
Abstract
Primary immunodeficiency diseases (PIDs) are monogenic inborn immune disorders. Of currently listed 555 genetic inborn errors of immunity (IEI) in 10 IUIS categories, PIDs range from life-threatening severe combined immunodeficiency (SCID) to milder antibody deficiencies. PIDs increase mortality risk and demand genetic/immunological testing. In Iran, high consanguinity rates may warrant influence of genetic factors. Diagnostic challenges due to heterogeneity necessitate application of advanced tools like whole exome sequencing (WES). This study was carried out on Eastern Iran’s PIDs and similar approaches should be employed worldwide in populations that are highly consanguineous. This study focused on 99 patients from Eastern Iran clinically diagnosed with PIDs who were referred for genetic evaluations between 2016 and 2025. Genetic analyses involved DNA extraction from blood samples, WES using Illumina platforms, and in-house bioinformatic pipelines for variant identification and classification. Sanger sequencing and co-segregation studies further validated findings. Medical records, family histories, and pedigrees were thoroughly analyzed. Also we have delineated syndromic and non-syndromic PID disorders. The significant findings were as follows: Finding of 47 novel disease-causing variants; High consanguinity rates (76%) correlated with an 82.8% diagnostic yield and a mortality rate of 8% amongst patients. SCID-associated mutations, as the most common discovered PIDs, found in 16 cases; other common disorders were AR LRBA LOF, AR ATM LOF, and AR EPG5 (possibly hypomorphic LOF). Additionally, certain mutations may link to pregnancy loss which may need further functional studies. Around 70% of unresolved cases were syndromic. The most frequently suspected pathogenic variants (n = 5), all novel and in homozygosity with matching phenotype, were noted in EPG5 and linked to Vici syndrome. In highly consanguineous populations, WES may reach a definite or highly probable genetic diagnosis in over 80% of cases, and that some IEIs may be associated with pregnancy loss.
Similar content being viewed by others

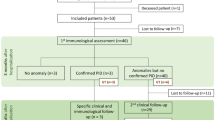

Introduction
Primary immunodeficiency diseases (PIDs) are monogenic inborn errors of immunity (IEIs) and categorized into 10 groups. There are currently 555 IEIs caused by inherited defects in one or more components of the immune system1,2. These disorders present with diverse clinical features. PIDs may exhibit severe forms such as severe combined immunodeficiency (SCID) cause life-threatening infections, while milder conditions like milder antibody deficiencies may appear later in life. PIDs also increase the risk of malignancies, particularly lymphomas, and their clinical variability often requires genetic and immunological testing for accurate diagnosis and management3,4.
Around the world, a growing number of people are found to be affected by PIDs, posing significant challenges to their daily lives. In Iran, an overall consanguinity rate of 38.6%, studied in 12 ethnic minorities/religious populations highlights the significant genetic factors contributing to PIDs5. Lower respiratory tract infections in PIDs are the most common remarkable diagnosis and one tenth of these patients undergo allogeneic hematopoietic stem cell transplantation6. The differential diagnosis shares overlapping clinical and immunological features; genetic testing are critical for distinguishing these conditions7.
Diagnostic difficulties arise from multiple factors, including genetic and phenotypic heterogeneity, variable penetrance and age of onset, as well as the presence of intronic, de novo, and mosaic mutations. Additionally, variable modes of inheritance and functional variability even within the same gene—complicate interpretation. For instance, multimorphic changes observed in genes such as CEBPE and IRF4 are recognized in the latest IUIS classification and contribute to this complexity2. Further mechanisms, including epistasis, sporadic inheritance, and monoallelic expression, add layers of intricacy to genetic diagnoses. Acknowledging these factors highlights the multifaceted nature of interpreting genetic data and underscores the importance of comprehensive analysis in clinical settings. Figure 1 illustrates the frequencies of different genetic inheritance patterns. However, the inheritance pattern of some IEIs is not clearly understood8,9.
Since genetic structure of populations may vary, causative alleles and manifestation of genetic diseases may be different across populations. Unveiling the specific genetic patterns of PIDs in Eastern Iran benefits the local community and adds valuable insights to the understanding of immunodeficiency disorders. The current study aimed to implement a more comprehensive retrospective analysis on patients presented with various forms of PIDs who were referred to a genetic center for molecular diagnosis using whole-exome sequencing (WES). Reanalyzing medical records of patients, the study sought to present both prevalent and novel causative variants.
Materials and methods
Study design and patient cohort
This retrospective study was conducted on 99 unrelated patients diagnosed with PIDs from Eastern Iran (with a population of ~ 9 million populations). Patients were referred to the Next Generation Genetic Polyclinic (Mashhad, Iran) between 2016 and 2025 for genetic evaluation.
Demographic data were collected. All methods were performed in accordance with the relevant guidelines and regulations. A comprehensive review of patients’ medical records, family histories, and pedigrees was conducted. As an exclusion criterion, this study focuses on PIDs rather than the broader spectrum of IEIs. Although some autoinflammatory and autoimmune IEIs—such as inflammasome-mediated diseases and type I interferonopathies including APECED/APS-1—can achieve complete remission with targeted biologic therapies, these responses are variable and highly disease-specific. In contrast, the PID group included here exhibits more uniform immunological defects and treatment trajectories, allowing for more consistent evaluation of clinical outcomes.
Clinical assessment and phenotypic classification
Upon clinical admission, patients evaluated for infection history, immunoglobulin levels, and other immunological parameters. Flowcytometry applied to evaluate the specific cell populations and subpopulations. Demographic details were documented. Based on clinical features and laboratory findings, patients were classified into phenotypic categories following the IUIS classification of PIDs. This study also carefully distinguishes between syndromic and non-syndromic PID disorders to facilitate genotype–phenotype correlation analysis.
DNA extraction and exome sequencing analysis
WES was performed on DNA extracted (SimBioLab, Iran) from peripheral blood samples or fetal tissue where applicable. Genomic DNA was enzymatically fragmented and underwent whole exome capturing by SureSelect Human All Exon v6, v7 and v8 kits. Generated libraries were created with Illumina compatible adaptors and sequenced on an Illumina platform (HiSeq 4000) to yield an average coverage depth of ~ 100X (Macrogen, South Korea). Evaluation was focused on coding exons along with flanking ± 10 intronic bases within the captured region.
Variant validation and segregation analysis
To verify the accuracy of candidate causal variants, PCR and Sanger sequencing (ABI 3100 capillary sequencer; Applied Biosystems) were used. The Glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH) was used as an internal control. Primers were designed using Primer3 plus, UCSC in-Silico PCR and primer-BLAST. Co-segregation analysis was performed in available family members to confirm the inheritance pattern and correlation with clinical phenotypes. Subsequent data was analyzed using SnackVar software version 2.4.3 ( https://github.com/Young-gonKim/SnackVar). The PCR, Gap PCR, MLPA (MRC-Holland, Amsterdam, The Netherlands) and Array CGH methods (2 × 400 K microarray kit; Agilent Technology) were applied to identify exon-level copy number variants (CNV). Abnormalities were confirmed by real-time PCR (qPCR)10.
Bioinformatics and in silico analysis
An end-to-end in-house bioinformatics pipeline was applied. All reads were aligned against the UCSC hg38 human reference genome using the burrow wheeler aligner (BWA). The pipeline uses tools for exclusion of low-quality reads (Fastp software), marking duplicates, base quality score recalibration (BQSR), base calling, filtration of variants before annotation and filtration after annotation by wANNOVAR and Franklin11. Variants are classified on the basis of multiple available databases including NCBI 1000 genomes, NHLBI Exome Sequencing Project, Exome Aggregation Consortium (ExAC), and HGMD® and in accordance with American College of Medical Genetics and Genomics (ACMG) guidelines into pathogenic, likely pathogenic, or variants of uncertain significance (VUS)12. Different criteria were employed to filter variants including known genes involved in PIDs, a Minor Allele Frequency (MAF) of less than 0.01, as well as the location and impact of the variant on protein function. Subsequently, the candidate variants were evaluated against clinical features to validate the findings. Finally, inherited CNV were detected by ExomeDepth package in R13.
Ethical considerations
The study was approved by the ethical committee of Ferdowsi University of Mashhad (IR.UM.REC.1404.059), and informed consent was obtained from all participants or their legal guardians. The Declaration of Helsinki was considered to perform this study including privacy protection of participants and the confidentiality of their personal information.
Results
Clinical details of patients
Candidate variants were identified in 82 patients (82.8%), including 38 non-syndromic (~ 46%) and 44 syndromic (~ 53%) PIDs, and the genetic etiology of 17 patients (17.1%; ~ 29% (n = 5) non-syndromic and ~ 70% (n = 12) syndromic PIDs) remained unresolved, as shown in Table 1. At the time of the study, 7.8% had unfortunately passed away.
Out of 82 genetically resolved patients, 44 cases were males (53%), 31 were females (37.8%) and 7 cases were fetus with unknown gender (8.5%). The mean (± SD) age at diagnosis was 6.5 ± 7.3 years. Figure 2 illustrates the frequency of different phenotypes among all 99 patients with PIDs. Key observations indicate that the most frequent feature is recurrent infections; other highly frequent features include hypogammaglobulinemia, elevated immunoglobulin E, and respiratory infections respectively, which are more common among syndromic cases. On the other hand, antibody deficiency (IgG, IgA and IgM levels) was more prevalent phenotypes among non-syndromic cases.
The geographical dispersion of patients with PID (n = 21) in specific areas of Mashhad city has been observed from southeast to northeast of the city. Except two members of a family with defect in IKBKB gene (c.201-1G > A), similar genetic variants were not detected. After recurrent infections, elevated immunoglobulin E is more common clinical feature among those with PID in this area.
Molecular findings in the syndromic PIDs
Out of 44 resolved syndromic PIDs cases, WES identified 20 previously documented and 24 novel variants in 31 genes associated with PIDs, as presented in Table 2. These novel variants consisted of missense (8/24; 33.3%), small deletions (7/24; 29.1%), large deletions (2/24; 8.3%), splicing defects (5/24; 20.8%), small insertions (1/24; 4.1%) and nonsense (1/24; 4.1%). Based on in silico prediction methods, one novel variant were classified as pathogenic (P), 13 variants as likely pathogenic (LP) and 10 variants as VUS; however, by the time, VUS variants are likely to be resolved. Twenty cases of syndromic PIDs (35.7%) with novel variant exhibited autosomal recessive (AR) inheritance. Cases with hemizygote and heterozygote inheritance pattern consist 1 and 3 respectfully. Among syndromic PIDs, the EPG5 gene was the most frequently mutated genes (n = 4).
Molecular findings in the non-syndromic PIDs
In this study, WES was conducted on 38 patients with non-syndromic PIDs, revealing 15 previously reported and 23 novel variants, as detailed in Table 3. The novel variants were confirmed via Sanger sequencing and were found to be co-segregated with the phenotype. These variants included missense (14/23; 60.8%), small deletions (1/23; 4.3%), nonsense (0/23; 0%), small insertions (2/23; 8.6%), duplication (1/23; 4.3%) and splicing defects (3/23; 13%). Among them, 1 was computationally predicted as pathogenic, 8 were LP variant, while the remaining variants were classified as VUS (n = 14), which are likely expected to be resolved by the time. The majority of these unreported variants associated with the non-syndromic PIDs were present at the true homozygous state in the probands (n = 17, 74%), followed by the heterozygous state (n = 2, 8.7%) and hemizygote (n = 4, 17.3%).
Spectrum of genetic defects based on the IUIS classes
Genes causing PID affecting both cellular and humoral immunity representing the largest proportion (26.8%, 22/82), followed by combined immunodeficiencies with syndromic features (24.3%, 20/82). Notably, defects in intrinsic and innate immunity (14.6%, 12/82) and autoimmune/autoinflammatory disorders (8.5%, 7/82) also contribute significantly. In contrast, complement deficiencies and bone marrow failure genes are rare (Figure 3).
Potential immunogenetic drivers of recurrent miscarriage
A hypothesis-generating observation suggests a possible association between certain variants and miscarriage risk in affected families. Table 4 illustrates details of five families with a history of miscarriage and at least one affected child. A family with nonsense variant in LRBA (p.Arg317*) had already experienced two idiopathic miscarriage cases and a deceased child. Similarly, CARMIL2, might be associated with three miscarriages due to a splice-site variant (c.871 + 1G > T) but one affected child was alive. The frameshift variant in SPINK5 (p.Glu584fs), may tie to three miscarriages, the RAG2 missense variant (p.Gly44Arg); and the CARD9 nonsense variant (p.Gln295*) were observed in family with one idiopathic miscarriage. Further functional studies are needed to confirm the role of these mutations in miscarriage.
Discussion
This study yielded novel findings regarding the genetic diagnosis of PIDs in an Eastern Iranian cohort. The application of WES resulted in a high diagnostic yield of 82.8% (82/99), considerably exceeding rates reported in prior studies, such as Ripen et al. (46.7%)14. This high yield likely reflects the elevated rate of consanguinity (75/99; 75.7% cases) in our population, facilitating the identification of autosomal recessive variants. This genetic investigation revealed approximately ~ 49% (23/47) novel variants across 18 genes associated with non-syndromic PIDs and around 51% (24/47) novel variants in 19 genes related to syndromic PIDs (Tables 2 and 3). Syndromic cases are usually more challenging to be resolved. In this study a higher proportion of unresolved cases were also syndromic (12/17, ~ 70%). The presence of unborn cases in resolved cases highlights the importance of prenatal or early genetic testing in PIDs. Resolved cases had a higher proportion of affected relatives, indicating that family history may aid in genetic diagnosis (Table 1).
Clinically, recurrent infections were the predominant presentation, consistent with prior observations by Thalhammer et al., who reported infections as the primary initial presentation in 68% of their cohort15. Syndromic cases frequently exhibiting hypogammaglobulinemia and elevated IgE whereas non-syndromic cases predominantly exhibited antibody deficiencies involving IgG, IgA, and IgM.
The clustering of PIDs in Mashhad mostly without shared genetic variants suggests that the clustering is driven by heterogeneous genetic backgrounds. However, two members of the same family—one of whom was a fetus—were diagnosed with the c.201-1G > A mutation in the IKBKB gene. Following prenatal diagnostic confirmation, the parent opted for termination of the pregnancy. Early deaths (7.8%) highlight the need for timely intervention, as early detection can prevent premature mortality.
The most common PIDs found in eastern Iranian patients focusing on a syndromic gene of EPG5. In this gene, four distinct novel VUS mutations, associated with Vici syndrome, were observed (Table 2). We have applied reclassification according to ACMG guideline for these variants.
Our analysis identified genetic mutations in T-B- and T + B- SCID in 16.1% (16/99) of cases. We detected several novel variants—RAG1 (c.2893G > C), RAG2 (c.130G > A; c.200G > T), ADA (c.289 T > G), JAK3 (c.184 + 1G > T), IL7R (c.358delA), and a large DCLRE1C deletion (c.14939810_14954096del)—alongside known mutations in RAG1, NHEJ1, ADA, IL7R, CD3E, and IL2RG. Erman et al. also reported novel mutations in RAG1, JAK3, and IL2RG, including a novel homozygous mutation in RAG1 (c.2259G > A) and two previously unknown mutations in JAK3 (c.1898G > A and c.1383_1384insG)16. Both studies emphasize the genetic heterogeneity of SCID. Two other common disorders are LRBA deficiency and AT, each observed in three patients. This study identified various types of CVID, including three novel genetic variants, with an overall prevalence of 6% (6/99 patients) (Tables 2 and 3).
AT cases harbored two previously and a novel pathogenic variant (Table 2). In the retrospective study by Sweta Das et al., which included 20 patients diagnosed with AT, WES identified 23 variants of which ten were novel17. The EPG5 gene is a key autophagy gene causes Vici syndrome. The associated variants were mentioned earlier. In a study describing the genetic data of 50 patients with Vici syndrome, the most common mutation in their study was c.1007A > G p.Gln336Arg which accounts for ~ 10% of pathogenic mutations in EPG5 gene18. Another study with ancestry data showed that this mutation may be associated with Ashkenazi descent19.
The classification of our findings based on IUIS demonstrated that the most frequent diagnoses were Class I (26.8%) and Class II (24.3%), followed by Class VI (14.6%) (Figure 3). This aligns partially with Al-Herz et al. (264 patients), where Class I (35.2%) and Class II (24%) dominated, though their rates were notably higher20. In contrast, Sheikhbahaei et al. (197 patients) reported Class III (PAD, 25.4%) and Class V (Phagocytic defects, 23.8%) as most prevalent, with Class II (9.6%) being far less common21. The elevated Class VI in our cohort (14.6%) highlights the heterogeneity of PID epidemiology across populations.
Additionally, the association of variants in LRBA, SPINK5, CARMIL2, CARD9, and RAG2 with recurrent miscarriages in affected families cautiously suggests an intriguing link between immune dysfunction and pregnancy loss that warrants further study (Table 4). Phan et al. identified two novel compound heterozygous stop-gain mutations in LRBA were c.1933C > T (p.R645X) and c.949C > T (p.R317X). The proband’s brother died at 16 years old with chronic immune thrombocytopenic purpura, the mother also had an idiopatic miscarriage after the proband. Rare studies support a plausible biological link between IEI and 22. Hence, further studies with larger cohorts are needed to validate these associations and functional studies are necessary to support the association between these variants and miscarriage.
Prenatal diagnosis (PND) identified homozygous pathogenic or likely pathogenic variants in multiple critical genes, emphasizing the clinical value of integrating WES into reproductive counseling and management. (Tables 2 and 3; asterisked cases). El Hawary et al. used PND to identify homozygous pathogenic variants in RAG, RAG2, NCF1, CYBA, IL10RB, and IL10RA genes in 12 Egyptian fetuses (34.3%) with IEI, highlighting the importance of prenatal genetic testing23.
The classification of several variants in this study has changed over time with advances in genomic technologies and updated ACMG criteria. Many were initially labeled as VUS but were later reclassified as new evidence emerged (Tables 2 and 3). This underscores the dynamic nature of variant interpretation and the need for ongoing reassessment to improve diagnostic accuracy and patient care. Although WES offers high diagnostic yield, especially in heterogeneous populations, it still requires careful variant filtering to distinguish pathogenic from benign changes24,25.
We also acknowledge the inherent limitations of single-center pilot studies in accurately representing the epidemiological landscape of PIDs within a specific geographical region. Such studies often involve a limited sample size and may not capture the full diversity of patient demographics and clinical presentations, leading to potential biases in the reported distribution rates. Furthermore, the findings from a single center may not be generalizable to broader populations. Ultimately, while single-center studies can offer valuable preliminary insights, they should be interpreted with caution and supplemented by larger, more diverse research efforts to help improving clinical practice and making related policy. A further limitation is the exclusion of autoinflammatory and autoimmune IEIs, where responses to targeted biologics are highly variable, whereas the PID cohort presents more consistent clinical patterns, enabling clearer analysis.
Conclusion
In summary, our study highlights the effectiveness of using WES as a practical and efficient method for comprehensive genotyping of PIDs. With a diverse and reasonably sized sample (n = 99), this research provides valuable insights into the genetic architecture of these diseases. It emphasizes the heterogeneous nature of PIDs, particularly in populations with high consanguinity rates. The identification of novel variants through this approach significantly enhances our understanding of the pathogenesis and distribution patterns of PIDs in the eastern Iranian population. Furthermore, the discovery of the novel variants holds great potential for advancing molecular diagnostics, improving genetic counseling, refining gene therapy strategies, and enhancing subtype classification of PIDs, especially in complex cases. Moreover, findings help genetic counselors by highlighting the mutations most common in this population, and they provide a basis for diagnostic tools and healthcare plans tailored to the region. Most importantly, they can improve care in pediatric immunology and genetic counseling centers, where children are often diagnosed late due to limited awareness and testing options.
Data availability
The datasets generated and analyzed during the current study are available in the ClinVar repository [accession number = SCV006301721—SCV006301793]. Moreover, data related to this study can be accessible upon reasonable request from corresponding author.
References
Bousfiha, A. et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J. Clin. Immunol. 40, 66–81 (2020).
Poli, M. C. et al. Human inborn errors of immunity: 2024 Update on the classification from the International Union of Immunological Societies Expert Committee. J. Hum. Immunol. (2025).
Rodriguez, J. A. et al. Imaging features of primary immunodeficiency disorders. Radiol. Cardiothorac. Imaging 3, e200418 (2021).
Krishna, S., Srinivasan, R., Prakash, A., Bhattad, S. & Indumathi, C. Clinical and laboratory profile of children with primary immunodeficiency-Perspective from a developing country. Curr. Med. Res. Pract. 11, 262–268 (2021).
Saadat, M., Ansari-Lari, M. & Farhud, D. Short report consanguineous marriage in Iran. Ann. Hum. Biol. 31, 263–269 (2004).
Nepesov, S. et al. Diagnosis of primary immunodeficiency diseases in pediatric patients hospitalized for recurrent, severe, or unusual infections. Allergol. Immunopathol. (Madr.) 50, 50–56 (2022).
Yazdani, R., Aghamohammadi, A. & Rezaei, N. Application of Flow Cytometry in Predominantly Antibody Deficiencies. Endocrine, Metabolic & Immune Disorders-Drug Targets (Formerly Current Drug Targets - Immune, Endocrine & Metabolic Disorders) 21, 647–663 (2021).
Tangye, S. G. et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J. Clin. Immunol. 42, 1473–1507 (2022).
Kalia, S. S. et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2. 0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 19, 249–255 (2017).
Kim, Y.-G. et al. SnackVar: an open-source software for Sanger sequencing analysis optimized for clinical use. J. Mol. Diagn. 23, 140–148 (2021).
De Summa, S. et al. GATK hard filtering: tunable parameters to improve variant calling for next generation sequencing targeted gene panel data. BMC Bioinf. 18, 57–65 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–423 (2015).
Plagnol, V. et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 28, 2747–2754 (2012).
Ripen, A. M. et al. A single-center pilot study in Malaysia on the clinical utility of whole-exome sequencing for inborn errors of immunity. Clin. Exp. Immunol. 206, 119–128 (2021).
Thalhammer, J. et al. Initial presenting manifestations in 16,486 patients with inborn errors of immunity include infections and noninfectious manifestations. J. Allergy Clin. Immunol. 148, 1332–1341 (2021).
Erman, B. et al. Investigation of genetic defects in severe combined immunodeficiency patients from Turkey by targeted sequencing. Scand. J. Immunol. 85, 227–234 (2017).
Das, S. et al. Exploration of clinical and genetic findings in Ataxia-Telangiectasia (AT) patients from the Indian subcontinent. Eur. J. Med. Genet. 66, 104766 (2023).
Kane, M. S. et al. Aberrant splicing induced by the most common EPG5 mutation in an individual with Vici syndrome. Brain 139, e52–e52 (2016).
Byrne, S. et al. EPG5-related Vici syndrome: A paradigm of neurodevelopmental disorders with defective autophagy. Brain 139, 765–781 (2016).
Al-Herz, W. et al. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front. Immunol. 9, 3146 (2019).
Sheikhbahaei, S. et al. Gene mutations responsible for primary immunodeficiency disorders: A report from the first primary immunodeficiency biobank in Iran. Allergy Asthma Clin. Immunol. 12, 1–11 (2016).
Christiansen, O. B. Immunogenetic determinants in recurrent miscarriage–evidence from genetic-epidemiological research. Am. J. Reprod. Immunol. 48, 143–143 (2002).
El Hawary, R. E. et al. Genetic testing in Egyptian patients with inborn errors of immunity: A single-center experience. J. Clin. Immunol. 42, 1051–1070 (2022).
Platt, C. D. et al. Efficacy and economics of targeted panel versus whole-exome sequencing in 878 patients with suspected primary immunodeficiency. J. Allergy Clin. Immunol. 147, 723–726 (2021).
Simon, A. J. et al. Whole exome sequencing (WES) approach for diagnosing primary immunodeficiencies (PIDs) in a highly consanguineous community. Clin. Immunol. 214, 108376 (2020).
Acknowledgements
The authors thank the Next Generation Genetic Polyclinic (Mashhad, Iran), Mashhad University of Medical Sciences (especially the Department of Medical Genetics and Molecular Medicine), and all contributors who supported this work, including patients and specialists who referred them to the center.
Transparency statement
The authors affirm that this manuscript is an honest, accurate, and transparent account of the study being reported. No important aspects of the study have been omitted, and any discrepancies from the planned study have been explained.
Author information
Authors and Affiliations
Contributions
M S: Conceptualization, Methodology, Writing – Original Draft Preparation, Data Analysis, Data Curation, Formal Analysis, Visualization; Z N: Writing – Review & Editing, Project Administration, Supervision; H A: Supervision, Validation; A P: Supervision, Writing – Review & Editing; E Gh K: Data Curation; M H A: Clinical Managment; N M: Clinical Managment . All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
There were no ethical issues related to this study. This study was approved by the ethical committee of Ferdowsi University of Mashhad, Mashhad, Iran (IR.UM.REC.1404.059).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Salehi, M., Ahanchian, H., Karimiani, E.G. et al. Genetic and epidemiological patterns of primary immunodeficiency diseases in Eastern Iranian patients. Sci Rep 16, 6993 (2026). https://doi.org/10.1038/s41598-026-35604-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-35604-4
