line represents the pattern observed for Acarbose bound to the target protein.
Exploring atomic flexibility: root mean square fluctuations
The internal dynamics of the protein were evaluated by calculating the RMSF of the C-alpha atoms of the protein, providing valuable insights into flexibility and stability. As depicted in (Fig. 5), all the complexes show similar binding patterns providing understanding regarding the functionalities and interactions with target protein. Slighter fluctuations were observed in the loop regions spanning residues 120–240 of Domain IV of the protein. The loop regions are however, inherently associated with structural flexibility. Notably, even with fluctuations occurring in certain regions, the active site of the protein remained stable during the simulation compared to the reference inhibitor. The RMSF analysis revealed that the binding of the five selected compounds markedly reduced the fluctuations thereby possessing the stabilization effect on the protein. The average fluctuations recorded for the test Compounds I-VI were 1.53 ± 0.90, 1.57 ± 0.75, 1.37 ± 0.63, 1.47 ± 0.73, 1.33 ± 0.57, and 1.33 ± 0.56 Å respectively compared to the acarbose presenting with the value of 1.33 ± 0.60 Å. In summary, these findings contribute to a better insight into the molecular interactions between the protein and the binding partner, that could guide the future strategies aimed at targeting GTF.

The per residue fluctuation profile of the selected compounds—Compound I (red), Compound II (pink), Compound III (cyan), Compound IV (green), Compound V (orange), and Compound VI (violet)—within the protein’s binding site over the course of simulation, offering insights into their respective degrees of flexibility. The black line represents the pattern observed for Acarbose bound to the target protein.
MMPBSA: Estimation of binding free energies
The quantification of binding energy for the small ligands to biological receptors is an essential aspect of studies involving molecular interaction. The calculations mainly involve molecular mechanics energies, commonly employing the Poisson-Boltzmann (MM/PBSA) and generalized born (MM/GBSA) approaches and are often integrated with molecular dynamics simulations. Herein the current study, the ΔG bind for the selected antibiofilm inhibitors was evaluated via the MM/PBSA approach to assess their binding affinities of tested ligands. To evaluate the binding free energy, the snapshots from the terminal stages were extracted, and the resulting free energy values along with other components are provided in (Table 3). The highest affinity was attained for Compound V featuring the values of −24.20 kcal/mol. Furthermore, the detailed analysis of individual energy components contributing to the binding process was also computed. Energy decomposition analysis found that non-electrostatic interactions, i.e., van der Waals (Evdw) and electrostatic interactions (Eele), were positively adding to binding, while polar terms were generally unfavorable in all test compounds. Evdw and Eele in Compound V were found to be −36.29 and −30.23 kcal/mol, respectively. Time-dependent hydrogen bond analysis (Fig. S4) also reflected the ability of these compounds to interact with major binding pocket residues, thereby enhancing ligand stability in the pocket. To further understand ligand-protein interactions, per-residue energy decomposition analysis was performed employing the MM/PBSA method. The analysis provides a quantification of individual residue contribution towards overall binding stability, and this aids in the identification of major stabilizing interactions. The results, shown in (Fig. S5), suggest that residues, Arg475, Asp481, Asp588, Asp909, Asn862, Gln592, Gln960, His587, Leu434, Trp517 and Tyr430, have a significant contribution towards ligand binding, affirming their role in inhibitor recognition and stability.
This comprehensive analysis illuminates the energetic mechanisms driving the molecular recognition between the selected ligands and the target protein, providing valuable insights for future workup and successive optimization.
Principal component analysis and free energy landscape
Principal Component Analysis, also called PCA, is an important mathematical algorithm used in structural biology that helps in reducing the high conformational space of proteins to the concise set of PCs, each capturing the most variance. These principal components then become the representatives of complex data systems. In molecular dynamics, PCA helps unveil the functional motions of proteins by focusing on the major components and reducing the protein’s conformational space. Further, it helps to identify crucial folding and unfolding patterns of proteins, providing insights into the underlying mechanisms that govern protein functions, which is important to understand the cellular processes. To examine the energetics of the generated conformational space, additional FEL analysis was performed. This study examined the dynamic behavior of acarbose-bound GTFSI and hits-bound GTFSI, using PCA-FEL by extracting the dominant motions from structure ensembles. The visual representation of the observed PCs for the systems under study is presented in (Fig. 6a–g). The cumulative variance and temporal values of PCs are displayed in Figs. S6 and S7). According to the PCA plot, GTFSI bound to Compound II showed the highest variability in the values of PC1 (−100 to 50) thus indicating relatively more dynamic behavior along the axis. The stability contour observed at the terminal of the simulation is depicted in (Fig. 6c). The binding of Compound I triggered pronounced structural alterations along PC2 (Fig. 6b). Global minimum and stable PCs, along both the first and second axis (PC1 = PC2 = 0), in the case of Compound III and V indicated that their binding restricts the intrinsic flexibility that is evident from the condensed contours (Fig. 6d, f). On the other hand, although the same pattern was observed for the reference compound, final scattering points favor pronounced conformational fluctuations compared to others (Fig. 6a).

Scatter plot of principal components along with a free energy landscape observed for the GTFSI bound to (a) Acarbose, (b) Compound I, (c) Compound II, (d) Compound III, (e) Compound IV, (f) Compound V, and (g) Compound VI.
Despite the merits of computational methods like DFT, molecular docking, and MD simulations, there are limitations that need to be recognized. Although our docking methodology included protein flexibility, it could still fall short in recognizing the dynamic character of enzyme-inhibitor interactions on long time scales. Likewise, MD simulations, while useful in structural stability modeling, are constrained by simulation length and might fail to represent long-term conformational changes in physiological conditions. Further, while binding free energy calculations are informative, they are based on approximations and do not consider all possible entropic contributions.
Moreover, the compounds that were tested here have been proven experimentally to inhibit antimicrobial activity in culture-based assays but their direct enzymatic inhibition of GTFs is yet to be investigated. Future research involving enzyme-based inhibition assays as well as in vivo models will be crucial to further establish their mechanism of action and therapeutic utility.
Conclusion
Dental caries, a biofilm-mediated bacterial disease, affects a significant portion of the global population. Targeting GTFs has been shown to effectively inhibit the production of extracellular polysaccharides, which are crucial for biofilm formation. A deeper understanding of these molecular events facilitates the development of selective and potent inhibitors. Building on our previous work, this study provides a detailed characterization of the binding modes of selected GTF inhibitors using computational approaches, including density functional theory, molecular docking, and molecular dynamics simulations.
All tested compounds demonstrated stable binding, engaged key residues within the active site, and restricted target site mobility relative to the reference inhibitor. Stability parameters aligned with free energy calculations and conformational analysis, further validating the observed interactions. Among the candidates, Compound V exhibited the strongest affinity toward GTFSI, with an average deviation of 2.07 ± 0.30 Å and the lowest free energy (−24.02 kcal/mol). Reduced fluctuations along the PC axis and the formation of stable energy basins further supported its binding efficiency.
This study enhances our understanding of the molecular basis of biofilm inhibition and provides a computational framework for future research. To build upon these findings, experimental validation of the identified inhibitors through, in vitro and in vivo assays is recommended to confirm their efficacy against S. mutans biofilms. Additionally, future studies could explore structural modifications to optimize binding affinity and specificity. The integration of machine learning models for predictive inhibitor design and high-throughput screening could further accelerate the discovery of novel anti-biofilm agents, ultimately contributing to improved therapeutic strategies for dental caries prevention.
Materials and methods
Protein structure preparation and refinement
The crystal structure of S. mutans GTF in complexed with standard inhibitor, Acarbose was retrieved from the RSCB Protein Data Bank38 under the accession ID of 3AIC with a resolution of 3.0 Å39. The protein is complex with inhibitor acarbose and features a homomeric structure comprising A-H chains. All chains were inspected with UCSF Chimera version 1.17.140 to address atom correction, eliminate alternate location of residues partial charges issues, addition of hydrogen atoms, and address any missing residues. The G chain was found to be ideal fulfilling all parameters, thus retained. Unnecessary heteroatoms and the water molecules were removed using Molecular Operating Environment (MOE) version 201941. The optimization was done for lone pairs and the addition of hydrogens was done with the Protonate 3D module, followed by minimizing the energy constraints and assignment of the partial charges according to AMBER10EHT force field42. The gradient value was set to 0.1 kcal/molA2, while the rest of the parameters were kept at default.
Preparation of ligands
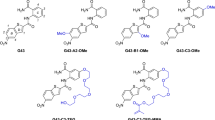
To explore the binding mechanisms, the data of the six antibiofilm inhibitors with identified activity was collected (Fig. 7). The 2D structures of the compounds were constructed via ChemBioDraw Ultra version 13.043. The structures were imported to MOE for structure preparation, concerning their ionization, tautomeric variations, and protonation states. default. The MMFF94X force field was implemented to obtain an energy-minimized conformation, keeping the dielectric constant and RMS gradient threshold of 1 and 0.1 kcal/mol/Å respectively44. The compounds after initial preparation underwent additional optimization discussed in the succeeding section.

The 2D structural representation of heterocyclic scaffolds against Streptococcus mutans’s biofilm.
Density functional theory
Density functional theory (DFT) is a computational technique based on quantum mechanics that is extensively applied in chemistry, physics, and materials science to calculate the electronic and nuclear structure of atoms, molecules, and many-body systems, especially in their ground state. DFT calculations were employed using Gaussian 16 software45. The geometries of all the compounds were optimized using B3LYP exchange-correlation functional and 6-31G (d, p) basis set. The structural verification as a true minimum was validated using frequency analysis. Moreover, Frontier molecular orbital analysis and global reactivity descriptors were thoroughly researched. Single-point energy calculations were carried out at the same level, and global reactivity features were investigated to analyze both the chemical reactivity and inhibitory activity of the compounds.
Koopmans’s theorem was used to calculate different electronic properties such as electronegativity (χ), electron affinity (Ea), ionization energy (IE), global chemical softness (σ), and hardness (η)46,47.
Molecular Docking
Molecular docking evaluates potential conformations attained by any ligand within the macromolecular binding sites and the subsequent determination of binding scores. The structures in their optimized conformations were subjected to molecular docking studies. Initially, benchmarking via redocking experiment was performed to validate the docking protocol and software, in terms of reproducing the crystal pose of the native ligand. The docking grid was placed at the active site based on the known inhibitor binding site of the ligand-bound protein to ensure precise ligand placement. The prepared protein structure was provided as an input, and the cognate ligand was docked at the targeted site. The optimized docking process used the triangle matcher conformational placement algorithm, while ASE and Affinity dG scoring functions were used for estimation. To ensure flexibility of the target site, an induced fit protocol was employed. After benchmarking, test compounds were docked into the protein, and the best-scoring pose was chosen for evaluation48. Interaction diagrams of protein-ligand complexes were drawn using UCSF Chimera.
Molecular dynamics simulations
All-atom molecular dynamics simulations were conducted in a comprehensive manner using the PMEMD algorithm with CUDA acceleration on AMBER 2249. This method was utilized to gain atomic-level insights into the binding interactions of the identified antibiofilm inhibitors. The antechamber50 and tleap modules were used to create the topologies for the target protein complexes obtained from the docking experiment. The Amber ff19SB force field51 was assigned to the protein as it has a higher accuracy for modeling protein side-chain and backbone interactions, so it is perfectly suited for biomolecular simulations. The GAFF252 force field was used for small molecules because it is dedicated to organic molecules, providing good parameterization of ligand-protein interactions as well as consistent behavior within the Amber framework. These selections enable a realistic and reliable simulation of protein-ligand interactions. The six systems were immersed in a solvent environment generated with TIP3P53, an explicit water model, inside a periodic box with atoms of the protein separated by at least 10 Å. Counter ions, sodium (Na+) in the present case were added to the systems to preserve the neutrality. The SHAKE technique was used to constrain the electrostatic interactions, while the particle mesh ewald (PME)54, calculate long-range interactions. The energy minimization was performed in a step-wise manner, where the restraint was lifted gradually, to mitigate unfavorable interactions and probable steric conflicts. All systems were then equilibrated by NVT (temperature: 300 K) and NPT (pressure: 1 bar) ensembles with the help of Langevin dynamics and Berendsen coupling55. A production run of 100 ns was performed with an integration time step of 2 fs. The results were analyzed using CPPTRAJ modules56, while VMD57 was used for visualizing dynamic trajectories and Xmgrace58 was utilized for graphs plotting.
Binding free energy calculations
The molecular mechanics Poisson Boltzmann surface area (MMPBSA) method is a popular and established method for reliable estimation of protein-ligand complex binding free energies from MD simulation snapshots59. Using the MM/PBSA approach, incorporated into AMBER 22, the computation of free energy was done on a collection of 500 frames. Further, various energy components involved in binding were also computed.
The calculation was done using the following equation:
The molecular mechanics energy differences in the gas phase are symbolized as ΔEMM, while −TΔS and ΔGsolv represent conformational entropic contributions and desolvation free energy with ligand binding, respectively. Further, ΔEMM comprises ΔEelec (electrostatics), ΔEvdW (Van der Waals), and ΔEinternal (bond, dihedral, and angle) energies. Further, ΔGpol and ΔGnp represent polar and nonpolar solvation energies, respectively.
Principal component analysis
Large datasets are very difficult to interpret; however, there is a useful technique called principal component analysis (PCA) which acts as a dimensionality reduction technique without losing any important information. The PCA explains the direction and magnitude of the variance in the dataset. The dataset variables are converted into a new set of variables, named principal components (PCs), which are orthogonal (perpendicular) projections of the data onto a lower-dimensional space. The PCA of the simulated systems was performed with the help of the MD analysis tool to explore the conformational ensembles attained by the protein upon ligand binding60,61.
Free energy landscape
The free energy landscape (FEL) of protein-ligand complexes can be obtained by employing a conformational sampling method that facilitates the exploration of different conformations near the native structure. In the present study, the trajectories obtained from molecular simulations were selected for sampling purposes. The two-dimensional picture of the FEL describes the probability of a system to be in a given state, characterized by a value qα of some variables of interest (reaction coordinates). The FEL is calculated according to the following equation:
Here, k is the Boltzmann constant, T is the simulation temperature, P(qα) is the estimated probability density function from an MD data histogram, and Pmax(q) is the probability of the most probable state. FEL calculations were performed using the gmx_sham module of GROMACS62.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.
References
Das, A. et al. Biofilm modifiers: the disparity in paradigm of oral biofilm ecosystem. Biomed. Pharmacother. 164, 114966 (2023).
Li, Y. et al. Engineered biomaterials trigger remineralization and antimicrobial effects for dental caries restoration. Molecules 28 (17), 6373 (2023).
Abou Neel, E. A. et al. Demineralization–remineralization dynamics in teeth and bone. Int. J. Nanomed. 4743–4763. (2016).
Cheng, L. et al. Expert consensus on dental caries management. Int. J. Oral Sci. 14 (1), 17 (2022).
Matsui, R. & Cvitkovitch, D. Acid tolerance mechanisms utilized by Streptococcus mutans. Future Microbiol. 5 (3), 403–417 (2010).
Zhang, J., Sardana, D., Wong, M., Leung, K. & Lo, E. Factors associated with dental root caries: a systematic review. JDR Clin. Transl. Res. 5 (1), 13–29 (2020).
Liu, T. et al. Interspecies interactions between Streptococcus mutans and Streptococcus agalactiae in vitro. Front. Cell. Infect. Microbiol. 10, 344 (2020).
Khan, S. et al. Antibacterial and antibiofilm potential of chlorophyllin against Streptococcus mutans in vitro and. Silico Antibiot. 13 (9), 899 (2024).
Soro, A. S., Lamont, R. J., Egland, P. G., Koo, H. & Liu, Y. Dental caries. Molecular Medical Microbiology 915–930. (Elsevier, 2024).
Koo, H., de Nino, P., Schobel, B., Vacca Smith, A. & Bowen, W. Influence of cranberry juice on glucan-mediated processes involved in Streptococcus mutans biofilm development. Caries Res. 40 (1), 20–27 (2005).
Thimothe, J., Bonsi, I. A., Padilla-Zakour, O. I. & Koo, H. Chemical characterization of red wine grape (Vitis vinifera and Vitis interspecific hybrids) and pomace phenolic extracts and their biological activity against Streptococcus mutans. J. Agric. Food Chem. 55 (25), 10200–10207 (2007).
Koo, H. et al. Inhibition of Streptococcus mutans biofilm accumulation and polysaccharide production by apigenin and tt-farnesol. J. Antimicrob. Chemother. 52 (5), 782–789 (2003).
Koo, H., Rosalen, P. L., Cury, J. A., Park, Y. K. & Bowen, W. H. Effects of compounds found in propolis on Streptococcus mutans growth and on glucosyltransferase activity. Antimicrob. Agents Chemother. 46 (5), 1302–1309 (2002).
Pandit, S., Song, K. Y. & Jeon, J. G. Withania somnifera attenuates acid production, acid tolerance and extra-cellular polysaccharide formation of Streptococcus mutans biofilms. Am. J. Chin. Med. 42 (01), 157–171 (2014).
Nguyen, P. T. M., Falsetta, M. L., Hwang, G., Gonzalez-Begne, M. & Koo, H. α-Mangostin disrupts the development of Streptococcus mutans biofilms and facilitates its mechanical removal. PLoS One 9 (10), e111312. (2014).
Rajabnia, R., Ghasempour, M., Gharekhani, S., Gholamhoseinnia, S. & Soroorhomayoon, S. Anti-Streptococcus mutans property of a Chitosan: containing resin sealant. J. Int. Soc. Prev. Commun. Dentist. 6 (1), 49–53 (2016).
Koo, H. et al. Effects of apigenin and tt‐farnesol on glucosyltransferase activity, biofilm viability and caries development in rats. Oral Microbiol. Immunol. 17 (6), 337–343 (2002).
Koo, H. et al. Apigenin and tt-farnesol with fluoride effects on S. mutans biofilms and dental caries. J. Dent. Res. 84 (11), 1016–1020 (2005).
Branco-de-Almeida, L. S. et al. Effects of 7-epiclusianone on Streptococcus mutans and caries development in rats. Planta Med. 77 (01), 40–45 (2011).
Murata, R. M. et al. Inhibition of Streptococcus mutans biofilm accumulation and development of dental caries in vivo by 7-epiclusianone and fluoride. Biofouling 26 (7), 865–872 (2010).
Greenberg, M., Urnezis, P. & Tian, M. Compressed mints and chewing gum containing magnolia bark extract are effective against bacteria responsible for oral Malodor. J. Agric. Food Chem. 55 (23), 9465–9469 (2007).
Falsetta, M. L. et al. Novel antibiofilm chemotherapy targets exopolysaccharide synthesis and stress tolerance in Streptococcus mutans to modulate virulence expression in vivo. Antimicrob. Agents Chemother. 56 (12), 6201–6211 (2012).
Kim, D. et al. Cranberry flavonoids modulate cariogenic properties of mixed-species biofilm through exopolysaccharides-matrix disruption. PLoS One 10 (12), e0145844. (2015).
Batoni, G., Maisetta, G. & Esin, S. Therapeutic potential of antimicrobial peptides in polymicrobial biofilm-associated infections. Int. J. Mol. Sci. 22 (2), 482 (2021).
Kalelkar, P. P., Riddick, M. & García, A. J. Biomaterial-based antimicrobial therapies for the treatment of bacterial infections. Nat. Reviews Mater. 7 (1), 39–54 (2022).
Singh, A. et al. Bacterial biofilm infections, their resistance to antibiotics therapy and current treatment strategies. Biomed. Mater. 17 (2), 022003 (2022).
Zhang, Q., Ma, Q., Wang, Y., Wu, H. & Zou, J. Molecular mechanisms of inhibiting glucosyltransferases for biofilm formation in Streptococcus mutans. Int. J. Oral Sci. 13 (1), 30 (2021).
Nijampatnam, B., Nadkarni, D. H., Wu, H. & Velu, S. E. Antibacterial and antibiofilm activities of Makaluvamine analogs. Microorganisms 2 (3), 128–139 (2014).
Nijampatnam, B., Casals, L., Zheng, R., Wu, H. & Velu, S. E. Hydroxychalcone inhibitors of Streptococcus mutans glucosyl transferases and biofilms as potential anticaries agents. Bioorg. Med. Chem. Lett. 26 (15), 3508–3513 (2016).
Zhang, Q. et al. New small-molecule inhibitors of dihydrofolate reductase inhibit Streptococcus mutans. Int. J. Antimicrob. Agents 46 (2), 174–182 (2015).
Zhang, Q. et al. Structure-based discovery of small molecule inhibitors of cariogenic virulence. Sci. Rep. 7 (1), 5974 (2017).
Atta, L. et al. Virtual screening, synthesis and biological evaluation of Streptococcus mutans mediated biofilm inhibitors. Molecules 27 (4), 1455 (2022).
Yuan, Y., Deng, J. & Cui, Q. Molecular dynamics simulations Establish the molecular basis for the broad allostery hotspot distributions in the Tetracycline repressor. J. Am. Chem. Soc. 144 (24), 10870–10887 (2022).
Baidya, A. T. et al. Mechanistic insight into the Inhibition of choline acetyltransferase by proton pump inhibitors. ACS Chem. Neurosci. 14 (4), 749–765 (2023).
Chen, Y. et al. Five similar Anthocyanidin molecules display distinct disruptive effects and mechanisms of action on Aβ1–42 protofibril: A molecular dynamic simulation study. Int. J. Biol. Macromol. 256, 128467 (2024).
Shanmugam, M. et al. Synthesis, characterization, and antiproliferative and apoptosis inducing effects of novel s-triazine derivatives. New J. Chem. 42 (3), 1698–1714 (2018).
Nakamura, K., Yamagishi, S., Matsui, T. & Inoue, H. Acarbose, an alpha-glucosidase inhibitor, improves insulin resistance in fructose-fed rats. Drugs Under Exp. Clin. Res. 31 (4), 155–159 (2005).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28 (1), 235–242 (2000).
Ito, K. et al. Crystal structure of Glucansucrase from the dental caries pathogen Streptococcus mutans. J. Mol. Biol. 408 (2), 177–186 (2011).
Pettersen, E. F. et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25 (13), 1605–1612 (2004).
Molecular Operating Environment (MOE); Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC & Canada H3A 2R7 (2019).
Case, D. A. et al. Amber 10. (2008).
Narayanaswamy, V. K., Rissdörfer, M. & Odhav, B. Review on CambridgeSoft chembiodraw ultra 13.0 V. Int. J. Theor. Appl. Sci. 5, 45–49 (2013).
Halgren, T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17 (5-6), 490–519 (1996).
Frisch, M. et al. Gaussian 16 (Gaussian, Inc., 2016).
Zhan, C. G., Nichols, J. A. & Dixon, D. A. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies. J. Phys. Chem. A. 107 (20), 4184–4195 (2003).
Luo, J., Xue, Z. Q., Liu, W. M., Wu, J. L. & Yang, Z. Q. Koopmans’ theorem for large molecular systems within density functional theory. J. Phys. Chem. A. 110 (43), 12005–12009 (2006).
Czestkowski, W. et al. Structure-based discovery of high-affinity small molecule ligands and development of tool probes to study the role of chitinase-3-like protein 1. J. Med. Chem. (2024).
Case, D. A. et al. J. Chem. Inf. Model. 63 (20), 6183–6191. (2023).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Antechamber: an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 222 (1), (2001).
Tian, C. et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16 (1), 528–552 (2019).
He, X., Man, V. H., Yang, W., Lee, T. S. & Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 153 (11). (2020).
Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105 (43), 9954–9960 (2001).
Essmann, U. et al. G. A smooth particle mesh Ewald method. J. Chem. Phys. 103 (19), 8577–8593 (1995).
Berendsen, H. J., Postma, J., Van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81 (8), 3684–3690 (1984).
Roe, D. R. & Cheatham, T. E. III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9 (7), 3084–3095 (2013).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14 (1), 33–38 (1996).
Turner, P. & XMGRACE Version 5.1. 19. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology. (2005).
Ranade, S. D. et al. Design, synthesis, molecular dynamics simulation, MM/GBSA studies and Kinesin spindle protein inhibitory evaluation of some 4-aminoquinoline hybrids. Comput. Biol. Chem. 105, 107881 (2023).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32 (10), 2319–2327 (2011).
Gowers, R. J. et al. MDAnalysis: a Python package for the rapid analysis of molecular dynamics simulations; Los Alamos National lab. (LANL) (2019).
Maisuradze, G. G., Liwo, A. & Scheraga, H. A. Relation between free energy landscapes of proteins and dynamics. J. Chem. Theory Comput. 6 (2), 583–595 (2010).
Acknowledgements
The authors express their gratitude to the Researchers Supporting Project (RSPD2025R994) at King Saud University, Riyadh, Saudi Arabia, for their support.
Author information
Authors and Affiliations
Contributions
L.A. performed the research work and wrote the manuscript. A.R.S., M.M., and S.M. validated the methodology and wrote the manuscript. M.N.A. and A.A. reviewed the manuscript. Z.U.H. provided the resources and supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Atta, L., Siddiqui, A.R., Mushtaq, M. et al. Molecular insights into antibiofilm inhibitors of Streptococcus mutans glucosyltransferases through in silico approaches. Sci Rep 15, 14160 (2025). https://doi.org/10.1038/s41598-025-98927-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-98927-8
