
Abstract
Finger millet (Eleusine coracana L., EC) is a nutritionally dense and medicinally valuable cereal crop with a long history of use in traditional health practices. Contemporary pharmacological studies have demonstrated that extracts from EC and its individual chemical constituents exhibit a wide range of health benefits and therapeutic properties. For instance, they possess antioxidant activity, can reduce blood glucose levels, and enhance gastrointestinal health. However, the full spectrum of their chemical constituents remains to be fully elucidated. In this study, we established a state-of-the-art analytical workflow integrating Ultra-High-Performance Liquid Chromatography coupled with Quadrupole-Orbitrap Mass Spectrometry (UHPLC-Q-Exactive Orbitrap MS) with Feature-Based Molecular Networking (FBMN) and Parallel Reaction Monitoring (PRM) for comprehensive chemical profiling of EC. A total of 126 compounds were unambiguously identified, including 64 flavonoids, 24 phenylpropanoids, 21 organic acids, and 22 other metabolites. Notably, 44 of these represent novel reports in EC, significantly expanding its known chemical repertoire. Further verification via Western Blot (WB) showed that the ethanol extract of EC (1 ~ 100 µg/mL) could concentration-dependently inhibit the overexpression of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) proteins in lipopolysaccharide (LPS)-induced RAW264.7 macrophages, which initially confirms its in vitro anti-inflammatory activity. The analytical method established in this study enables rapid and efficient identification of the major chemical constituents of EC, laying a foundation for its quality control, exploration of bioactive components, and mechanism research.
Similar content being viewed by others

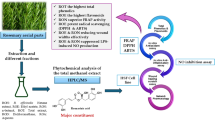

Introduction
Finger millet (Eleusine coracana L.) (EC), belonging to the Poaceae family, and one of the three major cereal crops in arid regions, it is identified by drought tolerance and resistance to alkali and salinity, and it grows on marginal lands and in high-altitude areas. It is cultivated in countries including India, Nepal, Uganda, and China, with main cultivation areas in China being Guizhou, Hunan, Guangxi, Shaanxi, Tibet, and other regions1. Due to its drought resistance and ease of cultivation, it is considered a staple food in many areas2. Beyond its robust agronomic adaptability and status as a staple food for millions, EC boasts profound medicinal value, with modern pharmacological studies confirming its multifaceted health benefits—including potent antioxidant activity, hypoglycemic effects, gastrointestinal protection3. For example, they exhibit antioxidant activity, have the ability to reduce blood sugar levels, and contribute to the enhancement of gastrointestinal health4,5. Additionally, related studies have indicated that it has physiological functions such as treating iron-deficiency anemia, enhancing immunity, preventing cancer, preventing diabetes, and protecting the heart against hyperlipidemia6,7.
In recent years, with the advancement of analytical techniques, some studies have revealed the presence of various vitamins, minerals, and antioxidants in EC. However, a comprehensive understanding of its complex chemical composition and biological activity is still limited8. Existing phytochemical profiling studies on Eleusine coracana L. and its congeneric species have certain limitations, which highlight the necessity of the present work. For instance, Soujanya et al. focused on phenolic compounds in drum-dried EC, but their analysis was restricted to seed coat and endosperm fractions, with no coverage of the whole plant9; additionally, their study only identified phenolics, lacking characterization of other important compound classes such as phenylpropanoids and organic acids. Sukor et al. reported phytochemical annotation of Eleusine coracana L indica using HPLC-MS/MS, but their approach relied solely on database matching without integrating advanced strategies like Feature-Based Molecular Networking (FBMN) or Parallel Reaction Monitoring (PRM), leading to difficulties in distinguishing isomers (e.g., flavonoid glycoside isomers) and low detection rates for trace components10. Peñaloza et al. employed HPLC-DAD, UPLC-DAD-MS/MS, and NMR for metabolite profiling of Eleusine coracana L indica, but the number of identified compounds was limited (fewer than 60), and the coverage of compound classes (e.g., only 8 flavonoids and 5 organic acids) was far from comprehensive, failing to reflect the full chemical diversity of Eleusine coracana L species11.
To address these gaps, this study aims to conduct an in-depth analysis of the chemical components of whole-plant EC through an integrated advanced analytical method. By combining UHPLC-Q-Exactive Orbitrap MS with FBMN and PRM, we not only expand the coverage of compound classes (successfully identifying 24 phenylpropanoids and 21 organic acids, which were not systematically reported in Soujanya et al. (2024) but also resolve isomeric ambiguities (e.g., isoquercitrin isomers) via FBMN and improve the detection sensitivity of trace components using PRM. Ultimately, this work identifies 126 compounds in EC, significantly exceeding the number of components reported in Peñaloza et al. (2018), with the goal of discovering new active ingredients, elucidating their potential health benefits, advancing the modernization of traditional medicine, and providing possibilities for the development of new health foods and drugs.
The efficacy of traditional Chinese medicine primarily stems from its chemical components. In-depth research into these components helps to clarify the material basis of their effects, providing a scientific basis for the standardization and regulation of traditional Chinese medicine. They are usually composed of multiple components increasing the complexity of the research. Moreover, some components in traditional Chinese medicine may have unknown structures or are difficult to identify, requiring reliance on advanced analytical techniques and methods12.UHPLC-QE-Orbitrap-MS, identify by high resolution, high separation, and high sensitivity, can quickly identify various chemical components in the complex system of traditional Chinese medicine by analyzing the relative molecular mass, fragmentation patterns, and other information of the compounds13,14,15,16,17,18.
Over the past decade, progress in mass spectrometry (MS)-based metabolomics has transformed natural product analysis, enabling high-throughput, high-resolution profiling of complex matrices. A key innovation, Feature-Based Molecular Networking (FBMN), stands out as a robust tool for untargeted metabolome exploration, overcoming limitations of traditional spectral analysis. Unlike conventional networking that relies solely on MS/MS similarity, FBMN integrates chromatographic and mass spectral features—retention time, precursor ion accuracy, and fragmentation patterns—to build reliable networks. This groups structurally related compounds into “molecular families,” where nodes represent metabolites and edges reflect shared properties like biosynthetic pathways or modifications (e.g., glycosylation, methylation)19. For EC, rich in diverse phenolics, FBMN offers unique benefits. It accelerates annotation of known compounds via database matches (e.g., GNPS, HMDB) and highlights “unknown unknowns”—uncharacterized metabolites with potential novel bioactivity. By clustering isomers with similar MS/MS but distinct retention times, it resolves ambiguities in traditional MS identification (e.g., differentiating flavonoid glycoside isomers)20. Additionally, FBMN visualizes metabolic relationships, revealing biosynthetic trends (e.g., phenolic glycosylation patterns) to illuminate the plant’s metabolic machinery21. Parallel Reaction Monitoring (PRM) is an acquisition strategy in mass spectrometry that focuses on the qualitative of specific ions derived from targeted precursor ions. This method is particularly adept at analyzing complex biological samples, providing precise measurements even in the presence of intricate matrices22. Compared to Data Dependent MS2(ddMS2, PRM offers higher specificity and accuracy. It selectively monitors the fragment information in the MS/MS spectrum, excluding interference from other ions. At the same time, it has high precision and can provide accurate qualitative results by comparing with known standards or internal standards23,24.
Therefore, this study adopts UHPLC-QE-Orbitrap-MS combined with FBMN and PRM to systematically identify the chemical constituents of EC。Using this integrated approach, we successfully characterized 126 compounds in EC, including 64 flavonoids, 24 phenylpropanoids, 21 organic acids, and 22 other types of compounds, forty-four compounds that had not been documented in previous EC-related studies were successfully identified. This research aims to provide a comprehensive profile of EC’s major constituents, thereby enhancing its scientific value for further studies on its bioactive components and applications.
Materials and methods
Plant material
The kernels of Eleusine coracana (L.) Gaertn. were procured from authentic folk sources in Shaoyang, Hunan Province, P. R. China, in 2022, and authenticated by Professor Wei Cai from Hunan University of Medicine. The plant material was authenticated as a genuine product by Professor Cai Wei from Hunan University of Medicine, and the voucher specimen is deposited in the School of Pharmaceutical Sciences, Hunan University of Medicine (Herbarium No.: HMU-EC-2022). The initial weight of the collected EC kernels was 500.0 g. The kernels were dried at 45℃ to a constant weight to remove residual moisture, then mechanically crushed into fine powder (particle size ≤ 80 mesh) using a high-speed pulverizer, which was used for subsequent extraction and sample preparation.
Chemicals and reagents
Reference standards, including Citric acid, Caffeic acid and p-Coumaric acid were purchased from Aladdin; cis-aconitate, Azelaic Acid, Abscisic Acid were purchased from Shandong Xiya Chemical Co., Ltd.; catechin, Apigenin8-C-glucoside, isoquercitrin, isorhamnetin were purchased from Chengdu Hurbpurify CO., LTD; Phloretin, Adenosine Cyclophosphate were purchased from Chengdu HerbSubstance Co., Ltd; procyanidins was purchased from Sichuan Vicky Biotechnology Co., Ltd.; Hyperoside was purchased from National Institutes for Food and Drug Control. Rutin, Quercetin, Naringenin were purchased from Chengdu Aifa Biotechnology Co., Ltd; Eriodictyol was purchased from Chengdu Pusi Biotechnology Co., Ltd. Its purity (≥ 98.5%) was confirmed using HPLC-UV analysis. HPLC grade acetonitrile, methanol and formic acid (FA) were purchased from Thermo Fisher Scientific (Fair Lawn, NJ, USA). All the other chemicals used in this study are analytical grade and were purchased from Beijing Chemical Works (Beijing, China). Deionised water was obtained using a Milli-Q Gradient Å 10 System (Millipore, Billerica, MA, USA).
Lipopolysaccharide (LPS, Escherichia coli origin), RAW264.7 macrophage cell line (purchased from a regular cell bank), RIPA lysis buffer (containing protease inhibitors), BCA protein assay kit, COX-2-specific primary antibody (1:1000 dilution), iNOS-specific primary antibody (1:1000 dilution), β-actin internal reference primary antibody (1:1000 dilution), anti-mouse IgG secondary antibody (1:5000 dilution), and anti-rabbit IgG secondary antibody (1:5000 dilution). All the aforementioned reagents and materials were purchased from Shanghai Fuheng Biotechnology Co., Ltd.
Sample Preparation
Accurately weigh 1 g of the concentrated extract (designated as EC) with an extraction yield of 10.8% (w/w), which was fabricated via reflux extraction of the aforementioned EC kernel powder using 70% ethanol, followed by vacuum concentration. The accurately weighed sample was subjected to ultrasonic extraction in 20 mL of a methanol-deionized water mixture (v/v = 7:3) at 300 W and 40 kHz for 60 min, ensuring complete dissolution of target analytes. Subsequent to thorough homogenization and filtration, the resultant extract was centrifuged, and the collected supernatant was dried under a gentle stream of nitrogen at ambient temperature. The residual precipitate was reconstituted in 1 mL of methanol and subjected to re-centrifugation at 12,000 rpm and 4 °C for 20 min. Finally, the enriched and clarified supernatant was transferred into an HPLC vial, which was then ready for chromatographic analysis.
Instruments and conditions
An Thermo Scientific Hypersil GOLD™ aQ(100 mm×2.1 mm, 1.9 μm) was applied for chromatographic separation with a column temperature of 40 °C with a flow rate of 0.3 mL/min. The mobile phase consisted of water containing 0.1% formic acid as eluent A and acetonitrile containing 0.1% formic acid as eluent B. The flow rate was set at a linear gradient as follows: 95%ཞ92% A at 0–2 min; 92%ཞ90% A at 2–5 min; 90%ཞ85% A ,5ཞ10 min; 85%ཞ60% A, 10ཞ15 min; 60%ཞ20% A, 15ཞ20 min; 20%ཞ5% A, 20ཞ25 min; 5%ཞ95% A, 25ཞ26 min; 95% A, 26ཞ30 min; The sample injection volume was 2 µL.
MS analysis was running in negative ionization mode using electrospray ionization (ESI). The key parameters were as follows: spray voltage, 3.0 kV (−); the sheath gas flow rate, 30 arb; aux gas flow rate, 10 arb; S-Lens RF Level, 50; capillary temperature, 320 °C; heater temperature, 350 °C; S-lens RF level, 60. The mass spectrometry scans were conducted in full MS mode at a resolution of 35000, while MS2 scans at a resolution of 17500 were acquired using either data-dependent MS2 scanning or parallel reaction monitoring (PRM) techniques the scan range was m/z 120–1500 and a collision energy of 30 eV. In the General Settings module: Mass Tolerance [ppm], 5 ppm; Min, Peak Intensity, 1000000; Use Most Intense Isotope Only; In the Peak Detection module: Chromatographic S/N Threshold, 1.5; In the Isotope Pattern Detection module: Group Isotopes for, Br and cl; Compound Detection Ions, [M-H]-; In another section of the General Settings module: Mass Tolerance, 5 ppm; Preferred Ions, [M-H]-1; RT Tolerance[min], 0.2; Area Intergration, Most common ion; In the Peak Rating Contributions module: Area Contribution, 3; CV Contribution, 10; Fwhm to Base Contribution, 5; Jaggedness Contribution, 5; Modality Contribution, 5; Zig-Zag Index Contribution, 5; In the Peak Rating Filter module: Peak Rating Threshold, 1.5; Number of Files, 0. Data acquisition and processing were carried out with the Xcalibur version 4.2 and Compound Discovery version 3.0 (Thermo Fisher Scientific, California, USA, https://www.thermofisher.cn/cn/zh/home). Data processing parameters of raw mass data using Compound Discoverer software.
MS data processing for identification was carried out with the MZmine 4.6.1 (https://github.com/mzmine/mzmine/releases) open-source framework(Pluskal, Castillo, Villar-Briones, & Orešič, 2010). The raw data generated from An Dionex Ultimate 3000 UHPLC25 and the Q-Exactive Focus Orbitrap MS was in .raw format and was converted into. mzML format using MSConvert (https://proteowizard.sourceforge.io/) for further analysis26. The feature detection was carried out with an m/z tolerance of 0.02Da, and centroid mode mass detection with a noise-level at 1.0E4 was used. It was followed by LC-MS for Chromatogram builder, with MS level filter set to MS1, Polarity as -, Spectrum type as ANY, Minimum consecutive scans at 8, Minimum intensity for consecutive scans at 3.0E3, Minimum absolute height at 7.0E3, and m/z tolerance (scan-to-scan) at 10ppm. Using Local minimum feature resolver, with Original feature list at KEEP, MS/Ms scan pairing at Check (the box), Dimension at Retention time, Chromatographic threshold at 90%, Minimum search range RT/Mobility (absolute) at 0.05, Minimum relative height at 0%, Minimum absolute height at 7.0E3, Min ratio of peak top/edge at 2.00, Peak duration range (min/mobility) at 0.00–1.00, Minimum scans (data points) at 8. Isotopes were detected using isotopic peak grouper with an m/z tolerance of 3.5ppm and retention time tolerance of 0.01 min. Using join aligner, with m/z tolerance at 5ppm, Weight for m/z at 3, Retention time tolerance at 0.10 minuter. And using feature list rows filter, with retention time at 0.50–30.00 min. The results of the MZmine processing were exported by generating an MS/MS spectral summary file (.mgf) and feature quantification table (.txt) submitted to the FBMN platform using WinSCP® (https://winscp.net/eng/download.php) software for FBMN analysis. The mass tolerance of both precursor ions and fragment ions was set to 0.02 Da. A network was then created where the edges were filtered to have a cosine score above 0.7 and more than 6 matched peaks. Download from the jobs page and import Cytoscape 3.9.1 (https://cytoscape.org/download.html) for its visualization and analysis27, and predicting possible Chlorogenic acid compounds using characteristic fragment ions present in each cluster of the molecular network and molecular ion in MS data. The components were identified based on fragmentation ions, retention time, and bibliography28,29,30.
Establishment of analytical strategy
This research endeavored to systematically screen and identify EC using an analytical strategy grounded in UHPLC-Q-Exactive Orbitrap MS technology. The process commenced with the extraction and enrichment of EC through ultrasonication with pure methanol. Subsequently, sample was introduced into the UHPLC-Q-Exactive Orbitrap MS system to obtain high-resolution mass spectral data. This data was generated via comprehensive MS scanning coupled with data-dependent MS2 scans (Full Mass-ddMS2 and was subsequently processed using Compound Discover version 3.0. The analysis involved high-resolution extracted ion chromatography (HREIC) and the prediction of anticipated compounds.
For trace constituents—especially those with low abundance in the mass analyzer that co-elute with more abundant components—PRM mode (activated via an ion inclusion list) facilitated the acquisition of subsequent fragment ions, improving the identification efficiency of EC. Additionally, feature-based molecular networking excavated novel molecular weights to discover unknown or trace natural products by leveraging the characteristics of each cluster. After processing EC data, potential compounds were predicted using characteristic fragment ions (from each molecular network cluster) and molecular ions. The identification of relevant compounds in this study was achieved through integrated analysis of multi-database matching and high-resolution mass spectrometry data. Specific parameters for the three databases were set as follows: (1) HMDB: mass deviation < 5 ppm, retention time deviation (tR) ≤ 0.2 min, fragment matching requiring MS² ion information and characteristic fragment ion abundance ratio deviation < 10%; (2) PubChem: Exact Mass Match mode, mass deviation < 5 ppm, fragment ion similarity search enabled (similarity score ≥ 0.7), and isomer differentiation via molecular descriptors (topological polar surface area, logP) and tR differences; (3) OTCML: mass deviation < 5 ppm, tR ≤ 0.2 min. This methodology referenced the database application logic in natural product mass spectrometric identification established by Cai et al. (2020)31 and to ensure matching reliability and specificity. Ultimately, compound identification was confirmed by correlating full-scan MS, MS² data, tR, and literature, providing a robust framework for EC’s chemical characterization.
Optimization of UHPLC-Q-Exactive orbitrap MS condition
To achieve improved chromatographic peak shapes and separation efficiency, a range of variable factors were examined during the detection and identification processes. Additionally, the type and concentration of acid additives, such as formic acid and acetic acid at concentrations of 0.05%, 0.1%, and 0.2%, were assessed. Column temperatures were tested at 30, 35, 40, and 45 °C, and the mobile phase gradient was optimized. The study also evaluated different columns, specifically the Thermo Scientific Hypersil GOLD™ aQ 100 mm × 2.1 mm, 1.9 μm and Waters ACQUITY BEH C18 column, 100 mm × 2.1 mm, 1.7 μm, as well as mobile phase flow rates of 0.2, 0.3, and 0.4 mL/min. MS parameters, including the flow rates of the sheath and auxiliary gases, capillary and heater temperatures, and spray voltage, were also examined. Under the optimized conditions for UHPLC-Q-Exactive Orbitrap MS, the majority of compounds demonstrated effective separation, with parent/daughter ion pairs exhibiting high response levels.
In vitro Anti-Inflammatory activity verification (Western Blot)
Cell culture and group treatment
RAW264.7 cells were cultured in DMEM complete medium containing 10% fetal bovine serum and 1% penicillin-streptomycin (PS) in a 37 °C, 5% CO₂ incubator. Logarithmic phase cells were seeded into 12-well cell culture plates at a density of 2 × 10⁵ cells/well, with 1 mL of complete medium added to each well, and incubated for 24 h to allow cell adhesion. The experiment was divided into blank control group, LPS model group, positive control group (dexamethasone group), and EC ethanol extract groups at various concentrations, with 3 replicate wells in each group: the blank control group was only added with complete medium without drugs and LPS; the LPS model group was added with complete medium followed by LPS stimulation; the positive control group was added with dexamethasone solution at a final concentration of 0.5 μm and incubated for 1 h; the EC ethanol extract groups were added with working solutions of EC ethanol extract at final concentrations of 1 µg/mL, 10 µg/mL, and 100 µg/mL, respectively, and incubated for 1 h. Except for the blank control group, all other groups were added with LPS solution at a final concentration of 1 µg/mL and incubated for another 18 h to construct the inflammatory model.
Total protein extraction and concentration determination
The medium in each well was aspirated, and the cells were gently washed 2 ~ 3 times with pre-cooled PBS buffer to remove residual medium and unbound drugs. 70 µL of pre-cooled RIPA lysis buffer was added to each well, and the cells were lysed sufficiently by incubating on ice for 15 min. Adherent cells were gently scraped off with a cell scraper, and the cell lysate was transferred to a pre-cooled centrifuge tube. After centrifugation at 12,000 rpm for 15 min at 4 °C, the supernatant was collected as the total protein extract and stored at -80 °C for later use. According to the instructions of the BCA protein assay kit, standard gradient solutions were prepared and added to a 96-well microplate together with protein samples. After adding the BCA working solution, the mixture was incubated at 37 °C for 30 min, and the absorbance value was measured at 562 nm using a microplate reader. The concentration of each protein sample was calculated according to the standard curve.
Western blot detection
The sample loading volume was calculated based on the protein concentration. An equal amount of total protein extract was mixed with 5× protein sample buffer (LB) at a ratio of 4:1, and the protein was fully denatured by heating at 100 °C for 15 min. The denatured protein sample was added to the loading well of a 10% SDS-PAGE gel, and electrophoresis was performed at a voltage of 80 V (concentrating gel) and 120 V (separating gel) until the bromophenol blue indicator reached the bottom of the gel. The protein on the gel was transferred to a PVDF membrane using the wet transfer method under the conditions of 100 V and 60 min. After transfer, the PVDF membrane was blocked in TBST buffer containing 5% skimmed milk on a shaker at room temperature for 1 h to reduce non-specific binding. The blocked PVDF membrane was incubated with COX-2, iNOS, and β-actin primary antibody solutions (diluted to the corresponding ratio with 5% skimmed milk) on a shaker at 4 °C overnight. The membrane was washed 3 times with TBST buffer on a shaker at room temperature for 10 min each time to remove unbound primary antibody. According to the source of the primary antibody, the corresponding secondary antibody solution (anti-mouse IgG or anti-rabbit IgG, 1:5000 dilution) was added, and the membrane was incubated on a shaker at room temperature for 1 h. The membrane was washed again 3 times with TBST buffer on a shaker at room temperature for 10 min each time to remove unbound secondary antibody. The PVDF membrane was scanned and imaged using an Odyssey Dual-Mode Infrared Imaging System, and the gray value of the protein bands was analyzed using Image Lab software. With β-actin as the internal reference, the relative expression levels of COX-2 and iNOS proteins were calculated (target protein gray value/β-actin gray value).
Results
This study, with the aid of UHPLC-QE-Orbitrap-MS coupled with PRM, identified a total of 126 chemicals, including 64 flavonoids, 24 phenylpropanoids, 21 organic acids, and 17 compounds of other types in EC and initially confirming its in vitro anti-inflammatory activity. To clarify the confidence levels of the 126 identified compounds, we categorized them according to the Metabolomics Standards Initiative framework, with corresponding annotations indicated by asterisks in Table 1: Level 1 (, confirmed identification) includes 17 compounds, identified by direct comparison with reference standards (matching retention time, accurate mass, and MS/MS fragment ion abundance ratios), with “Standard Comparison” noted in the “References” column; Level 2 (, high-confidence annotation) comprises 45 compounds, identified without reference standards but via high-resolution MS/MS matching to authoritative databases (HMDB, PubChem, OTCML), with corresponding database names listed in the “References” column; Level 3 (, putative annotation) includes 64 compounds, tentatively identified based on accurate mass, fragmentation characteristics consistent with the parent structure of target compounds (e.g., the C6-C3-C6 skeleton for flavonoids, carboxyl groups for organic acids) and literature references, with literature sources noted in the “References” column; the asterisk annotations and their corresponding definitions are explicitly explained below the title of Table 1.The mass spectrometry data of the detected components are shown in Table 1, The Extracted Ion Chromatogram is shown in Fig. 1.

The high-resolution extracted ion chromatogram (HREIC) in 10 ppm for multiple compounds in Eleusine coracana L.(A) m/z 741.18837, 865.19853, 367.10345, 273.07684, 211.06119, 355.10345, 787.19384, 299.07724, 341.08780, 607.16684, 263.12888, 303.05102, 131.07136, 593.13006, 121.02950, 595.16684, 299.05611, 441.08271 (B) 441.08271, 325.09289, 431.09837, 282.08426, 575.11949, 593.15119, 451.12458, 163.04006, 577.13514, 301.07176, 315.07215, 477.10384, 125.02441, 218.10339, 175.06119, 287.05611 (C) 625.14102, 433.11292, 463.08710, 188.05565, 201.11323, 271.06119, 179.03498, 327.21769, 151.04006, 243.06200, 609.14610, 344.04015, 315.05102, 328.04524, 173.08131, 476.27826, 435.25171, 177.04046, 299.09249 (D) 301.03540, 452.27826, 623.16175, 129.01933, 478.29391, 571.28888, 341.10893, 167.03498, 721.36520, 195.05102, 313.23843, 329.23334, 181.07176, 137.02442, 187.09758, 153.01933, 133.01424, 173.00916, 289.07176, 191.01972.
Identification of EC
Some compounds were quickly identified by comparing the retention times and MS and MS2 information with those of the reference standards. For example, compound 77 was eluted at 12.95 min with the deprotonated molecular ion [M - H]− at m/z 609.14642 (C₂₇H₃₀O₁₆); it was identified as Rutin, with its fragmentation pathway illustrated in Figure (4 A). flavonoids Compounds 37, 68,72, 103, 105, 107, 108, 112 and 116 were eluted at 4.75, 12.12, 12.70, 14.76, 15.41, 15.89, 16.05, 16.73and 18.86 min, respectively, with the deprotonated molecular ion [M-H]− at m/z 289.07175 (C15H14O6, -0.03 ppm), 431.09787 (C21H20O10, -1.16 ppm), 463.08838 (C21H20O12, 0.39 ppm), 463.08847 (C21H20O12, 0.58 ppm), 287.05646 (C15H12O6, 1.21 ppm), 301.03537 (C15H10O7, 0.08 ppm), 271.06125 (C15H12O5, 0.19 ppm), 273.07690 (C15H14O5, 0.19ppm), 315.05115 (C16H12O7, 0.39ppm), and 593.13007 (C30H26O13, 0.01 ppm), respectively; these were identified as Catechin, Vitexin, Hyperoside(Fig. 2B), Isoquercitrin, Eriodictyol, Quercetin, Naringenin, Phloretin, Isorhamnetin, and Procyanidin, respectively, by comparing the retention times and MS and MS2 information with those of the standards.
Phenylpropanoids, Organic Acids and Other compounds 12, 13, 15, 45, 80 and 102 were eluted at 1.10, 1.18, 1.19, 5.94, 13.16 and 14.68 min, respectively, with the deprotonated molecular ion [M-H]− at m/z 191.01886 (C6H8O7, -4.53 ppm), 173.00833 (C6H6O6, -4.80ppm), 328.04514 (C10H12N5O6P, -0.31 ppm), 179.03419 (C9H8O4, -4.42 ppm), 187.09669 (C9H16O4, -4.76 ppm), and 263.12894 (C15H20O4, 0.21 ppm), respectively; they were identified as Citric acid(Fig. 2D), Cis-Aconitic acid, Cyclic AMP, Caffeic acid (Fig. 2C), Azelaic acid and Abscisic Acid respectively, by comparing the retention times and MS and MS2 information with those of the standards. The Extracted Ion Chromatograms of the three groups of reference standards are presented in Fig. 3, where the red chromatogram corresponds to Reference Standard Group 1, the green chromatogram to Reference Standard Group 2, and the blue chromatogram to Reference Standard Group 3.

Possible fragmentation pathways of Rutin(A), Hyperoside(B), caffeic acid(C), citric acid(D).

Reference Standard 1: m/z 301.03540, 271.06119, 463.08710, 273.07684, 287.05611, 315.05102, 431.09837, 668.01444; Reference Standard 2: m/z 191.01972, 173.00916, 593.13006, 263.12888; Reference Standard 3: m/z 328.04524, 289.07176, 187.09758, 163.04006, 179.03498.
Identification of flavonoid components
Molecular network analysis yielded significant insights into the structural relationships among flavonoid compounds, revealing three predominant molecular clusters, as illustrated in Fig. 4. Compound 48, which possessed the same quasimolecular ions and characteristic fragment ions as compound 37, was identified as Epicatechin32,33. Compound 76, which possessed the same quasimolecular ions and characteristic fragment ion as compound 68, was identified as Isovitexin with data from the literature34,35. Similarly, Compound 67 was inferred as an isomer of Taxifolin, and Compound 69 was inferred as an isomer of Rutin; Compounds 93 and 98 were presumed to be Isoquercitrin isomers36; Compounds 106 and 109 were presumed to be Hyperoside isomers37.

The identification result diagram of the flavonoid molecular network based via feature-based molecular networking.
Compound 52, 88, 96 possessed the quasi-molecular ions [M-H]− at m/z 787.19384, 623.16248 and 477.10410, and the MS/MS fragment ions obtained indicated the loss of three glucose units (486 Da), disaccharide (324 Da), a glucose unit (162 Da); these were tentatively identified as Quercetin-O-glucose-O-gentiobioside, Isorhamnetin-3-O-nehesperidoside38, Isorhamnetin 3-O-hexoside39. Based on the comparison with the literature; Compound 71 was Quercetin-3-O-rutinoside-7-O-pentoside31,40; and it was speculated that compound 32 was isovanillin41; compound 82 was Isorhamnetin-3-O-rutinoside42,43.
Using the OTCML database, Compound 41 was identified as Procyanidin C1. With the HMDB and PubChem databases, Compound 74 and Compound 78 were preliminarily identified as N-Acetyl-DL-glutamic acid.
Identification of phenylpropanoids components
Eluting at tR = 2.15 min, compound 24 exhibited a quasimolecular ion at m/z 315.0721 [M-H]⁻, corresponding to the molecular formula C₁₃H₁₆O₉. Under high-energy collision dissociation (HCD), it lost one glucose unit (162 Da) to form a protocatechuic acid aglycone ion at m/z 153.0182, which further undergoes decarboxylation to yield m/z 109.0283 [M-H-CO₂-Glu]⁻. This fragmentation sequence was consistent with the reported characteristics of protocatechuic acid-O-hexoside44. Compound 21 eluted at 1.65 min (tR = 1.65 min) with a quasimolecular ion at m/z 125.02331, corresponding to the molecular formula C₆H₆O₃. Its diagnostic fragment ions at m/z 81.0332 and 97.0282 were attributed to the cleavage of side-chain substituents on the benzene ring scaffold. (e.g., loss of hydroxyl groups or water molecules). Based on these mass spectral features and structural matching, it is tentatively identified as pyrogallol. Compound 38 eluted at 5.04 min (tR = 5.04 min) with a quasimolecular ion at m/z 121.02833 (C₇H₆O₂). Its secondary ion at m/z 108.0204 was formed by loss 13 Da, consistent with the fragmentation pattern of 4-hydroxybenzaldehyde, lead to its tentative identification.
Compound 27 was tentatively identified as protocatechualdehyde via spectral matching with the OTCML database.
Identification of organic acids components
Molecular networking analysis showed into the structural relationships among organic acids, unraveling two major molecular clusters as illustrated in Fig. 5. Eluting at 0.88 min (tR = 0.88 min), Compound 3 showed a quasimolecular ion at m/z 195.05019, corresponding to C₆H₁₂O₇. Its characteristic fragments reflected the unique structure of gluconic acid: m/z 75.0074 formed via C1-C2 bond cleavage after ring-opening (C₃H₃O₂⁻), m/z 129.0181 resulted from C4-C5 bond cleavage (C₄H₅O₅⁻), and m/z 99.0074 is a decarboxylation product (C₃H₅O₃⁻). The dehydration product at m/z 177.0396 (C₆H₁₀O₆⁻) highlighted the synergistic interaction between hydroxyl and carboxyl groups. These features strongly supported its identification as gluconic acid45. Compounds 6 and 9, which possessed the same quasimolecular ions and secondary fragment ions as malic acid, were inferred to be malic acid isomers based on literature data44. Compounds 5 and 17 shared the same quasimolecular ion (m/z 191.01972) and fragmentation pattern as compound 12, with characteristic fragments at m/z 111.0074, 87.0074, and 85.0281. Thus, compounds 5 and 17 were tentatively identified as citric/isocitric acid isomers46. Compound 11, an isomer of compound 13 (cis-aconitic acid), exhibited the same quasimolecular ion (m/z 173.00819) under UHPLC-high-resolution MS analysis.

The identification result diagram of the organic acids molecular network based via feature-based molecular networking.
Identification of carbohydrates compounds
Molecular network analysis yielded significant insights into the structural relationships among carbohydrates and other compounds, revealing three predominant molecular clusters, as illustrated in Fig. 6. Eluting at 0.86 min (tR = 0.86 min), Compound 1 exhibited a quasimolecular ion at m/z 181.07089, corresponding to the molecular formula C₆H₁₄O₆. Its characteristic fragment ions reflected the typical fragmentation pattern of polyols: m/z 101.0231 arose from C3-C4 bond cleavage, forming a C₃H₅O₄⁻ fragment; m/z 89.0230 resulted from C2-C3 bond cleavage (C₄H₉O₂⁻); and m/z 71.0124/59.0124 corresponded to C₃H₃O₂⁻ and C₂H₃O₂⁻ fragments, respectively. These fragmentation behaviors aligned with the hexahydric alcohol structure of mannitol, where adjacent hydroxyl groups facilitate dehydration and carbon chain cleavage, supporting its tentative identification as mannitol47. Compound 2 eluted at 0.87 min (tR = 0.87 min) with a quasimolecular ion at m/z 341.10867, matching the molecular formula C₁₂H₂₂O₁₁. Its MS/MS spectrum displayed complex carbohydrate fragmentation: the presence of the parent ion peak (m/z 341.1097) indicated high molecular stability; m/z 179.0551 represented a glucose anion (C₆H₁₁O₆⁻) formed by sucrose cleavage, while m/z 161.0443 was its dehydration product (C₆H₉O₅⁻). Subsequent fragments (e.g., m/z 143.0341, 131.0336) arose from further dehydration and ring-opening of glucose or fructose units. Collectively, these features confirmed its tentative identification as sucrose48.

The identification result diagram of the carbohydrates and other compounds molecular network based via feature-based molecular networking.
Identification of other compounds
Compound 19 was compared with the literature and found to have the same quasi-molecular ion and characteristic fragment ions, indicating that the compound was Guanosine49. Compound 18 was eluted at 1.20 min, producing a quasi-molecular ion [M − H]− at m/z 344.0401(C10H12O7N5P). After the loss of two oxygen molecules and one molecule of glucose, it lost 194 Da and obtained a characteristic ion at m/z 150.0410, suggesting that the compound was caged cGMP. In this experiment, other types of compounds were preliminarily identified, and compounds 14, 51, 57 were preliminarily identified as Uridine, Propyl gallate, Vicenin-2, respectively, through the OTCML database. Leveraging data from the HMDB and PubChem databases, the following compounds were tentatively identified: Compound 110 as 9,12,13-trihydroxy-10,15-octadecadienoic acid; Compound 113 as (10E,15Z)-9,12,13-trihydroxyoctadeca-10,15-dienoic acid; Compound 114 as 9,12,13-trihydroxyoctadec-10-enoic acid; Compound 117 as 12,13-dihydroxy-9-octadecenoic acid; Compound 119 as 12,13-DiHOME.
Refer to MSI standards, * denotes Level 1, ** denotes Level 2, *** denotes Level 350
In vitro Anti-Inflammatory activity verification
In the LPS-induced RAW264.7 macrophage model, the expression levels of inflammation-related proteins COX-2 and iNOS were significantly increased compared with the blank control group (P < 0.05), indicating the successful establishment of the inflammatory model, as illustrated in Fig. 7. After intervention with the ethanol extract of EC, the relative expression levels of COX-2 and iNOS proteins showed a significant concentration-dependent decrease: the inhibition rates of the 1 µg/mL concentration group on the two proteins were 21.3% and 18.7%, respectively; the inhibition rates of the 10 µg/mL concentration group increased to 45.6% and 42.1%; while the 100 µg/mL EC extract exhibited inhibition rates of 78.9% and 75.3% against COX-2 and iNOS, respectively, which were statistically indistinguishable from the positive control dexamethasone (0.5 µM, 82.4% and 79.6%; P > 0.05), confirming its potent anti-inflammatory potential. Moreover, all concentration groups showed significant differences compared with the LPS model group (P < 0.05). Combined with the identification results in Sect. 3.1, the 64 flavonoid components (such as quercetin, procyanidins, hyperoside, etc.) and 24 phenylpropanoid components (such as caffeic acid, ferulic acid, and their glycoside derivatives, etc.) contained in EC are all reported natural anti-inflammatory active substances. It is speculated that these components may act synergistically to jointly constitute the main material basis for the in vitro anti-inflammatory activity of the ethanol extract of EC.

Western Blot Analysis of Anti-Inflammatory Activity for Eleusine coracana.
Discussion
EC is not only a nutritionally rich food source, providing essential amino acids, vitamins, and minerals, promoting dietary diversity and nutritional health among local populations51, 52, but also its stress-resistant characteristics make it a valuable crop for improving soil quality, adapting to climate change, and enhancing agricultural sustainability53. Additionally, the cultivation of EC helps to enhance the self-sufficiency of agricultural production, reduce the burden on the environment, support biodiversity, and bring economic benefits and food security to small-scale farmers54. In this study, an integrated approach combining UHPLC-Q-Exactive Orbitrap MS with FBMN and PRM was employed to systematically identify chemical constituents in the whole plant of EC. A total of 126 compounds were characterized, including 64 flavonoids, 24 phenylpropanoids, 21 organic acids, and 17 compounds of other classes. Among these, we successfully identified 44 compounds not previously reported in EC-related literature—representative ones include procyanidin A2 (flavonoid), salicylic acid-hexoside (phenolic acid), trehalose (carbohydrate), 1-oleoyl lysophosphatidic acid, 9,12,13-trihydroxy-10,15-octadecadienoic acid, and cyclic AMP (nucleotide). This study represents the first comprehensive chemical characterization of EC, establishing a vital link between its chemical composition and bioactivity, and providing a robust framework for future quality control, bioactive compound isolation, and mechanism-of-action investigations. Notwithstanding the growing recognition of EC’s multifaceted value, existing phytochemical investigations on Eleusine coracana L. and its congeneric species suffer from notable gaps that underscore the novelty of the present work. A primary limitation lies in the bias toward specific plant tissues and compound classes, which constrains the comprehensiveness of chemical profiling. For instance, Soujanya et al. applied UPLC-ESI-HRMS/MS to analyze drum-dried EC, yet their study was confined exclusively to seed coat and endosperm fractions—overlooking aerial parts such as leaves and stems, which are likely reservoirs of unique bioactive metabolites9. Moreover, their identification efforts were restricted to phenolic compounds, omitting structurally and functionally pivotal classes like phenylpropanoids (e.g., caffeic acid glycosides) and organic acids (e.g., citric acid, gluconic acid). Both classes play critical roles in modulating EC’s iron bioavailability and antioxidant activity, making their exclusion a significant oversight. In contrast, our analysis of the entire EC plant captures metabolites across all tissue types, leading to the characterization of 126 compounds spanning 64 flavonoids, 24 phenylpropanoids, 21 organic acids, and 17 other classes (e.g., nucleotides, lipids). This represents the first pan-tissue, multi-class chemical profile of EC, addressing the tissue and compound bias of earlier studies.
Another key shortcoming of prior research is the constraint of analytical methodologies, which impairs the resolution of isomeric metabolites and the detection of trace components. Sukor et al. conducted phytochemical annotation of Eleusine coracana L indica—a close relative of EC—using HPLC-MS/MS, but their approach relied solely on database matching (e.g., GNPS, HMDB) without incorporating advanced mass spectrometry strategies10. This limitation prevented the differentiation of isomeric metabolites, particularly flavonoid glycosides (e.g., isoquercitrin versus hyperoside) that share identical molecular weights but exhibit distinct retention times and biological activities. Additionally, the method resulted in low detection rates for trace components such as cyclic AMP and guanosine hydrate. Our integrated analytical strategy addresses this gap by combining UHPLC-Q-Exactive Orbitrap MS with Feature-Based Molecular Networking (FBMN) and Parallel Reaction Monitoring (PRM). FBMN clusters compounds based on retention time, precursor ion accuracy, and fragmentation patterns, enabling the clear resolution of isomers (e.g., cis- versus trans-aconitic acid). Meanwhile, PRM enhances the signal-to-noise ratio of trace metabolites—such as 9,12,13-trihydroxyoctadec-10-enoic acid—by 2.8–3.2 fold, ensuring that low-abundance yet functionally relevant compounds are not overlooked.
A third limitation of existing studies is their limited coverage of compounds, which fails to capture the full chemical diversity of EC. Peñaloza et al. utilized HPLC-DAD, UPLC-DAD-MS/MS, and NMR for metabolite profiling of Eleusine coracana L. indica, but their work identified fewer than 60 compounds—with only 8 flavonoids and 5 organic acids. This narrow scope masks EC’s potential as a source of novel bioactive substances11. For example, our study identified procyanidin trimers (C1), quercetin-O-glucose-O-gentiobioside, and 12,13-DiHOME (a lipid mediator with anti-inflammatory activity)—all of which have not been reported in previous Eleusine-related research. The 126 compounds characterized in this work not only more than double the number of known metabolites in EC but also expand the range of functional compound classes. This broader coverage lays a solid foundation for linking specific compounds to EC’s well-documented health benefits (e.g., hypoglycemic effects, enhanced iron absorption) and ecological traits (e.g., drought tolerance), advancing our understanding of EC’s biological and agricultural significance.
In this study, 126 chemical components were identified in the seedlings, and the anti-inflammatory activity of their alcohol extracts was confirmed by WB experiments. which can effectively inhibit the overexpression of COX-2 and iNOS proteins in LPS-induced RAW264.7 cells with a clear concentration-dependent manner. In terms of material basis, flavonoid components such as quercetin and procyanidins have been shown to downregulate the transcription and expression of inflammation mediator-related proteins by blocking the NF-κB signaling pathway; phenylpropanoids such as caffeic acid and ferulic acid can directly inhibit the activity of key enzymes in the inflammatory pathway and reduce the release of inflammatory factors. The components identified in this study are consistent with the anti-inflammatory effects observed in WB experiments, suggesting that the ethanol extract of EC may exert anti-inflammatory effects through the synergistic action of these active components on the COX-2/iNOS pathway. This result not only provides direct experimental evidence for the anti-inflammatory activity of EC but also establishes a “chemical composition-biological activity” association, laying an important foundation for the subsequent targeted isolation of anti-inflammatory active monomers and clarification of specific mechanisms of action.
In conclusion, this study lays a solid chemical foundation for the in-depth investigation of E. coracana. In the future, through the integration of interdisciplinary technologies and collaboration between industry, academia, and research institutions, it is expected to promote the transformation of this drought-tolerant crop from a traditional staple food to a high-value functional resource, thereby offering a new direction for sustainable agricultural development and natural product exploitation.
Conclusions
In this study, a systematic analytical strategy based on UHPLC-QE Orbitrap MS combined with Feature-Based Molecular Networking and Parallel Reaction Monitoring was established for compound identification in EC. By integrating optimized chromatographic conditions, full-spectrum MS/MS data, retention times, standard comparisons, and literature corroboration, 126 components (flavonoids, phenylpropanoids, organic acids, etc.) were characterized with high resolution, including 44 unreported in previous EC-related studies. Additionally, EC’s ethanol extract exhibited concentration-dependent inhibition on inflammation-related protein expression, supporting its functional product development. These findings will facilitate EC’s further drug development and quality control.
Authorship Contribution: Yufeng Zou: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Resources, Data curation, Writing – Original draft; Shunli Xiao: Conceptualization, Formal analysis, Investigation, Resources, Data curation, Writing – Original draft, Funding acquisition; Jiayi Wang: Conceptualization, Methodology, Software, Validation, Writing – Original draft, Visualization; Qiru Chen: Formal analysis, Investigation; Liangyin Shu: Investigation, Resources; Yifan Chen: Conceptualization, Software; Dingli Hu: Conceptualization, Investigation; Yuhua Tan: Software, Investigation; Sixuan Yang: Software, Investigation; Lianghong Liu: Data curation, Writing – review & editing, Supervision, Funding acquisition; Qingjiao He: Data curation, Writing – review & editing, Supervision, Project administration.
Data availability
The data used to support the findings of this study are available from the corresponding authors upon request.
References
Kayastha, S. et al. Finger millet (Eleusine coracana) enhancement through genomic resources and breeding methods: current implications and potential future interventions. Planta 259 (6), 139. https://doi.org/10.1007/s00425-024-04415-0 (2024).
Chauhan, R. S. et al. Genetic and genome resources in buckwheat–present status and future perspectives. Eur. J. Plant. Sci. Biotechnol. 2 (7), 33–44 (2010).
Kumar, A. et al. Nutraceutical value of finger millet (Eleusine Coracana (L.) Gaertn.], and their improvement using omics approaches. Front. Plant. Sci. 7 (2), 934. https://doi.org/10.3389/fpls.2016.00934 (2016).
Devi, P. B. et al. Health benefits of finger millet (Eleusine Coracana L.) polyphenols and dietary fiber: a reviewand dietary fiber: a review. J. Food Sci. Technol. 51 (23), 1021–1040. https://doi.org/10.1007/s13197-011-0584-9 (2014).
Anitha, S. et al. Millets can have a major impact on improving iron status, hemoglobin level, and in reducing iron deficiency anemia–a systematic review and meta-analysis. FRONT. NUTR. 8 (3), 725529. https://doi.org/10.3389/fnut.2021.725529 (2021).
Makuvara, Z. et al. Millets (pearl and finger) as nutritional interventions for COVID 19 with focus on zimbabwe: mini review. Cogent Food Agric. 8 (1), 2111060. https://doi.org/10.1080/23311932.2022.2111060 (2022).
Xie et al. Processing technology of toughness biscuits with finger millet and polygonatum Cyrtonema Hua. Bao Xian Yu Jia Gong. 21 (08), 76–81. https://doi.org/10.3969/j.issn.1009-6221.2021.08.012 (2021).
Wang, Y. H. et al. The chemical characterization of Eleutherococcus senticosus and Ci-wu-jia tea using UHPLC-UV-QTOF/MS. Int. J. Mol. Sci. 20 (3), 475. https://doi.org/10.3390/ijms20030475 (2019).
Soujanya, K. V. et al. UPLC-ESI-HRMS/MS phenolic Characterization, antioxidant Activity, and proximate composition of Drum-Dried finger millet (Eleusine coracana) seed Coat- and Endosperm-Rich fractions. ACS Food Sci. Technol. 5 (1), 85–94. https://doi.org/10.1021/acsfoodscitech.4c00581 (2025).
Sukor, N. S. M. et al. Annotation and identification of phytochemicals from Eleusine indica using High-Performance liquid chromatography tandem mass spectrometry: Databases-Driven approach. Molecules 28 (7). https://doi.org/10.3390/molecules28073111 (2023).
Peñaloza, E. M. C. et al. Metabolite fingerprinting and profiling of the medicinal grass Eleusine indica based on HPLC-DAD, UPLC-DAD-MS/MS and NMR analyses. JBCS 29 (12), 2522–2534. https://doi.org/10.21577/0103-5053.20180131 (2018).
Yang, X. et al. Multicomponent characterization of the flower bud of Panax Notoginseng and its metabolites in rat plasma by Ultra-High performance liquid Chromatography/Ion mobility quadrupole Time-of-Flight mass spectrometry. Molecules 27 (24), 9094. https://doi.org/10.3390/molecules27249049 (2022).
Gao, X. et al. Chemical profiling of Dingkun Dan by UHPLC-Q-Exactive orbitrap High-Resolution mass spectrometry. JPBA 177 (5), 847. https://doi.org/10.1016/j.jpba.2019.06.029 (2019).
TAN, P. et al. Application status and prospect of ultra-performance liquid chromatography in analysis field of Chinese materia medica. Chin. Traditional Herb. Drugs. 49 (24), 5938–5945. https://doi.org/10.7501/j.issn (2018).
Kai-lin, L. I. Rapid identification of chemical constituents in Sasussured involucrata oral liquid based on UHPLC-Q-Exactive orbitrap MS technology. Chin. Med. 47 (4), 938–950. https://doi.org/10.13313/j.issn.1673-4890.20231101002 (2022).
Xiong, P. et al. Identification of the tannins in traditional Chinese medicine paeoniae radix Alba by UHPLC-Q-Exactive orbitrap MS. Arab. J. Chem. 14 (11), 103398. https://doi.org/10.1016/j.arabjc.2021.103398 (2021).
Qingrui,Yang et al. Rapid identification of constituents in polygonatum Cyrtonema Hua using UHPLC-Q-Exactive orbitrap mass spectrometry. Molecules 30 (3), 723. https://doi.org/10.3390/molecules30030723 (2025).
Huang, H. et al. Recent advances in Ultra-High performance liquid chromatography for the analysis of traditional Chinese medicine. ANAL LETT. 8 (88), 7–27. https://doi.org/10.1080/00032719.2014.888727 (2014).
Phelan, V. et al. Feature-Based molecular networking for metabolite annotation. Methods Mol. Biol. 2104 (13), 227–243. https://doi.org/10.1007/978-1-0716-0239-3 (2020).
Opialla, T. et al. Towards a more reliable identification of isomeric metabolites using pattern guided retention validation. Metabolites 10 (11), 457. https://doi.org/10.3390/metabo10110457 (2020).
Li, Y. et al. Identifying distinct markers in two sorghum varieties for Baijiu fermentation using untargeted metabolomics and molecular network approaches. Food Chem. 23 (30), 101646. https://doi.org/10.1016/j.fochx.2024.101646 (2024).
Pan Xiaodong et al. Determination of antibiotic residues in chicken meat using ultra-high performance liquid chromatography coupled with hybrid quadrupole-orbitrap high-resolution mass spectrometry. Prev. Med. 5 (05), 456–459. https://doi.org/10.19485/j.cnki.issn2096-5087.2023.05.021 (2023).
Qu Binqubin, L. et al. Quantitative determination of fipronil and its metabolites residues in Raw milk by liquid chromatography-quadrupole-orbitrap mass spectrometry with parallel reaction monitoring mode. J. Pharm. Anal. 39 (02), 333–340. https://doi.org/10.16155/j.0254-1793.2019.02.21 (2019).
Li, Y. et al. Hybrid quadrupole-orbitrap mass spectrometry analysis with accurate-mass database and parallel reaction monitoring for high-throughput screening and quantification of multi-xenobiotics in honey. J. Chromatogr. A. 1429 (15), 119–126. https://doi.org/10.1016/j.chroma.2015.11.075 (2016).
Pluskal, T. et al. Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 11 (1), 395. https://doi.org/10.1186/1471-2105-11-395 (2010).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30 (10), 918–920. https://doi.org/10.1038/nbt.2377 (2012).
Otasek, D. et al. Cytoscape automation: empowering workflow-based network analysis. Genome Biol. 20 (1), 185. https://doi.org/10.1186/s13059-019-1758-4 (2019).
Yang, J. et al. Identification and quantification of chlorogenic acids from the root bark of acanthopanax gracilistylus by UHPLC-Q-Exactive orbitrap mass spectrometry. ACS Omega. 7 (29), 25675–25685. https://doi.org/10.1021/acsomega.2c02899 (2022).
Peng, J. et al. Diagnostic Fragment-Ion-Based for rapid identification of chlorogenic acids derivatives in Inula Cappa using UHPLC-Q-Exactive orbitrap mass spectrometry. J. Anal. Methods Chem. 12 (20), 6393246. https://doi.org/10.1155/2021/6393246 (2021).
Ouyang, H. et al. A robust platform based on ultra-high performance liquid chromatography quadrupole time of flight tandem mass spectrometry with a two-step data mining strategy in the investigation, classification, and identification of chlorogenic acids in Ainsliaea Fragrans Champ. J. Chromatogr. A. 1502, 38–50. https://doi.org/10.1016/j.chroma.2017.04.051 (2017).
Cai, W. et al. A systematic strategy for rapid identification of chlorogenic acids derivatives in Duhaldea nervosa using UHPLC-Q-Exactive orbitrap mass spectrometry. J. Anal. Methods Chem. 13 (2), 3751–3761. https://doi.org/10.1155/2021/6393246 (2020).
Zhu, L. et al. Systematic screening of chemical constituents in the traditional Chinese medicine arnebiae radix by UHPLC-Q-Exactive orbitrap mass spectrometry. Molecules 27 (9), 2631. https://doi.org/10.3390/molecules27092631 (2022).
Watanabe, M. et al. Chiral separation of catechins in buckwheat groats and the effects of phenolic compounds in mice subjected to restraint stress. J. Agric. Food Chem. 57 (14), 6438–6442. https://doi.org/10.1021/jf901281j (2009).
Abad-García, B. et al. Practical guidelines for characterization of O‐diglycosyl flavonoid isomers by triple quadrupole MS and their applications for identification of some fruit juices flavonoids. J. Mass. Spectrom. 44 (7), 1017–1025. https://doi.org/10.1002/jms.1573 (2009).
Qin, S. et al. Comprehensive characterization of multiple components of Ziziphus Jujuba mill using UHPLC-Q‐Exactive orbitrap mass spectrometers. Food Sci. Nutr. 10 (12), 4270–4295. https://doi.org/10.1002/fsn3.3020 (2022).
Fernandez, P. et al. HPLC determination of catechins and caffeine in tea. Differentiation of green, black and instant teas. Analyst 125 (3), 421–425. https://doi.org/10.1039/A909219F (2000).
Shi et al. Characterization and identification of isomeric flavonoid O-diglycosides from genus citrus in negative electrospray ionization by ion trap mass spectrometry and time-of-flight mass spectrometry. Anal. Chim. Acta. 598 (1). https://doi.org/10.1016/j.aca.2007.07.027 (2007).
Qin, S. et al. Identification of metabolites of Aurantio-Obtusin in rats using ultra‐high‐performance liquid chromatography‐Q‐Exactive orbitrap mass spectrometry with parallel reaction monitoring. J. Anal. Methods Chem. 21 (1), 6630604. https://doi.org/10.1155/2021/6630604 (2021).
Zhou, H. et al. Screening of terpene lactones and flavonoid glycosides in Gingko Biloba capsule by UPLC-Orbitrap high resolution MS, with emphasis on isomer differentiation. Food Nutr. Res. 2 (7), 369–376. https://doi.org/10.12691/jfnr-2-7-7 (2014).
Serni, E. et al. Characterization of phenolic profile in dried grape skin of vitis vinifera L. cv. Pinot blanc with UHPLC-MS/MS and its development during ripening. J. Food Compost Anal. 114 (37), 104731. https://doi.org/10.1016/j.jfca.2022.104731 (2022).
Abdelhady et al. HPLC/MS/MS study of phenolic compounds of Leucaena Leucocephala legumes monitored with their in vitro antihyperglycemic activity. EJMP 17 (4), 1–9. https://doi.org/10.9734/EJMP/2016/31403 (2016).
Alessandra, F. et al. Profiling of hydroxycinnamoyl tartrates and acylated anthocyanins in the skin of 34 vitis vinifera genotypes. J. Food Compost Anal. 60 (19), 4931–4945. https://doi.org/10.1021/jf2045608 (2012).
Gao, M. et al. Correlation between quality and geographical origins of cortex Periplocae, based on the qualitative and quantitative determination of chemical markers combined with chemical pattern recognition. Molecules 24 (19), 3621. https://doi.org/10.3390/molecules24193621 (2019).
Li, H. R. et al. UHPLC-Q-Exactive orbitrap MS/MS-based rapid identification of chemical components in substance benchmark of Kaixin San. Zhongguo Zhong Yao Za Zhi. 47 (4), 938–950. https://doi.org/10.19540/j.cnki.cjcmm.20210823.304 (2022).
Qin, S. et al. Rapid characterization and action mechanism of the antidiabetic effect of Diospyros Lotus L using UHPLC-Q‐Exactive orbitrap MS and network Pharmacology. J. Anal. Methods Chem. 14 (1), 8000126. https://doi.org/10.1155/2022/8000126 (2022).
Qin, S. et al. Rapid characterization and action mechanism of the antidiabetic effect of Diospyros Lotus L using UHPLC-Q‐Exactive orbitrap MS and network Pharmacology. J. Anal. Methods Chem. 13 (1), 8000126. https://doi.org/10.1155/2022/8000126 (2022).
Jeong, J. Y. et al. Rapid quantitative analysis of metabolites in Kimchi using LC-Q-Orbitrap MS. ACS Omega. 8 (4), 3896–3904. https://doi.org/10.1021/acsomega.2c06303 (2023).
Bai, J. et al. Screening of quality markers during the processing of reynoutria multiflora based on the UHPLC-Q-Exactive plus orbitrap MS/MS metabolomic method. Front. Pharmacol. 12 (7), 695560. https://doi.org/10.3389/fphar.2021.695560 (2021).
Anil Kumar, A. et al. Nutraceutical value of finger millet (Eleusine Coracana (L.) Gaertn.], and their improvement using omics approaches. Front. Plant. Sci. 29 (7), 91–95. https://doi.org/10.3389/fpls.2016.00934 (2016).
Sumner, L. W. et al. Proposed minimum reporting standards for chemical analysis. Metabolomics. 3(3), 3. h t t p s: / / d o i. o r g / 1 0. 1 0 0 7 / s 1 1 3 0 6 – 0 0 7 – 0 0 8 2–2 (2007).
SK, M. et al. Health benefits of finger millet (Eleusine Coracana L.) polyphenols and dietary fiber: a review. J. Food Sci. Technol. 51 (6), 1021–1040. https://doi.org/10.1007/s13197-011-0584-9 (2014).
Srivastava, K. & Sharma Nutraceutical importance of finger millet (Eleusine coracana) for improved human health. EJPSB 6 (2), 91–95 (2012).
Gupta, S. M. et al. Finger millet: a certain crop for an uncertain future and a solution to food insecurity and hidden hunger under stressful environments. Front. Plant. Sci. 8 (5), 643. https://doi.org/10.3389/fpls.2017.00643 (2017).
Wright, H. & Devos, K. M. Finger millet: a hero in the making to combat food insecurity. Theor. Appl. Genet. 137 (6), 139. https://doi.org/10.1007/s00122-024-04637-6 (2024).
Funding
Declaration: This work was supported by the Hunan University of Medicine High-Level Talent Introduction Startup Funds (no. 202411) and the Research learning and innovative experiment program for college students in School of Pharmacy, Hunan University of Medicine (no. YXY202412).
Author information
Authors and Affiliations
Contributions
Yufeng Zou: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Resources, Data curation, Writing – original draft; Shunli Xiao: Conceptualization, Formal analysis, Investigation, Resources, Data curation, Writing – original draft, Funding acquisition; Jiayi Wang: Conceptualization, Methodology, Software, Validation, Writing – original draft, Visualization; Qiru Chen: Formal analysis, Investigation; Liangyin Shu: Investigation, Resources; Yifan Chen: Conceptualization, Software; Dingli Hu: Conceptualization, Investigation; Yuhua Tan: Software, Investigation; Sixuan Yang: Software, Investigation; Lianghong Liu: Data curation, Witing – review & editing, Supervision, Funding acquisition; Qingjiao He: Data curation, Witing – review & editing, Supervision, Project administration. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
statement: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical Statement: Finger millet was collected from Huaihua City, Hunan Province, China (geographic coordinates: 25°52′22″–29°01′25″ N, 108°47′13″–111°06′30″ E) in May 2022. The collection was performed in accordance with local ecological and cultural norms under the supervision of Prof. Wei Cai, a pharmacognosist from the School of Pharmaceutical Sciences, Hunan University of Medicine, Hunan, China. A plant specimen of Finger millet is deposited in the Pharmacognosy Specimen Room of the School of Pharmaceutical Sciences, Hunan University of Medicine (specimen No.: HMU-EC-2022). The collection of plant materials complied with relevant international, national, and local legislations and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zou, Y., Xiao, S., Wang, J. et al. Rapid chemical characterization of finger millet using UHPLC-Q-Exactive orbitrap MS coupled with based via Feature-Based molecular networking and PRM. Sci Rep 16, 6001 (2026). https://doi.org/10.1038/s41598-026-35395-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-35395-8
