
Abstract
Hepatocellular carcinoma (LIHC) is a major global health concern, ranking as the sixth most common cancer and the fourth leading cause of cancer-related mortality worldwide, highlighting the urgent need to develop reliable diagnostic tools, biomarkers, and therapeutic targets to improve early detection and reduce mortality; KAT2A, a histone lysine acetyltransferase, plays crucial roles in multiple biological processes including chromatin remodeling, transcriptional regulation, cell cycle control, and apoptosis initiation, yet its specific role in the initiation and progression of hepatocellular carcinoma remains poorly understood; in this study, we assessed KAT2A expression levels in liver hepatocellular carcinoma tissues using data from the TCGA database and further analyzed its association with immune cell infiltration through single-cell data analysis, followed by evaluating the relationship between KAT2A and patient prognosis using Kaplan–Meier survival analysis combined with Cox regression modeling, while also exploring its potential biological functions via Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses, and finally validating the impact of KAT2A on the proliferation and migration of hepatocellular carcinoma cells through in vitro experiments; our findings revealed that KAT2A is significantly upregulated in LIHC tissues and plays a key role in promoting the proliferation and migration of LIHC cells, suggesting that KAT2A may serve as a novel prognostic biomarker and a promising therapeutic target for immunotherapy in hepatocellular carcinoma.
Similar content being viewed by others


Introduction
LIHC holds the position of the sixth most common cancer worldwide and is responsible for the fourth highest number of cancer-related deaths globally. Annually, the disease is associated with approximately 841,000 new cases and results in around 782,000 fatalities1. A significant challenge in the management of LIHC arises from the fact that many patients are diagnosed when the cancer is in intermediate to advanced stages. This late diagnosis adversely affects treatment outcomes, as evidenced by a disheartening five-year survival rate of only 18%2. In recent years, immunotherapy has emerged as a pivotal strategy aimed at combatting LIHC. This treatment approach concentrates on the inhibition of immune checkpoints, thereby bolstering the capacity of immune cells to detect and eliminate cancer cells3. One of the dominant immune checkpoints in this context is programmed cell death protein 1 (PD-1), which is primarily present on activated T cells, B cells, and macrophages. PD-1 is known to suppress the activation of specific T cells when it binds to its ligands, PD-L1 and PD-L2. Notably, PD-L1 is often highly expressed on the surfaces of tumor cells. The interaction between PD-L1 on these tumor cells and PD-1 on immune cells facilitates tumor immune evasion, thereby impairing the ability of the immune system to eliminate cancerous cells4,5. Recent advancements in the field of LIHC treatment have focused on immunotherapies that target the PD-1/PD-L1 axis, as well as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)6,7. However, the application of anti-PD-1 and anti-PD-L1 inhibitors is not without complications; these treatments can lead to several immune-related side effects. Patients may experience increased levels of aspartate aminotransferase (AST), skin rashes, hypothyroidism, colitis, and pneumonia, with the risk of severe cases resulting in lasting complications8,9. Given these potential adverse effects, there is a critical need for ongoing research to identify new biomarkers and effective therapeutic targets that can improve treatment outcomes for patients with LIHC.
KAT2A, commonly known as GCN5, is identified as the first histone lysine acetyltransferase that enables the acetylation of histone 3-lysine 9 (H3K9) and histone 3-lysine 14 (H3K14)10. This enzyme plays a pivotal role in various biological processes, including chromatin remodeling, transcription regulation, the cell cycle, and apoptosis11,12,13,14. Moreover, KAT2A participates in the acetylation of lysine residues in non-histone proteins such as p5315, PGC-1α11, CDC612, cyclin A14, and Myc16, which are actions that promote tumor cell proliferation, migration, and invasion. Importantly, KAT2A exhibits significant overexpression in a range of cancers, including breast cancer, non-small cell lung carcinoma17, pancreatic cancer18, colon cancer19, nasopharyngeal carcinoma20, and renal cell carcinoma21.
In this research study, we undertook a comprehensive analysis of the expression levels of KAT2A, exploring its relationships with a variety of clinicopathological characteristics. To achieve this, we utilized multiple databases, including TIMER22, UALCAN23, TCGA24, and GEO25. These resources provided invaluable insights into the expression patterns of KAT2A and allowed us to correlate these patterns with clinical features relevant to patient outcomes. In addition to examining the expression level of the KAT2A gene, we also studied its co-expressed genes, which allows us to further investigate the potential molecular mechanisms of KAT2A involved in hepatocellular carcinoma. Furthermore, our study also conducted an in-depth investigation into the association between KAT2A and immune infiltration in hepatocellular carcinoma. Through the TIMER database and the CIBERSORTx database26, we successfully revealed the correlation mechanisms between the KAT2A gene and multiple immune cells, thereby more clearly elucidating the interaction pattern between KAT2A and the tumor microenvironment. Furthermore, we further verified the relationship between the expression of the KAT2A gene and immune infiltration through single-cell analysis, thereby accurately identifying the association between KAT2A-highly expressing cells and specific immune cell subsets27,28. Our findings suggest that KAT2A possesses the potential to serve as a prognostic biomarker specifically for LIHC. Additionally, KAT2A may represent a promising target for immunotherapy, opening new avenues for treatment strategies aimed at enhancing patient outcomes in hepatocellular carcinoma therapy.
Methods
Data download
Gene expression matrix data (in TPM format) and corresponding sample clinical data of Liver Hepatocellular Carcinoma (LIHC) were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). The expression values were then subjected to log2(value + 1) transformation. For the single-cell dataset, GSE146115 was retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), which comprises 4 HCC samples, 3200 cells, and 27,227 genes. The immunohistochemical staining profile of KAT2A in LIHC tissues was obtained from the HPA database29 (https://www.proteinatlas.org/).
Analysis of KAT2A in pan-cancer
The TIMER database (https://cistrome.shinyapps.io/timer/) integrates sample data of 32 types of tumors and their adjacent non-tumor tissues from The Cancer Genome Atlas (TCGA). We utilized this database to analyze the expression levels of KAT2A in pan-cancer and adjacent non-tumor tissues. Meanwhile, we investigated the expression levels of KAT2A in various normal tissues using the GTEx database30 (https://gtexportal.org/home/). Subsequently, the HCCDB database31 (http://lifeome.net/database/hccdb2) was used to further explore the differential expression of KAT2A between adjacent non-tumor tissues and tumor tissues in different hepatocellular carcinoma datasets.
UALCAN database analysis
The UALCAN database (https://ualcan.path.uab.edu/) is an online resource specifically designed for cancer data analysis and exploration. It encompasses a vast amount of data from institutions such as The Cancer Genome Atlas (TCGA), the Clinical Proteomic Tumor Analysis Consortium (CPTAC), and the Children’s Brain Tumor Tissue Consortium (CBTTC). With this powerful data access capability, researchers can identify potential biomarkers and deeply analyze gene expression patterns and survival rate characteristics associated with different cancer types. We utilized this platform to conduct a systematic study on the mRNA and protein expression levels of the KAT2A gene in liver hepatocellular carcinoma (LIHC).
Survival analysis
According to the median expression value of the KAT2A gene in the gene expression matrix of liver hepatocellular carcinoma (LIHC), patients were divided into a high-expression group and a low-expression group. The survival package (version 3.8-3) in R software32, version 4.5.1 was utilized to perform survival analysis and Cox regression analysis on the clinical data and expression matrix of LIHC from The Cancer Genome Atlas (TCGA) database. Subsequently, the survminer package (version 0.5.1) was used to generate Kaplan-Meier (KM) survival curves. Meanwhile, the rms package (version 8.0–0) was employed for the drawing of Cox regression tables.
ROC diagnostic curve
The pROC package (version 1.19.0.1) in R software (version 4.5.1) was used to analyze the gene expression matrix of liver hepatocellular carcinoma (LIHC) from the TCGA database. Subsequently, the ggplot2 package (version 4.0.0) was applied to plot the ROC diagnostic curve.
Differential expression analysis
The limma package (version 3.64.1) was used to perform differential expression analysis between the high and low KAT2A expression groups. Subsequently, the ggplot2 package (version 4.0.0) was employed to generate volcano plots of differential expression. Next, 125 genes associated with the differential expression of KAT2A were filtered out based on the criteria of |logFC| > 1.5 and p < 0.05. Thereafter, the clusterProfiler package (version 4.16.0) was utilized to conduct Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)33 enrichment analyses on these 125 genes. Finally, the ggplot2 package (version 4.0.0) was used for visualization of the analysis results.
Transcription factor analysis
We obtained information on transcription factors related to KAT2A from four authoritative databases. These databases include the ENCODE database34 (https://www.encodeproject.org), the KnockTF database35 (https://bio.liclab.net/KnockTFv2), the hTFtarget database36 (https://guolab.wchscu.cn/hTFtarget), and the ChEA database37 (http://amp.pharm.mssm.edu/lib/chea.jsp). All of these databases provide abundant information on transcription factors and their interactions. The data extracted from these sources play a crucial role in deeply understanding the complex regulatory mechanisms involving KAT2A, thus enriching the overall understanding in this research field.
Immune infiltration analysis
The CIBERSORT.R script and the LM22 signature matrix file (“LM22.txt”), which contains gene signatures for 22 immune cell types, were obtained from the CIBERSORTx website26 (https://cibersortx.stanford.edu/). A deconvolution algorithm38 was subsequently applied to estimate immune cell abundance in the gene expression matrix of KAT2A high- and low-expression groups. During the analysis, the permutation parameter was set to 1000 to compute p-values and confidence intervals, and quantile normalization (QN = T) was enabled to standardize gene expression data across samples.
Single-cell analysis
The downloaded single - cell dataset was processed using the Seurat package (version 5.3.1) to create a Seurat object39. Only genes expressed in at least 3 cells and cells expressing at least 200 genes were retained. To screen high - quality cells, the following criteria were applied: (1) The number of genes detected in each cell was greater than 200 and less than 6000; (2) The proportion of mitochondrial genes in each cell was less than 20%. Subsequently, data scaling and UMAP dimensionality reduction were performed, and the UMAP results were visualized. Then, marker genes of cell types required for annotation were obtained by accessing the CellMarker database40 (http://xteam.xbio.top/ACT/). Annotation was carried out according to the expression of marker genes among different clusters. Next, the CellChat package (version 1.6.1) was used to construct and visualize the inter-cellular communication network.
Cell culture
For the purposes of this study, specific cell lines including LM3, Huh7, and PLC/PRF/5 were acquired from the China Cell Bank, which is based in Shanghai, China. These cell lines were cultivated using DMEM medium, which is a product of Gibco, located in Waltham, Massachusetts, USA. The DMEM was supplemented with 10% fetal bovine serum sourced from Ausbian in Australia, and it also contained 1% penicillin-streptomycin solution, which is also manufactured by Gibco. Throughout the culturing process, the cells were maintained in a controlled incubator environment, featuring an atmosphere composed of 5% CO2 at a stable temperature of 37 degrees Celsius.
Transfection
The synthesis of KAT2A siRNA was executed by GenePharma, a company based in Shanghai, China. To facilitate the introduction of this siRNA into the cells, Lipo8000, a product from Beyotime also located in Shanghai, China, was employed in accordance with the manufacturer’s guidelines. Following this transfection process, the successful establishment of KAT2A knockdown cells was confirmed 72 h later, marking a significant advancement in the experiment.
Western blotting
The cells were subjected to a process of homogenization to break them down into a uniform mixture. This step is crucial as it allows for the accurate assessment of protein concentration, which was evaluated using a bicinchoninic acid assay. Following this initial analysis, the proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), a technique that allows for the resolution of proteins based on their molecular weight. Once separation was complete, the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane, which is essential for subsequent immunodetection processes.After the transfer, the membrane was incubated in a blocking solution composed of 5% bovine serum albumin (BSA) diluted in Tris-buffered saline (TBST) containing 0.1% Tween 20. This blocking step, which lasted for one hour at room temperature, is important to prevent non-specific binding of antibodies to the membrane. The membrane was then incubated overnight at a temperature of 4 °C with primary antibodies specific to the proteins of interest: anti-β-actin at a dilution of 1:3000 and anti-KAT2A at a dilution of 1:1000, both sourced from Proteintech in Chicago, IL, USA. Following this incubation period, the membrane was rinsed thoroughly with TBST to remove any unbound primary antibodies.In the next stage, the membrane was treated with secondary antibodies for a duration of 1.5 h at a dilution of 1:3000. This step is crucial as secondary antibodies bind to the primary antibodies, thereby amplifying the signal for detection. To visualize the protein bands on the membrane, the Enhanced Chemiluminescent detection kit from Biosharp, Beijing, China, was employed, facilitating the observation of the protein bands resulting from the electrophoresis and transfer process.
Proliferation assays
The methods utilized to evaluate the proliferative rate of cell lines with KAT2A knockdown consisted of two main approaches: colony formation assays and the CCK8 assay, specifically utilizing the product from Yeasen, China (catalog number 40203ES76). For the CCK8 assay, a precise procedure was followed where 2000 cells were first seeded into each well of a 96-well plate and then incubated at 37 °C. After this incubation period, the CCK8 reagent was introduced into each well, and the incubation was extended for an additional four hours to allow for the reaction to occur. Following this incubation, the optical density at a wavelength of 450 nm was measured using a microplate reader, specifically the ReadMax 1200 from Flash, Shanghai, China, which provided a quantitative assessment of cell viability based on the amount of formazan produced.In complement to the CCK8 assay, colony formation assays were employed to further assess the proliferative capability of the cancer cells. In this method, 200 cancer cells were plated in six-well plates, with the culture medium being refreshed every other day to maintain optimal growth conditions. After a 2-week incubation period, the resulting colonies needed to be visualized for analysis. This was achieved by fixing the colonies with paraformaldehyde, which preserves the cellular structures, followed by staining with crystal violet. This staining process allowed for the clear observation of the colonies formed, providing a visual representation of the proliferative ability of the KAT2A-knockdown cell lines.
Migration assays
The capability of cell migration was assessed through the use of a scratch assay. This particular technique was conducted using a 200 µL pipette within 60 mm culture dishes, after the cells had proliferated to reach a confluence of 90–100%. Images of the wound area were captured at two distinct time points: immediately at the beginning of the experiment (0 h) and after a 48-hour incubation period. For this imaging, an Olympus IX70 inverted microscope was employed to ensure high-quality visual documentation of the cell migration process.
Statistical analysis
All statistical analyses in this study were performed using R (The R Project for Statistical Computing), version 4.5.1, available at https://www.r-project.org/. Basic statistical functions relied on its built-in core packages, with no need for additional installation of extension packages. Comparisons of intergroup differences were conducted using the independent samples t-test41. Statistical significance was determined using a threshold of p < 0.05, where * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.
Results
KAT2A is highly expressed in LIHC
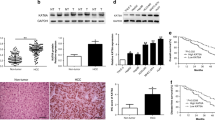
To investigate the expression significance of KAT2A in different cancer types, we performed a detailed analysis of KAT2A mRNA transcription levels using the TIMER database. It was found that KAT2A expression was significantly higher in most cancer types compared with adjacent non-tumor groups, and this result highlighted the potential role of KAT2A in tumorigenesis, especially in Liver Hepatocellular Carcinoma (Fig. 1A). Notably, KAT2A expression was significantly decreased in renal squamous cell carcinoma (RSCC) compared with normal tissues, suggesting that there may be a unique regulatory mechanism in this cancer type. Subsequently, we evaluated KAT2A expression levels in various normal tissues using the GTEx database, and the results showed that they were within the expected physiological range (Fig. 1B). For further in-depth investigation, we analyzed KAT2A expression levels in various hepatocellular carcinoma datasets using the HCCDB database. The results showed that in a broader patient population, KAT2A expression levels were significantly increased in 8 LIHC samples when compared with adjacent normal tissues, which supports the view that KAT2A may be involved in the progression of this specific cancer type (Figs. 1C and D).

Expression levels of KAT2A across different types of tumors and healthy tissues. (A) KAT2A expression levels found in a variety of healthy and cancerous tissues. (B) KAT2A expression levels detected in normal tissue samples. (C) Data on KAT2A mRNA levels from LIHC samples. (D) Boxplot illustrating KAT2A mRNA expression levels in LIHC samples (* indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.)
Protein expression levels of KAT2A
In order to deepen our understanding of KAT2A expression patterns in LIHC, we utilized the UALCAN database to evaluate KAT2A expression levels. Our results demonstrated that the mRNA and protein levels of KAT2A in LIHC tissues were significantly elevated compared to normal tissues (Fig. 2A and B). Furthermore, immunohistochemical staining data from the HPA database29 indicate that the protein level of KAT2A in LIHC tissues is higher than in adjacent tissues (Fig. 2C).

Expression levels of KAT2A in LIHC. (A and B) RNA and protein expression of KAT2A in LIHC tissues and normal tissues in UALCAN database. (C) Immunohistochemical staining of KAT2A in LIHC tissues and normal liver tissues in HPA database.
KAT2A expression levels in clinically characterized samples
To further investigate the clinical significance of KAT2A expression in Liver Hepatocellular Carcinoma (LIHC), we conducted an in-depth analysis based on the UALCAN database, systematically examining the association between KAT2A expression levels and key clinical characteristics of LIHC patients. The present study focused on critical clinical parameters including pathological stage, histological grade, lymph node metastasis status, and histological subtype. The results showed that the transcriptional levels of KAT2A in LIHC patient tissues were significantly higher compared with the normal control group (Fig. 3A–D). This finding highlights a significant correlation between high KAT2A expression and LIHC progression, suggesting that KAT2A may play an important role in the disease process of LIHC.

The correlation between KAT2A expression and various clinicopathological characteristics in the UALCAN database was examined. Specifically, the influence of stage (A) grade (B) lymph node metastasis (C) histological subtypes (D) on KAT2A expression in LIHC was illustrated.
Survival analysis of KAT2A
To investigate the correlation between KAT2A expression level and Progression-Free Survival (PFI), we plotted Kaplan-Meier (KM) curves for PFI. The results showed that the high KAT2A expression group was significantly associated with poor PFI prognosis (Fig. 4A). Subsequently, to further explore the diagnostic sensitivity of KAT2A expression level for hepatocellular carcinoma, we generated receiver operating characteristic (ROC) diagnostic curves. The results demonstrated that the area under the curve (AUC) was 0.977, which further indicated that KAT2A expression level has high diagnostic value for hepatocellular carcinoma (Fig. 4B). Furthermore, when the KAT2A expression level was greater than 3.961, its diagnostic sensitivity increased significantly, and this level was generally higher than the expression levels in normal group samples (Fig. 4C). Finally, to explore the effects of age, clinical stage, and KAT2A expression level on PFI prognosis, we performed univariate and multivariate Cox regression analyses for the above factors. The results revealed that KAT2A, T stage, and M stage all had significant effects on PFI (p < 0.05), among which KAT2A showed the highest significance (Fig. 4D). This further confirmed that KAT2A has high prognostic value for PFI in patients with hepatocellular carcinoma.

KAT2A’s prognostic, diagnostic, and Cox regression analyses in LIHC. (A) Kaplan–Meier curve of PFI stratified by KAT2A expression. (B) ROC curve of KAT2A for LIHC diagnosis. (C) Violin plot of KAT2A expression distribution with optimal cutoff. (D) Univariate and multivariate Cox regression analyses of PFI-related factors including KAT2A and clinicopathological characteristics.
GO and KEGG analysis of KAT2A
To investigate the mechanism of action of KAT2A in Liver Hepatocellular Carcinoma (LIHC), we first screened for differentially expressed genes (DEGs) between KAT2A high-expression and low-expression groups using the limma package, and generated a volcano plot. Meanwhile, the top 5 most significant upregulated and downregulated genes were displayed respectively (Fig. 5A). The volcano plot intuitively shows the distribution of DEGs: red dots represent genes upregulated in the KAT2A high-expression group, blue dots represent downregulated genes, and gray dots indicate genes with no significant difference, clearly reflecting the distribution of expression differences among DEGs. Subsequently, based on the criteria of |logFC| > 1.5 and adjusted p-value < 0.05, a total of 125 DEGs were identified. Further hierarchical clustering of these 125 DEGs via a heatmap revealed distinct separation in gene expression patterns between the two groups of samples. This not only verified the reliability of DEG screening but also suggested that KAT2A may drive transcriptomic changes in LIHC (Fig. 5B). We then performed functional enrichment analysis on the 125 DEGs using the clusterProfiler package to clarify their biological functions. Gene Ontology (GO) enrichment analysis (Fig. 5C) showed that in terms of biological processes (BP), DEGs were mainly enriched in processes such as “cell proliferation”, “regulation of cell cycle progression”, and “response to growth factor stimulation”; in cellular components (CC), they were concentrated in “cytoplasmic matrix”, “cell nucleus”, and “plasma membrane”; in molecular functions (MF), they were significantly enriched in functions including “ATP binding”, “protein kinase activity”, and “growth factor receptor binding”—all of which are closely related to cancer cell growth and survival. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (Fig. 5D) further indicated that these DEGs were significantly involved in multiple cancer-related pathways, such as the “PI3K-Akt signaling pathway”, “MAPK signaling pathway”, and “cell cycle pathway”. These pathways have been widely confirmed to regulate the proliferation, migration, and drug resistance of LIHC cells. Collectively, these results suggest that KAT2A-related DEGs are functionally closely associated with key biological processes and signaling pathways in LIHC progression, providing a molecular basis for elucidating the prognostic and diagnostic roles of KAT2A in LIHC.

Differential genes of KAT2A and their functional enrichment analyses in LIHC. (A) Volcano plot of differentially expressed genes in high vs. low KAT2A expression groups. (B) Correlation heatmap of 125 differentially expressed genes. (C) Bar plot of Gene Ontology (GO) enrichment analysis. (D) Bar plot of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis.
Prediction of KAT2A key transcription factors
To dissect the transcriptional regulatory mechanisms of KAT2A in Liver Hepatocellular Carcinoma (LIHC), we first screened transcription factors associated with KAT2A using four public databases—ENCODE, HTF-target, KnockTF, and CHEA—and constructed transcriptional regulatory networks (Fig. 6A–D). There were differences in the potential interacting transcription factors of KAT2A identified by different databases: for instance, transcription factors screened from the ENCODE database are mainly involved in cell cycle regulation, while those from the HTF-target database focus on transcription factors related to oncogenic signaling pathways.
To identify core transcription factors universally associated with KAT2A, we performed cross-comparison of the transcription factor results from the four databases and generated a Venn diagram, ultimately identifying MYC as the only shared transcription factor (Fig. 6E). Subsequently, we further validated the correlation between their expression levels via Spearman correlation analysis and plotted a scatter plot (Fig. 6F). The results showed a significant positive correlation between KAT2A and MYC expression in LIHC samples (r = 0.254, p < 0.001). These findings suggest that MYC may promote LIHC progression by regulating KAT2A or through synergistic effects with it, providing a new direction for exploring the oncogenic pathways of KAT2A.

KAT2A-associated transcription factors and its expression correlation with MYC in LIHC. (A) Transcription factor network from ENCODE. (B) Transcription factor network from HTF-target. (C) Transcription factor network from KnockTF. (D) Transcription factor network from CHEA. (E) Venn diagram of transcription factors across four databases. (F) Scatter plot showing the correlation between KAT2A and MYC expression.
Analysis of KAT2A expression across the LIHC microenvironment at Single-Cell resolution
To dissect the cellular atlas of hepatocellular carcinoma and the expression pattern of KAT2A at the single-cell level, we analyzed the GSE146115 single-cell RNA sequencing dataset, which includes 4 hepatocellular carcinoma samples and 3200 cells. First, Uniform Manifold Approximation and Projection (UMAP) was used for cell clustering visualization, revealing 14 clusters (Fig. 7A). After stratification by sample, UMAP plots displayed the UMAP profiles of cells from the 4 samples(Fig. 7B). Cell types in each cluster were annotated based on classical marker genes, and the annotated UMAP plots were generated (Fig. 7C). A dot plot was used to show the expression levels of marker genes across various cell types, further validating the above annotations (Fig. 7D). In addition, we analyzed the proportion of each cell type among the 4 samples and generated a stacked bar chart. The results showed differences in the proportion of immune cells between samples, suggesting potential sample-specific differences in immune infiltration (Fig. 7E). To explore intercellular communication, we constructed a cell-cell interaction network (Fig. 7F), which revealed strong signal interactions originating from dendritic cells with plasma cells, cholangiocytes, endothelial cells, and stellate cells. To further highlight the ligand-receptor signal intensity, we plotted a cell-interaction intensity heatmap (Fig. 7G), where a deeper red color indicates stronger communication between ligand cells (y-axis) and receptor cells (x-axis). Subsequently, we focused on KAT2A and mapped its expression onto the UMAP plot (Fig. 7H). To further illustrate its expression levels across different cell clusters, we generated violin plots and heatmaps (Figs. 7I-J). The results showed that KAT2A is highly expressed in cholangiocytes, proliferating cells, dendritic cells, and stellate cells, with particularly prominent expression in dendritic cells. This suggests that KAT2A may play an important role in immune regulation or functional activity through dendritic cells and proliferating cells. Moreover, the high expression of KAT2A may be closely related to the ability of dendritic cells to transmit signals to other immune cells and stromal cells, potentially participating in the regulation of the local immune microenvironment. These findings provide cellular-level clues for subsequent investigations into the role of KAT2A in biliary diseases or tumor immunity.

single-cell RNA-seq analyses of hepatocellular carcinoma (HCC) from the GSE146115 dataset, encompassing cell clustering, annotation, and KAT2A expression profiles. (A) UMAP visualization of HCC scRNA-seq data from GSE146115. (B) UMAP stratified by sample group in GSE146115. (C) UMAP with cell type annotation from GSE146115. (D) Dot plot of marker gene expression across cell clusters in GSE146115. (E) Stacked bar plot of cell type distribution per sample in GSE146115. (F) A cellular ligand-receptor communication network constructed based on the GSE146115 dataset. (G) Heatmap of cellular ligand-receptor signal intensity in the GSE146115 dataset. (H) UMAP depicting KAT2A expression in GSE146115. (I) Violin plot of KAT2A expression levels by cell type in the GSE146115 dataset. (J) Heatmap of KAT2A expression by cell types in the GSE146115 dataset.
KAT2A and immune microenvironment in hepatocellular carcinoma
To elucidate the association between KAT2A expression and the immune microenvironment in hepatocellular carcinoma (HCC), we systematically analyzed the correlation between KAT2A expression and immune cell infiltration patterns using TCGA-LIHC data and immune profiling tools. We first evaluated the infiltration levels of 22 immune cell subsets in KAT2A high- and low-expression groups, which were visualized using a stacked bar plot (Fig. 8A). To further compare immune cell infiltration between the two groups, we generated box plots illustrating differential immune cell infiltration (Fig. 8B). The box plots revealed that the KAT2A high-expression group exhibited significantly increased infiltration of regulatory T cells (Tregs) and resting dendritic cells, while infiltration of memory B cells, M2 macrophages, and neutrophils was markedly reduced. These findings suggest that high KAT2A expression may suppress the infiltration of memory B cells, M2 macrophages, and neutrophils, thereby potentially promoting HCC cell proliferation and immune evasion. Furthermore, additional analysis indicated that KAT2A gene mutations were significantly associated with infiltration levels of various immune cells, including B cells, CD8 + T cells, CD4 + T cells, macrophages, neutrophils, and dendritic cells (Fig. 8C). Finally, correlation scatter plots were used to analyze the relationship between KAT2A expression and immune-related genes (Fig. 8D–I). The results showed that KAT2A was significantly positively correlated with memory B cells, regulatory T cells (Tregs), and resting dendritic cells, but negatively correlated with monocytes, M2 macrophages, and neutrophils. In summary, these results indicate that KAT2A may participate in shaping the immune microenvironment of HCC and could influence responses to immunotherapy.

Analysis of the correlation between KAT2A expression levels and immune cell infiltration. (A) Stacked bar plot showing the relative abundance of immune cells in KAT2A high- and low-expression groups. (B) Box plot illustrating the differences in immune cell infiltration levels between the two groups. (C) KAT2A genetic alterations were significantly associated with infiltration levels of various immune cells. (D–I) Scatter plots displaying correlations between KAT2A expression and immune cell infiltration levels. (* indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.)
KAT2A knockdown inhibited the proliferation and migration of LIHC cells in vitro
To clarify the functional role of KAT2A in hepatocellular carcinoma (HCC) cell biology, we first evaluated KAT2A protein expression in three HCC cell lines (PRF/PLC/5, Huh7, and LM3) via Western blot analysis (Fig. 9A). Quantitative analysis revealed that KAT2A expression was significantly higher in LM3 cells and Huh7 cells compared to PRF/PLC/5 cells (p < 0.01), so PRF/PLC/5 and Huh7 cells were selected for subsequent KAT2A knockdown experiments. We then transfected HCC cells with KAT2A small interfering RNA (si-KAT2A) to silence KAT2A expression, using a non-targeting siRNA as the negative control (si-NC). We confirmed the efficient knockdown of KAT2A by Western blotting. The protein level of KAT2A was significantly decreased in si-KAT2A-transfected PRF/PLC/5 and Huh7 cells compared to the si-NC group (Fig. 9B). Subsequently, a wound healing assay was conducted to evaluate cell migration ability (Fig. 9C). At 48 h post-wounding, the wound closure rate was significantly lower in si-KAT2A-transfected PRF/PLC/5 cells compared to the si-NC group (p < 0.001). Similarly, KAT2A knockdown also resulted in a reduced closure rate in Huh7 cells (p < 0.001). These results collectively indicate that KAT2A silencing inhibits the migration of HCC cells. Furthermore, Western blot analysis confirmed that the expression of key proteins regulating proliferation and migration was significantly modulated by KAT2A (Figure S1). Colony formation assays (Fig. 9D) were conducted to evaluate the clonogenic potential of HCC cells. After 14 days of culture, the number of colonies formed by PRF/PLC/5 cells in the si-KAT2A group was drastically reduced compared to the si-NC group (p < 0.001). A similar trend was observed in Huh7 cells, thereby supporting the conclusion that KAT2A contributes to the clonogenic ability of HCC cells and implicating it as a potential marker for long-term proliferative and tumorigenic potential. Cell proliferation was assessed using the CCK-8 assay (Fig. 9E). Throughout the 72-hour observation period, the optical density (OD) values at 450 nm were consistently lower in the si-KAT2A groups than in the si-NC groups for both cell lines (p < 0.001), further indicating that KAT2A knockdown suppresses the short-term proliferation of HCC cells. Collectively, these in vitro results demonstrate that KAT2A promotes the migration, short-term proliferation, and clonogenic potential of HCC cells, supporting its potential as a therapeutic target for HCC.

investigates KAT2A expression in HCC cell lines and the effects of its knockdown on cell migration, invasion, and proliferation. (A) Western blot and quantitative analysis of KAT2A expression in PRF/PLC/5, Huh7, and LM3 cells. (B) Western blot and quantitative analysis verifying KAT2A knockdown efficiency by si-KAT2A. (C) Wound healing assay and quantitative analysis of HCC cell migration after KAT2A knockdown. (D) Colony formation assay (plate clone assay) and quantitative analysis of HCC cell clonogenic potential. (E) CCK-8 assay of HCC cell proliferation over time after KAT2A knockdown. (* indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.)
Discussion
Hepatocellular carcinoma (LIHC) remains a global healthcare challenge with limited effective biomarkers for prognosis and immunotherapy response prediction. In this study, we systematically investigated the role of KAT2A in LIHC through a multi-dimensional approach, integrating public database mining, single-cell RNA sequencing (scRNA-seq) analysis, and in vitro experiments. Our findings collectively demonstrate that KAT2A is significantly upregulated in LIHC tissues, correlates with adverse clinical outcomes, drives malignant biological behaviors of LIHC cells, and modulates the immunosuppressive tumor microenvironment (TME)—providing novel insights into its potential as a prognostic biomarker and therapeutic target for LIHC.
The overexpression of KAT2A in liver hepatocellular carcinoma is consistent with its well-established oncogenic role in other malignant tumors. Previous studies have reported that KAT2A is upregulated in breast cancer, non-small cell lung carcinoma, and pancreatic cancer, where it promotes tumor cell proliferation and invasion through the mechanism of histone and non-histone acetylation. Our study extends this mechanism to the field of LIHC: based on multi-omics datasets from TCGA, TIMER, and HCCDB, we confirmed that the mRNA and protein expression levels of KAT2A in LIHC tissues were significantly higher than those in adjacent non-tumor/normal tissues, and its overexpression was significantly associated with advanced pathological stages and histological grades. Survival analysis further demonstrated that high KAT2A expression predicts a shorter progression-free interval (PFI) and exhibits superior diagnostic efficacy compared to some well-established LIHC biomarkers, supporting its potential as a clinically actionable indicator.
It should be specifically noted that our single-cell RNA sequencing analysis of the GSE146115 dataset revealed a cell-type-specific expression pattern of KAT2A: it was significantly enriched in cholangiocytes, proliferating cells, and most prominently, dendritic cells (DCs). As critical regulators of anti-tumor immunity, the maturation status and antigen-presenting capacity of DCs directly influence the activation of CD8 + T cells42. The high expression of KAT2A in DCs, combined with our observation of strong signal communication between DC-derived signals and cells such as plasma cells, cholangiocytes, endothelial cells, and stellate cells, all suggests that KAT2A may mediate DC-associated immune regulatory mechanisms in LIHC.
Our functional enrichment analysis results provide deep insights into the molecular pathways through which KAT2A drives LIHC progression. The 125 KAT2A-associated differentially expressed genes (DEGs) are mainly enriched in cancer-related biological processes and signaling pathways—pathways that have been fully confirmed to regulate the proliferation, migration, and drug resistance of LIHC cells. For instance, the PI3K-Akt signaling pathway can promote LIHC cell survival by activating downstream effectors such as mTOR43, while MAPK signaling drives tumor growth by mediating oncogenic stress responses44. Our in vitro experiments further validated this finding: KAT2A knockdown significantly inhibited the proliferative capacity (confirmed by colony formation assays and CCK8 assays) and migratory capacity (verified by scratch assay) of LIHC cells, thereby confirming the critical role of KAT2A in maintaining the malignant phenotype of LIHC.
At the transcriptional regulatory level, we identified MYC as the only core transcription factor that was consistently detected across cross-comparisons of four authoritative databases—ENCODE, HTF-target, KnockTF, and CHEA—and KAT2A and MYC exhibited a significant positive correlation in their expression in LIHC. As a key oncogenic driver in LIHC, MYC often promotes cell cycle progression and metabolic reprogramming through gene amplification or overexpression45. Our findings suggest two potential coexisting mechanisms: MYC may directly transactivate KAT2A transcription, or KAT2A may enhance MYC transcriptional activity through acetylation modification. Regardless of the mechanism, the discovery of the KAT2A-MYC regulatory axis adds a new dimension to the oncogenic regulatory network of LIHC and provides a theoretical basis for the development of dual-target therapeutic strategies.
Based on TCGA-LIHC data and immune analysis tools, it has been clarified that high expression of KAT2A is associated with increased infiltration of regulatory T cells (Tregs) and resting dendritic cells in LIHC, as well as decreased infiltration of memory B cells, M2 macrophages, and neutrophils. This remodeling of the immune cell composition may promote LIHC proliferation and immune escape by enhancing immunosuppression and weakening anti-tumor immune responses. Additionally, KAT2A gene mutations were found to correlate with infiltration of various immune cells, and its expression level shows positive correlations with immune-related genes such as memory B cells and Tregs, while exhibiting negative correlations with monocytes, among others. These findings further suggest that KAT2A may regulate the LIHC immune microenvironment through dual dimensions—expression level and gene mutations—providing indirect clues to its mechanism of action. From a clinical perspective, this association offers a new direction for LIHC immunotherapy, indicating that KAT2A could potentially serve as a predictive biomarker for treatment response or a therapeutic target.
Despite the comprehensive design of this study, there are several limitations that need to be addressed in future research. First, although we validated the expression pattern of KAT2A using multiple public databases, the reliance on retrospective data may introduce selection bias. Prospective, multi-center clinical cohorts are required to confirm the prognostic and diagnostic value of KAT2A in hepatocellular carcinoma. Second, the in vitro experiments were limited to three LIHC cell lines (LM3, Huh7, and PLC/PRF/5). In vivo validation through animal experiments is necessary to confirm the role of KAT2A in tumor growth and metastasis. Third, the sample size for single-cell RNA sequencing analysis was relatively small. Larger-scale single-cell datasets are needed to validate the cell type-specific expression of KAT2A and its impact on intercellular communication within the tumor microenvironment. Fourth, the molecular mechanism of the KAT2A-MYC axis remains speculative. Further studies should employ chromatin immunoprecipitation sequencing to verify whether MYC directly binds to the KAT2A promoter, and co-immunoprecipitation assays to determine whether KAT2A acetylates the MYC protein. Finally, this study did not explore the therapeutic potential of KAT2A. Future investigations should evaluate the efficacy of KAT2A inhibitors, either alone or in combination with immune checkpoint inhibitors, in LIHC models.
Based on the findings of this study, several promising research directions warrant future exploration. First, the development of KAT2A-targeted diagnostic tools could facilitate the early detection of hepatocellular carcinoma (LIHC), particularly in high-risk populations. Second, KAT2A may serve as a stratification biomarker for immune checkpoint inhibitor (ICI) therapy. Prospective trials are warranted to validate whether high KAT2A expression predicts improved treatment outcomes in LIHC patients receiving anti-PD-1/PD-L1 agents. Third, mechanistic studies should further investigate the role of KAT2A in the maturation and function of dendritic cells (DCs). Our single-cell RNA sequencing data suggest that DCs are a key cell type mediating KAT2A-related immune regulation. For instance, knocking down KAT2A expression in DCs could be employed to evaluate its impact on antigen presentation capacity and T cell activation.
Conclusion
This study demonstrates that KAT2A is a key oncogenic driver of hepatocellular carcinoma. It drives the progression of hepatocellular carcinoma by promoting malignant tumor cell behaviors, regulating critical cancer-related pathways, interacting with the MYC transcription factor, and shaping the immunosuppressive tumor microenvironment. These findings indicate that KAT2A can serve as a novel prognostic biomarker and potential therapeutic target for liver hepatocellular carcinoma, with particular significance in the field of immunotherapy. Addressing the limitations of this study through prospective clinical validation and in vivo mechanistic research is crucial for translating these findings into improved diagnosis and treatment strategies for patients.
Data availability
Data are available from the corresponding author upon reasonable request.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68 (6), 394–424 (2018).
Zheng, X. et al. Retraction Note: Forkhead box (FOX) G1 promotes hepatocellular carcinoma epithelial-Mesenchymal transition by activating Wnt signal through forming T-cell factor-4/Beta-catenin/FOXG1 complex. J. Exp. Clin. Cancer Res. 42 (1), 156 (2023).
Sharma, P. et al. The future of immune checkpoint therapy. Science 348 (6230), 56–61 (2015).
Zhu, A. X. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with Sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 19 (7), 940–952 (2018).
Han, Y. et al. PD-1/PD-L1 pathway: current researches in cancer. Am. J. Cancer Res. 10 (3), 727–742 (2020).
El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389 (10088), 2492–2502 (2017).
Qin, S. et al. Atezolizumab plus bevacizumab versus active surveillance in patients with resected or ablated high-risk hepatocellular carcinoma (IMbrave050): a randomised, open-label, multicentre, phase 3 trial. Lancet 402 (10415), 1835–1847 (2023).
Sonpavde, G. P. et al. Immune-related adverse events with PD-1 versus PD-L1 inhibitors: a meta-analysis of 8730 patients from clinical trials. Future Oncol. 17 (19), 2545–2558 (2021).
Ramos-Casals, M. et al. Immune-Related adverse events of immune checkpoint inhibitors. Ann. Intern. Med. 177 (2), ITC17–ITC32 (2024).
Kuo, M. H. et al. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 383 (6597), 269–272 (1996).
Kelly, T. J. et al. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J. Biol. Chem. 284 (30), 19945–19952 (2009).
Paolinelli, R. et al. Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat. Struct. Mol. Biol. 16 (4), 412–420 (2009).
Pai, C. C. et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat. Commun. 5, 4091 (2014).
Orpinell, M. et al. The ATAC acetyl transferase complex controls mitotic progression by targeting non-histone substrates. Embo J. 29 (14), 2381–2394 (2010).
Barlev, N. A. et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell. 8 (6), 1243–1254 (2001).
Patel, J. H. et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 24 (24), 10826–10834 (2004).
Chen, L. et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, Cyclin D1, and Cyclin E1 expression. J. Biol. Chem. 288 (20), 14510–14521 (2013).
Tong, Y. et al. KAT2A succinyltransferase activity-mediated 14-3-3ζ upregulation promotes β-catenin stabilization-dependent Glycolysis and proliferation of pancreatic carcinoma cells. Cancer Lett. 469, 1–10 (2020).
Guo, S. et al. KAT2A alleviates the glucose starvation-induced mitochondrial oxidative stress and ferroptosis to promote colon cancer progression. Apoptosis 30, 3149 (2025).
Wang, Y. et al. The LncRNA PVT1 regulates nasopharyngeal carcinoma cell proliferation via activating the KAT2A acetyltransferase and stabilizing HIF-1α. Cell. Death Differ. 27 (2), 695–710 (2020).
Guo, Y. et al. Oncogenic chromatin modifier KAT2A activates MCT1 to drive the glycolytic process and tumor progression in renal cell carcinoma. Front. Cell. Dev. Biol. 9, 690796 (2021).
Li, T. et al. TIMER: A web server for comprehensive analysis of Tumor-Infiltrating immune cells. Cancer Res. 77 (21), e108–e110 (2017).
Chandrashekar, D. S. et al. UALCAN: an update to the integrated cancer data analysis platform. Neoplasia 25, 18–27 (2022).
Tomczak, K. et al. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp. Oncol. (Pozn). 19 (1A), A68–77 (2015).
Clough, E. et al. NCBI GEO: archive for gene expression and epigenomics data sets: 23-year update. Nucleic Acids Res. 52 (D1), D138–D144 (2024).
Steen, C. B. et al. Profiling cell type abundance and expression in bulk tissues with CIBERSORTx. In: Methods in Molecular Biology. Springer, 2117135–2117157 (2020).
Wang, H. et al. Single-cell RNA sequencing reveals a reprogramming of hepatic immune cells and a protective role for B cells in MASH-driven HCC. Hepatol. Commun. 9 (5), e0668 (2025).
Hirsova, P. et al. Emerging Roles of T Cells in the Pathogenesis of Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Front. Endocrinol. (Lausanne). 12, 760860 (2021).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347 (6220), 1260419 (2015).
The Genotype-Tissue. Expression (GTEx) project. Nat. Genet. 45 (6), 580–585 (2013).
Lian, Q. et al. HCCDB: A database of hepatocellular carcinoma expression atlas. Genomics Proteom. Bioinf. 16 (4), 269–275 (2018).
Dessau, R. B. et al. ‘R--project for statistical computing. Ugeskr Laeger. 170 (5), 328–330 (2008).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951 (2019).
Jou, J. et al. The ENCODE portal as an epigenomics resource. Curr. Protoc. Bioinf. 68 (1), e89 (2019).
Feng, C. et al. KnockTF: a comprehensive human gene expression profile database with knockdown/knockout of transcription factors. Nucleic Acids Res. 48 (D1), D93–D100 (2020).
Zhang, Q. et al. hTFtarget: A comprehensive database for regulations of human transcription factors and their targets. Genomics Proteom. Bioinf. 18 (2), 120–128 (2020).
Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26 (19), 2438–2444 (2010).
Zheng, S. C. et al. A novel cell-type Deconvolution algorithm reveals substantial contamination by immune cells in saliva, buccal and cervix. Epigenomics 10 (7), 925–940 (2018).
Grabski, I. N. et al. Significance analysis for clustering with single-cell RNA-sequencing data. Nat. Methods. 20 (8), 1196–1202 (2023).
Zhang, X. et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47 (D1), D721–D728 (2019).
Kim, T. K. T test as a parametric statistic. Korean J. Anesthesiol. 68 (6), 540–546 (2015).
Fu, C. et al. Plasmacytoid dendritic cells cross-prime Naive CD8 T cells by transferring antigen to conventional dendritic cells through exosomes. Proc. Natl. Acad. Sci. U S A. 117 (38), 23730–23741 (2020).
Glaviano, A. et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer. 22 (1), 138 (2023).
Su, P. H. et al. Paired Box-1 (PAX1) activates multiple phosphatases and inhibits kinase cascades in cervical cancer. Sci. Rep. 9 (1), 9195 (2019).
Baluapuri, A. et al. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell. Biol. 21 (5), 255–267 (2020).
Funding
Affiliated Hospital of Guangdong Medical University Clinical Research Program Item: LCYJ2022B001.
Author information
Authors and Affiliations
Contributions
Z.X has made contributions in the aspects of conceptualization, formal analysis, validation, methodology, and drafting the initial version of the manuscript. J.T and J.L have contributed to validation, methodology, and writing the review and editing. S.H and J.X have made contributions in formal analysis. G.Z and J.Z have made contributions in validation, and methodology. X.T and G.L have contributed to conceptualization, validation. The data will be provided upon request.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, ZY., Tan, JH., Li, JX. et al. In vitro experiments and bioinformatic analyses implicate KAT2A in the occurrence and development of hepatocellular carcinoma. Sci Rep 16, 5737 (2026). https://doi.org/10.1038/s41598-026-36174-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-36174-1
