
Abstract
Cancer-associated fibroblasts (CAFs) are the dominant component of the tumor microenvironment (TME), which contributes to tumor progression. Aldo-keto reductase 1 family member C3 (AKR1C3) has been correlated with the development of various kinds of cancers. Nevertheless, the effect and mechanism of CAFs on AKR1C3 in cholangiocarcinoma (CCA) remain unelucidated. Q-PCR assay and IHC were conducted to detect the expression of AKR1C3 in CCA tissues. Subsequently, CCK8 assay, colony formation, crystal violet assay, apoptosis assay, glucose uptake, and lactate production assay were performed to investigate the effect of AKR1C3 on the biological function of CCA cells. The regulatory effect of CAFs on AKR1C3 was examined in CCA cells cultured with a CAF-conditioned medium. Finally, western blot, co-immunoprecipitation, and ubiquitination degradation kits were used to explore the potential molecular mechanism. AKR1C3 was overexpressed in CCA tissues compared to normal tissues. Patients with a high staining intensity score of AKR1C3 exhibited a poor overall survival time. AKR1C3 was found to enhance the proliferation, colony formation, drug resistance, and aerobic glycolysis of CCA cells. Moreover, CAFs modulate the overexpression of AKR1C3 in the onset and progression of CCA through the IL-6/STAT3 signaling pathway. This regulation occurs via the IL-6/STAT3/AKR1C3 signaling axis, indicating that targeting AKR1C3 could serve as a potential therapeutic strategy for patients with CCA.
Similar content being viewed by others


Abbreviations:
17β-HSD: 17β-hydroxysteroid dehydrogenase;
5-FU: 5-fluorouracil;
AKR1C3: Aldo-keto reductase 1 family member C3;
AR: androgen receptor;
CAFs: cancer-associated fibroblasts;
CCA: cholangiocarcinoma;
CCK8: Cell Counting Kit-8 assay;
CHX: cyclohexane;
CML: chronic myeloid leukemia;
Co-IP: Co-immunoprecipitation;
DDP: diaminodichloroplatinum;
ECAR: extracellular acidification rate;
ECL: enhanced chemiluminescence;
FBS: fetal bovine serum;
HCC: hepatocellular carcinoma;
IHC: Immunohistochemistry;
IL-6: Interleukin-6;
JAK: Janus kinase;
MAPK: mitogen-activated protein kinase;
OCR: oxygen consumption rate;
OPSCC: oropharynx squamous cell carcinoma;
PI3K/Akt: phosphatidylinositol 3 kinase/protein kinase B;
Q-PCR: Quantitative real-time reverse transcription PCR assay;
PBS: phosphate-buffered saline;
TME: tumor microenvironment.
Introduction
Cholangiocarcinoma (CCA) is a highly aggressive malignant tumor originating from the ductular epithelium of the biliary tree and can be divided into the extrahepatic type and the intrahepatic type1. Due to its anatomical location and histological characteristics, CCA is difficult to diagnose, and complete resection is challenging, which leads to a dismal survival rate and poor overall prognosis2. In addition, the worldwide incidence and mortality rates of CCA have increased over the past 20 years3. Therefore, identifying novel tumor-related molecules with practical and clinical significance is essential for improving the prognosis and therapeutic efficacy of patients with CCA.
The tumor microenvironment (TME) of CCA contains multiple molecules that participate in tumor progression4. As one of the major stromal components in TME, cancer-associated fibroblasts (CAFs) have been associated with cancer progression, drug resistance, and prognosis of CCA5. Previously, Thongchot et al. reported that CAF-derived Interleukin-6 (IL-6) led to unfavorable prognosis in CCA by affecting autophagy-associated chemoresponse6. Furthermore, Kittirat et al. indicated that CAFs promoted cell viability and enhanced gemcitabine resistance in CCA cells through the activation of IL-6/STAT3 signaling7. Collectively, CAFs might provide an effective strategy to diagnose and treat CCA.
Aldo-keto reductase 1 family member C3 (AKR1C3), also known as type 5, 17β-hydroxysteroid dehydrogenase (17β-HSD), belongs to the human aldo‑keto reductase type 1 C (AKR1C) family and is composed of four enzymes including AKR1C1-48. AKR1C3 is intimately linked with multiple cellular functions, such as the metabolism and biosynthesis of prostaglandin, steroid hormone, and retinoid metabolism9. Recent studies have revealed the vital role of AKR1C3 in cancer biology, leading to proliferation, invasion, migration, and drug resistance of tumor cells10. AKR1C3 has been shown to be significantly upregulated in multiple types of cancers, including hepatocellular carcinoma (HCC)11,12, oropharynx squamous cell carcinoma (OPSCC)13, chronic myeloid leukemia (CML)14, prostate cancer15, and choriocarcinoma16. Nevertheless, the effect and mechanism of CAFs on AKR1C3 in CCA remain poorly understood.
The current study aimed to investigate the expression of AKR1C3 in CCA tissues. AKR1C3 overexpression effectively enhanced the proliferation, colony formation, drug resistance, and aerobic glycolysis of CCA cells. Moreover, in vitro and in vivo experiments revealed that CAFs regulate the occurrence and development of CCA via the IL-6/STAT3/AKR1C3 signaling axis. Collectively, AKR1C3 might be a potential target for the diagnosis and treatment of CCA.
Materials and methods
Reagents
RPMI-1640, fetal bovine serum (FBS), penicillin-streptomycin, and phosphate-buffered saline (PBS) were purchased from Gibco (Carlsbad, CA, USA). 5-fluorouracil (5-FU), diaminodichloroplatinum (DDP), and Ab-IL-6 were obtained from MedChemExpress (Monmouth Junction, NJ, USA). Cyclohexane (CHX) was sourced from Sigma-Aldrich (Indianapolis, IN, USA). AKR1C3 knockdown and overexpression lentiviruses were bought from PPL (Public Protein/Plasmid Library, China). Antibodies against P-STAT3 (9145 S), T-STAT3 (30835 S), PCNA (2586 S), P-GP (13879 S), GLUT-1 (73015 S) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against β-tubulin (ab78078), SOCS1 (ab280886), and AKR1C3 (ab316848) were obtained from Abcam Ltd. (Cambridge, United Kingdom). Antibodies against SOCS3 (68631 S) and PFK-1 (55028-1-AP) were obtained from Proteintech. (Hubei, China). Goat anti-rabbit and anti-mouse secondary antibodies were purchased from Merck Millipore (Billerica, MA, USA). Co-immunoprecipitation (Co-IP) and apoptosis assay kits were bought from Promega (Beijing, China). The ubiquitination degradation kit was sourced from Abcam Ltd. (Cambridge, United Kingdom). The study was approved by the Ethics Committee of Fujian Medical University. Furthermore, all methods were conducted in accordance with relevant guidelines and regulations.
Cell culture and stable cell lines
Six human CCA cell lines (Sk-Cha-1, HuH28, RBE, TFK1, Mz-Cha-1, and QBC939) of different origins were donated from the relevant laboratories of the School of Life Sciences and School of Medicine, Xiamen University. HuH28 and RBE cells were derived from human intrahepatic bile ducts, while Sk-Cha-1, TFK1, and QBC939 cells originated from extrahepatic bile ducts. Mz-Cha-1 was cultured from gallbladder adenocarcinoma metastases that were mechanically dissociated. All human CCA cell lines were cultured in RPMI-1640 medium containing 10% FBS and 1% penicillin-streptomycin in humidified 5% CO2 at 37˚C. In addition, normal human intrahepatic biliary epithelial cells (HBd Epi, cat. no. HUM-CELL-0035; PriCells Biomedical Technology Co., Ltd., Wuhan, China) were cultured in complete PriCells epithelial cell medium (cat. no. MED-0001; PriCells Biomedical Technology Co., Ltd., Wuhan, China) according to the manufacturer’s protocol. Thereafter, QBC939 and TFK1 cells were transfected with AKR1C3 knockdown lentiviruses, and QBC939 cells were transfected with overexpression-AKR1C3 lentiviruses to construct downregulated and upregulated AKR1C3 CCA cells. Then, puromycin was used to screen stable infection cells, which were confirmed by Q-PCR and Western blot analysis.
The isolation and culture of CAFs
Primary CAFs were isolated from fresh cholangiocarcinoma tissues using the tissue explant outgrowth method with minor modifications. Tumor specimens were obtained with written informed consent from patients undergoing curative resection at the First Affiliated Hospital of Xiamen University. The study was approved by the Institutional Ethics Committee of the First Affiliated Hospital of Xiamen University (Approval number: XMYY-2022KYSB003) and conducted in accordance with the Declaration of Helsinki. Fresh tissues were immediately placed in ice-cold PBS containing 5% penicillin-streptomycin and transported to the laboratory within 1 h. Necrotic, fatty, and normal-appearing tissues were removed under a sterile biosafety cabinet. Tumor tissues were washed 5–6 times with PBS containing 5% penicillin-streptomycin, minced into 1–2 mm³ fragments, and the fragments were evenly seeded into 10 cm cell culture dishes. After 30 min attachment at 37 °C in 5% CO₂, 10 mL of CAF complete medium (DMEM supplemented with 20% FBS and 1% penicillin-streptomycin) was gently added. Dishes were left undisturbed for 5–7 days to allow fibroblast outgrowth. When cells reached 70–80% confluence, tissue fragments were removed, and fibroblasts were passaged using 0.25% trypsin-EDTA at a 1:3 ratio. CAF identity was confirmed by immunofluorescence staining. Cells were positive for α-SMA (14395-1-AP, Proteintech, rabbit polyclonal, 1:200) and negative for the epithelial marker CK19 (10712-1-AP, rabbit polyclonal, Proteintech, 1:100). Only α-SMA+/CK19- cultures were used as validated CAFs. Primary CAFs were successfully established from tumors of six CCA patients. Three independent isolates exhibiting robust and stable growth (passages 3–6) were selected for all functional experiments, and key results were verified in at least two independent isolates. For preparation of CAF-conditioned medium, CAFs at 90% confluence were washed twice with PBS and incubated in serum-free DMEM for 24–48 h. The supernatant was collected, centrifuged at 1000 ×g for 5 min to remove debris, and used immediately or stored at −80 °C.
Cell counting Kit-8 assay (CCK8)
Cell viability and drug susceptibility were evaluated using a CCK8 assay. The cell lines were seeded in 96 well plates and incubated at 37 °C in humidified 5% CO2 for 1, 2, 3, 4, or 5 days. Over the course of the experiment, 10 µl CCK8 was added to each well at each time point for 2 h at 37 °C. The optical density was detected at 450 nm using a microplate reader (model 680, Bio-Rad, Hercules, CA, USA). For the drug susceptibility assay, cells were treated with 5-FU or DDP for 48 h at the beginning of the cell culture.
Colony formation assay
Cells were seeded into 6-well plates at 1 × 103 cells per well in RPMI 1640 containing 10% FBS for 14 days to allow colony formation. The colonies were fixed using 4% paraformaldehyde and stained with 0.1% crystal violet for 30 min. After washing with PBS, the colonies were counted by visual inspection.
Crystal Violet assay
Cell survival and cell colony after drug treatment were assessed using the Crystal Violet Assay. For cell survival, the cells in the logarithmic growth phase were digested and inoculated into a 3.5 cm plate. After 5-FU treatment for 48 h, the cells were collected and incubated into the 48-well plate for 6 h. For cell colony, the cells were incubated into a 6-well plate for about 1 week. Subsequently, the cells were fixed with methanol for 15 min and stained with crystal violet for 15 min.
Glucose uptake and lactate production assay
Cells were cultured into 6-well plates overnight to prepare for the glucose uptake assay. The cells were then exposed to different media, namely low glucose medium, no glucose medium, and no glucose medium containing 2.5 µg/mL 2NBDG. The fluorescence value was measured using a flow cytometer. Lactate production was detected by measuring the amount of lactate in the media using a lactate colorimetric/fluorometric assay kit (Biovision, K607-100) following the manufacturer’s instructions.
Cell bioenergy tests
The extracellular acidification rate (ECAR) assay and oxygen consumption rate (OCR) assay were measured using Seahorse XFe96 Extracellular Flux Analyzer (Agilent, Palo Alto, CA, USA) according to the manufacturer’s instructions.
Apoptosis assay
CCA cells were cultured in 6-cm Petri dishes and were treated with 40 µM 5-FU for 48 h. Afterward, cells were trypsinized and stained with annexin V-FITC and PerCP according to the instructions of the AnnexinV-FITC apoptosis detection kit.
Immunohistochemistry (IHC)
The tissue microarray, which consisted of CCA tissues (n = 91) and paired paratumor tissue (n = 31) sections, was purchased from Shanghai Outdo Biotech Company (Cat No. HBiDC122Su01). The samples were deparaffinized with xylene, rehydrated in graded alcohol solution, and antigen-repaired with citric acid buffer under high temperature and high pressure; the endogenous peroxidase was quenched with 3% H2O2. Potential nonspecific binding was blocked using 1% BSA for 1 h at room temperature. Then, TMA was incubated with a primary antibody against AKR1C3, followed by treatment with secondary antibodies and DAB according to the manufacturer’s instructions (Maixin Biotech, Fuzhou, China). The staining frequency of AKR1C3 was semiquantitatively scored based on the percentages of positive cells, 0% = negative; 1–25% = +1; 26–50% = +2; and > 50% = +3. The low group included scores from - to ++, whereas the high group encompassed scores from + + to +++. All sections were reviewed independently and without bias by two expert pathologists.
Quantitative real-time reverse transcription PCR assay (Q-PCR)
TRIzol Reagent was used to extract total RNA from CCA cells or tissues, and the cDNA was reverse transcribed according to the manufacturer’s guidelines. Quantitative real-time polymerase chain reaction (PCR) was conducted using A SYBR Green PCR kit (TianGen, Beijing, China), and the samples were detected in 96-well plates with the ABI 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The primers for the qPCR reactions are listed in Supplemental Table 1.
Western blot analysis
Western blot analysis was conducted according to the manufacturer’s guidelines. Equal concentrations of protein were transferred onto a PVDF membrane and then incubated with the primary antibodies. Subsequently, the proteins were incubated with HRP-conjugated secondary antibodies, followed by enhanced chemiluminescence (ECL) detection. Western blot bands have been semi-quantified by densitometry using ImageJ software (NIH, Bethesda, MA, USA).
Co-immunoprecipitation (Co-IP)
Total proteins were extracted in RIPA lysis buffer containing a protease/phosphatase inhibitor. Then, the cell lysate samples were precipitated with anti-Flag or anti-HA antibodies according to the manufacturer’s guidelines. After washing with ice-cold PBS three times to remove unbound antibodies, the bound proteins were analyzed by western blot.
Ubiquitination degradation kit
For the ubiquitination assay, shCtrl-QBC939 and shAKR1C3-QBC939 cells were treated with 50 µM cyclohexane (CHX) at different times. The cells were lysed in Cell Lysis Buffer for western blot according to the manufacturer’s guidelines.
Tumor xenografts
shCtrl-QBC939, shAKR1C3-QBC939, and CAFs cells were expanded and cultured under the above conditions. QBC939 and CAF cells were mixed and injected at a 2:1 ratio. A sufficient number of cultured QBC939 cells and CAF cells were digested, counted, and added to the basic medium. After alcohol disinfection, 100µL mixed cell suspension was injected subcutaneously into the right groin of nude mice. All animals were housed in specific pathogen-free conditions at the Laboratory Animal Center of Xiamen University. All animal experiments were performed in accordance with the animal study protocols approved by the Animal Care and Use Committee of Xiamen University. All experimental procedures were done in compliance with the ARRIVE guidelines (PLoS Bio 8(6), e1000412,2010). Nude mice were originally obtained from the Cyagen Biosciences (Guangzhou) incorporated company and were maintained in the Laboratory Animal Center of Xiamen University. The drug sensitivity test was performed when the tumor nodule size reached 100 mm3. 5-FU (40 mg/kg) was injected into the abdomen every three days. The mental state, activity, growth state, and injection site of nude mice were monitored every day. Tumor formation was observed within one week after injection. The length and diameter of the tumor were measured and recorded with vernier calipers every 2 days, and the mice were weighed. When the tumor size reached 1000–1500 mm3, the mice were euthanized by CO2 and sacrificed through cervical dislocation. Then the xenografts were weighed and photographed.
Statistical analyses
All data were analyzed using SPSS 21.0 software (SPSS Inc., Chicago, IL, USA). The measurement data were expressed as mean ± standard deviation. Comparisons between two groups or multiple groups were conducted by t-test or one-way analysis of variance, respectively. The survival rate was calculated by the Kaplan-Meier method. In this study, P value < 0.05 was considered significant.
Results
Overexpression and clinical significance of AKR1C3 in CCA
The GEPIA database was used to investigate the relationship between AKR1C3 expression and CCA. The results revealed that AKR1C3 mRNA was upregulated in several cancer tissues compared to the corresponding normal tissues, including CCA (Fig. 1A). Furthermore, AKR1C3 protein expression was detected in CCA tissues by IHC. TMA demonstrated a significantly elevated expression of AKR1C3 in CCA tissues compared to normal tissues (Fig. 1B). Moreover, the Q-PCR results also showed that the mRNA expression of AKR1C3 was significantly increased in CCA tissues (C) compared with the matched adjacent normal tissues (N) of 13 CCA patients (Fig. 1C, P = 0.0029). These results indicated AKR1C3 upregulation in CCA tissues.
Additionally, the clinical significance of AKR1C3 in CCA was explored. As shown in Table 1, the staining intensity score of AKR1C3 protein showed a significant difference between different bile duct tissues, including CCA (C), paracancer tissues (P), and normal tissues (N) (Fig. 1D, P = 0.0001). Meanwhile, patients with a high AKR1C3 staining intensity score exhibited a shorter overall survival time (Fig. 1E, P = 0.0063). In addition, compared with normal tissues, the area under the curve (AUC) of AKR1C3 expression in the CCA patient cohort was 0.6976 (Fig. 1F, P = 0.0009). Clinicopathological analysis revealed that AKR1C3 overexpression was strongly correlated with tumor size, N stage, and TNM stages in CCA patients (Table 2).
In univariate Cox regression analysis, high AKR1C3 expression was significantly associated with poorer overall survival (HR = 1.80, 95% CI = 1.25–2.60, P = 0.002). Tumor size ≥ 4 cm (HR = 2.00, 95% CI = 1.30–3.07, P = 0.0015), advanced T category (T3/4 vs. T1/2; HR = 2.30, 95% CI = 1.45–3.64, P = 0.0005), positive nodal status (N1 vs. N0; HR = 2.50, 95% CI = 1.62–3.85, P < 0.0001) and higher TNM stage (III + IV vs. I + II; HR = 2.10, 95% CI = 1.40–3.16, P = 0.0004) were all also significantly correlated with worse survival. Age ≥ 60 years showed a non-significant trend toward poorer outcome (HR = 1.40, 95% CI = 0.95–2.08, P = 0.087), and neither gender nor histologic grade reached significance. In multivariate analysis, high AKR1C3 expression remained an independent predictor of reduced overall survival (HR 1.50; 95% CI 1.05–2.15; P = 0.025), together with tumor size ≥ 4 cm (HR 1.75; 95% CI 1.12–2.74; P = 0.015), T3/4 category (HR 1.90; 95% CI 1.18–3.05; P = 0.008), N1 stage (HR 2.10; 95% CI 1.35–3.26; P = 0.0012) and stage III/IV disease (HR 1.85; 95% CI 1.22–2.80; P = 0.0035) (Table 3).

Expression and clinical significance of AKR1C3 in CCA. (A) The mRNA expression levels of AKR1C3 in several cancer tissues such as CCA were analyzed from the GEPIA database. (B) Expression of AKR1C3 protein was identified by IHC on CCA tissue microarrays. (C) The expression levels of AKR1C3 mRNA in C and N tissues were assessed by Q-PCR. C, cancer; P, paracarcinoma tissues. (D) Staining intensity score of AKR1C3 protein in different bile duct tissues. T, tumor; P, paracancer tissues; N, normal tissues. (E) Kaplan–Meier survival analysis of AKR1C3 expression in CCA patients. (F) The receiver operating characteristic (ROC) curve of AKR1C3.
AKR1C3 promotes the proliferation and colony formation of CCA cells
The expression level of AKR1C3 was evaluated in six CCA cell lines (Sk-Cha-1, HuH28, RBE, TFK-1, Mz-Cha-1, and QBC939) and a normal biliary epithelial cell line HBd Epi to further confirm its aberrant expression. Significantly higher mRNA and protein expression levels of AKR1C3 were observed in six CCA cell lines compared to those of HBd Epi, as shown in Fig. 2A. Next, AKR1C3 was downregulated or upregulated by stably transducing with shAKR1C3-expressing or overexpression-AKR1C3 lentiviruses to determine the biological functions of AKR1C3 in CCA development. As shown in Fig. 2B, the protein and mRNA expression levels of AKR1C3 were decreased in shAKR1C3 CCA cells (QBC939 and TFK1) and increased in OE-AKR1C3 QBC939 cells compared to control cells. Interestingly, the CCK8 results revealed significantly inhibited proliferative capacities of QBC939 and TFK1 cells after AKR1C3 knockdown (Fig. 2C). In contrast, overexpression of AKR1C3 in QBC939 cells significantly increased cell survival. Moreover, the colony formation capacities of CCA cells were found to be markedly decreased by stable knockdown of AKR1C3 and were potently enhanced by overexpression (Fig. 2D). These results suggested that AKR1C3 promoted the proliferation and colony formation of CCA cells.

The effect of aberrant expression of AKR1C3 on the proliferation and colony formation of CCA cells. (A) The mRNA and protein expressions of AKR1C3 in six CCA cell lines (Sk-Cha-1, HuH28, RBE, TFK-1, Mz-Cha-1, and QBC939) and a normal biliary epithelial cell line HBd Epi were detected by Q-PCR and Western blot, respectively. (B) AKR1C3 knockdown and overexpression CCA cell models were measured by Q-PCR and Western blot. (C) Cell viability of cholangiocarcinoma (CCA) cell lines following AKR1C3 knockdown was assessed at 24, 48, 72, 96, and 120 h using CCK8 assays. Similarly, CCA cell line viability after AKR1C3 overexpression was evaluated at 24, 48, 72, and 96 h utilizing the same CCK8 assay methodology. (D) Colony formation assays were conducted after AKR1C3 knockdown and overexpression in CCA cells. The relative number of colonies was quantified.
AKR1C3 mediates drug resistance in CCA cells
The effect of AKR1C3 knockdown on the drug susceptibility of CCA cells was investigated in vitro. The results of CCK-8 assays indicated that AKR1C3 knockdown markedly enhanced the drug susceptibility to 5-FU and DDP of shAKR1C3-transfected cells compared to shCtrl-transfected cells in both QBC939 (Fig. 3A) and TFK1 lines (Fig. 3B). Consistent with this enhanced sensitivity, quantification of the dose-response curves revealed significantly lower IC50 values for both 5-FU and DDP in AKR1C3-knockdown cells compared to control cells in both cell lines (Supplementary Fig. 1). In addition, the cell survival and colony formation showed a significant decrease in QBC939 cells treated with different concentrations of 5-FU after AKR1C3 knockdown compared with shCtrl cells (Fig. 3C-D). As shown in Fig. 3E, AKR1C3 knockdown enhanced the apoptosis of QBC939 cells and TFK1 cells induced by 5‑FU.

Effect of altered AKR1C3 on the drug susceptibility of CCA cells. CCK-8 assay was performed in shAKR1C3-transfected or shCtrl-transfected QBC939 cells (A) and TFK1 cells (B) to evaluate the drug susceptibility to 5-FU and DDP. * P < 0.05; ** P < 0.01. Cloning formation assay was performed in QBC939 cells (C) and TFK1 cells (D) after shAKR1C3 knockdown treated with different concentrations of 5‑FU. (E) The proportion of apoptotic shAKR1C3 and shCtrQBC939 cells and TFK1 cells following treatment with 40 µM 5‑FU. * P < 0.05; ** P < 0.01; **** P < 0.001;**** P < 0.0001.
AKR1C3 enhances aerobic Glycolysis in CCA cells
Furthermore, the effect of AKR1C3 on aerobic glycolysis of CCA cells was evaluated. Lower concentrations of glucose and lactate were observed in the culture medium of QBC939 and TFK1 cells after AKR1C3 knockdown compared with those of control cells, as shown in Fig. 4A and B. Moreover, the extracellular acidification rate (ECAR) assay revealed that steady-state glycolysis flux and glycolytic capacity were attenuated in QBC939 and TFK1 cells following AKR1C3 knockdown (Fig. 4C). The oxygen consumption rate (OCR) assay also indicated that AKR1C3 knockdown decreased the basal respiration and ATP production of QBC939 and TFK1 cells (Fig. 4D). Hence, these results revealed that AKR1C3 enhanced aerobic glycolysis in CCA cells.

Effect of altered AKR1C3 on the glycolysis of CCA cells. The glycolysis levels of QBC939 and TFK1 cells after AKR1C3 knockdown were assessed by glucose uptake (A), lactate release (B), extracellular acidification rate (ECAR) (C), and oxygen consumption rate (OCR) (D). * P < 0.05; ** P < 0.01.
AKR1C3 promotes malignant biological behavior of CCA by activating the STAT3 signaling pathway
To investigate the molecular mechanisms underlying the AKR1C3 aberrant expression in CCA, western blot analysis was performed to detect the expression of proliferation, invasion, drug resistance, glycolysis, and STAT3 signaling-associated proteins. The results indicated that AKR1C3 knockdown decreased the protein expression levels of P-STAT3, PCNA, MMP-7, MMP-9, P-GP, GLUT-1, and PFK-1 in QBC939, which were effectively reversed by IL-6 treatment (Fig. 5A). Furthermore, the expression of the SOCSs family, which belong to STAT3 signaling pathway inhibitors, was detected to study the potential mechanism. As shown in Fig. 5B, the mRNA expression levels of the SOCSs family exhibited no significant differences in QBC939 cells after AKR1C3 knockdown. However, the SOCS1 protein expression showed a marked increase following stable knockdown of AKR1C3 and was potently suppressed by overexpression of AKR1C3 (Fig. 5C). Moreover, AKR1C3 knockdown inhibited the ubiquitination degradation of SOCS1 in QBC939 cells (Fig. 5D). As shown in Fig. 5E, AKR1C3 interacted with the SOCS1 protein in QBC939 cells. These results indicated that AKR1C3 regulated the malignant biological behavior of CCA via the activation of the STAT3 signaling pathway.

The molecular mechanism of STAT3 signaling pathway regulated by AKR1C3 in CCA. (A) After treatment with IL-6, the expression levels of proliferation, invasion, drug resistance, glycolysis, and STAT3 signaling-associated proteins were detected by Western blot in QBC939 cells. IL-6, 30 ng/ml. (B) The mRNA expression levels of the SOCSs family in QBC939 cells after AKR1C3 knockdown were detected by Q-PCR. * P < 0.05 (C) The protein expression levels of SOCS1 and SOCS3 in CCA cells after AKR1C3 knockdown and overexpression were measured by Western blot. (D) Western blot was performed to assess the ubiquitination degradation of SOCS1 in shAKR1C3- or shCtrl-transfected QBC939 cells treated with 50 µM cyclohexane (CHX) at different times. (E) Co-IP assay was conducted to investigate the protein interaction between AKR1C3 and SOCS1 in QBC939 cells.
Molecular mechanism of CAFs regulated the occurrence and development of CCA via the IL-6/STAT3/AKR1C3 signaling axis
The tumor microenvironment (TME) plays an essential role in tumor progression, and CAFs constitute the most essential components of the TME. Primary CAFs used in this study were validated by α-SMA-positive and CK19-negative immunofluorescence staining (Supplementary Fig. 2). Therefore, the effect of CAFs on CCA was assessed in this study. As shown in Fig. 6A, CAFs enhanced the mRNA expression levels of AKR1C3 in QBC939 and TFK1 cells. Moreover, CAFs increased the expression of AKR1C3 and P-STAT3 in QBC939 cells in a time-dependent manner at the protein level (Fig. 6B), which was mechanistically linked to CAFs’ time-dependent IL-6 secretion (Supplementary Fig. 3). As shown in Fig. 6C, the high expression of AKR1C3 and P-STAT3 mediated by CAFs was inhibited by Ab-IL-6, indicating that CAFs might secrete IL-6 to increase AKR1C3 protein expression, thereby activating the STAT3 signaling pathway. STAT3 inhibition with Stattic suppressed P-STAT3 and AKR1C3 expression (Supplementary Fig. 4). Moreover, CAFs were found to enhance the protein expression levels of PCNA, P-GP, GLUT-1, and PFK-1 in QBC939 cells, which were effectively reversed by Ab-IL-6 and AKR1C3 knockdown (Fig. 6D). In addition, CAFs were found to accelerate proliferation (Fig. 6E), glucose uptake (Fig. 6F, left), lactate release (Fig. 6F, right), cell colony (Fig. 6G) in CCA cells; however, these changes were markedly reduced after Ab-IL-6 treatment and downregulation of AKR1C3, respectively. These findings suggested that CAFs regulated the occurrence and development of CCA via the IL-6/STAT3/AKR1C3 signaling axis.

CAFs regulated the occurrence and development of CCA by activating the AKR1C3/STAT3 signaling axis. (A) The mRNA expression levels of AKR1C3 in QBC939 and TFK1 cells after co-incubation with CAFs were detected by Q-PCR. ** P < 0.01. (B) The protein expression levels of AKR1C3, P-STAT3, and T-STAT3 in QBC939 cells after co-incubation with CAFs at different times were measured by Western blot. (C) After CAF treatment with or without Ab-IL-6 and (or) AKR1C3 knockdown for 8 h, the expression of AKR1C3, P-STAT3, and T-STAT3 were detected by Western blot in QBC939 cells. (D) After treatment of CAFs with or without Ab-IL-6 and (or) AKR1C3 knockdown for 8 h, the expression of PCNA, P-GP, GLUT-1, and PFK-1 were detected by Western blot in QBC939 cells. The proliferation (E) and glycolysis levels (F) in QBC939 and TFK1 cells after treatment of CAFs with or without Ab-IL-6 and (or) AKR1C3 knockdown were assessed by CCK-8, glucose uptake, and lactate release, respectively. * P < 0.05, ** P < 0.01, *** P < 0.001. The cell colony (G) of QBC939 cells exposed to 40 µM 5-FU after treatment of CAFs with or without Ab-IL-6 and (or) AKR1C3 knockdown were detected by crystal violet staining. Ab-IL-6, 2ug/ml.
CAFs regulated the occurrence and development of CCA via the IL-6/STAT3/AKR1C3 signaling axis in vivo
The tumorigenic potential of AKR1C3 was further evaluated by establishing a xenograft tumor model in nude mice. As shown in Fig. 7A, the growth of shAKR1C3-QBC939 xenografts was slower than that of the shCtrl group. In addition, the mixed xenografts of CAFs and shAKR1C3-QBC939 cells indicated significantly slower growth and higher sensitivity to 5-FU than those of the control group (Fig. 7B). Higher CAF infiltration and higher expression levels of AKR1C3 and PCNA were observed in the CAF-QBC939 xenografts compared with the QBC939 xenografts (Fig. 7C). Additionally, significant reductions in AKR1C3, P-STAT3, PCNA, P-GP, GLUT-1, and PFK-1 were observed in the CAFs-shAKR1C3-QBC939 xenografts with or without the treatment of 5-FU (Fig. 7D). As shown in Fig. 7E, these results suggested that CAFs regulated the occurrence and development of CCA via the IL-6/STAT3/AKR1C3 signaling axis in vivo.

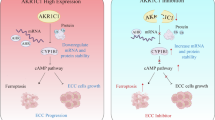
CAFs regulated the occurrence and development of CCA via the IL-6/STAT3/AKR1C3 signaling axis in vivo. (A) shAKR1C3-QBC939 cells and control cells were subcutaneously injected in nude mice to establish xenograft tumors. The representative tumors and their volume are depicted graphically. (B) The growth and sensitivity to 5-FU were detected in the mixed xenografts of CAFs and shAKR1C3-QBC939 cells. (C) HE and IHC staining were performed to examine CAF-QBC939 and QBC939 xenografts. (D) The protein expressions of AKR1C3, P-STAT3, PCNA, P-GP, GLUT-1, and PFK-1 in the CAFs-shAKR1C3-QBC939 xenografts with or without the treatment of 5-FU were detected by Western blot. (E) Schematic summary illustrating how CAF-derived IL-6 activates the STAT3/AKR1C3 axis in cholangiocarcinoma cells to drive tumor proliferation, chemoresistance, glycolysis, and metastatic potential.
Discussion
In recent years, a growing number of studies have reported the role of AKR1C3 as a marker of poor prognosis in various cancers, including HCC17. However, the relationship between AKR1C3 and CCA remains unknown. The present study revealed that AKR1C3 mRNA was elevated in CCA compared to the corresponding normal tissues from the GEPIA database, which was consistent with our Q-PCR results using fresh CCA tissues. Moreover, the protein expression levels of AKR1C3 were upregulated in CCA tissues compared to normal tissues. Notably, patients with a high AKR1C3 staining intensity score had a shorter overall survival time. Furthermore, clinicopathological analysis revealed that overexpression of AKR1C3 was strongly correlated with tumor size, N stage, and TNM stages in CCA patients. In addition, AKR1C3 enhanced the proliferation, colony formation, and drug resistance of CCA cells. These findings lend support to AKR1C3 being a vital oncogene in CCA.
Cancer is characterized by metabolic reprogramming, which enables tumor cells to fulfill the heightened energy needs necessary for quick growth, invasion, and spread. Similar to other tumors, CCA heavily relies on glucose metabolism. The shift from oxidative phosphorylation to glycolysis in CCA supports cancer cell growth and creates a beneficial environment for tumor development18. Changes in metabolic programming in CCA cells are intricate and might happen through mutations and epigenetic changes in oncogenes, tumor suppressor genes, signaling pathways, and glycolytic enzymes19. In this study, we found that AKR1C3 enhances aerobic glycolysis in CCA cells.
In terms of mechanism, several studies revealed the regulatory role of AKR1C3 in various cancers, which was mainly mediated by activating the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt) signal pathway. Zhu et al. discovered that AKR1C3 knockdown suppressed the MEK/ERK signaling pathways in HCC cells12. Notably, suppression of AKR1C3 or overexpression of AKR1D1 resulted in a significant reduction of androgen receptor (AR) and phosphorylated ERK1/2 levels. Consequently, AKR1C3 and AKR1D1 may serve as candidate diagnostic and prognostic biomarkers for HCC and offer potential therapeutic targets for its treatment. Wang et al. reported that PGD2 notably increased the expression of total Akt protein and Akt Ser473 phosphorylation in AKR1C2 and AKR1C3 stable transfectants. The use of a PI3K inhibitor (LY294002) reduced PGD2-induced cell proliferation in these transfectants. These findings indicate that AKR1C2 and AKR1C3 facilitate similar PGD2 conversion, leading to the buildup of proliferative signals via FP and PI3K/Akt pathways to enhance prostate cell growth20.
IL-6-mediated signal transducers and activators of transcription 3 (STAT3) activation were aberrantly sustained in various cancers21,22. Briefly, IL-6 can bind to the IL-6 receptor and trigger the related Janus kinase (JAK), stimulate phosphorylation, and then activate STAT3 to regulate downstream signals, which are involved in proliferation, invasion, metastasis, and drug resistance of tumor cells23. In recent years, a growing body of research has revealed that IL-6/STAT3 signaling plays a significant role in the development of CCA24. For example, Leng et al. revealed that Akirin2 was modulated by miR-490-3p and facilitated angiogenesis in CCA through the IL-6/STAT3/VEGFA signaling pathway25. Furthermore, Zheng et al. showed that gankyrin promoted tumor growth and metastasis by activating IL-6/STAT3 signaling in human CCA26. To uncover the mechanism underlying the effects of AKR1C3 on the malignant biological behavior of CCA cells, the involvement of IL-6/STAT3 was detected in CCA. Our findings indicated that AKR1C3 interacted with the SOCS1 protein in QBC939 cells, suggesting that AKR1C3 may enhance the malignant biological behavior of CCA through the activation of the STAT3 signaling pathway.
In addition, the TME plays a crucial role in tumor progression, and CAFs are the most essential components of the TME27. CAFs regulate the biology of tumor cells and other stromal cells by cell-cell contact, thereby affecting the initiation and development of cancer28. Clinical evidence has predominantly substantiated the tumor-promoting role of CAFs in CCA, highlighting the growing interest in further exploring this cellular population for therapeutic applications. Notably, a high abundance of α-SMA-positive CAFs has been correlated with increased tumor size, enhanced proliferation, and decreased survival rates in patients. Furthermore, recent research indicates that CAFs, in conjunction with other stromal components, may aid in the stratification of CCA patients29. Factors derived from CAFs that are involved in tumor development include TGFbeta, LIF, CXCL12, IL6, IGF1, and the MAPK and STAT3 signaling pathways. Our results indicated that CAFs regulated the occurrence and development of CCA via the IL-6/STAT3/AKR1C3 signaling axis in vitro and in vivo.
While these findings offer intriguing insights, our study is not without limitations. The mRNA expression levels of AKR1C3 are significantly higher in TFK1 and QBC939 cells compared to the other four CCA cell lines. As a result, TFK1 and QBC939 cells were chosen for further analysis. Notably, given that both TFK1 and QBC939 cells originate from extrahepatic bile ducts, it is crucial to validate the key findings across a broader range of cell lines to strengthen our conclusions.
In summary, our study underlined the importance of AKR1C3 in the progress of CCA. AKR1C3 was overexpressed in CCA tissues, which promoted the malignant biological behavior of CCA cells by activating the STAT3 signaling pathway. Furthermore, CAFs regulated the occurrence and development of CCA via the IL-6/STAT3/AKR1C3 signaling axis. Hence, AKR1C3 might be a potential biomarker and target molecule for CCA therapy.
Data availability
Raw data are available upon request to the corresponding author.
References
Razumilava, N., Gores, G. J. & Cholangiocarcinoma Lancet 383, 2168–2179, doi:https://doi.org/10.1016/s0140-6736(13)61903-0 (2014).
Blechacz, B. & Cholangiocarcinoma Current knowledge and new developments. Gut Liver. 11, 13–26. https://doi.org/10.5009/gnl15568 (2017).
Brindley, P. J. et al. Cholangiocarcinoma. Nat. Rev. Dis. Primers. 7, 65. https://doi.org/10.1038/s41572-021-00300-2 (2021).
Ying, F., Chan, M. S. M. & Lee, T. K. W. Cancer-Associated fibroblasts in hepatocellular carcinoma and cholangiocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 15, 985–999. https://doi.org/10.1016/j.jcmgh.2023.01.006 (2023).
Montori, M. et al. Cancer-Associated fibroblasts in cholangiocarcinoma: current knowledge and possible implications for therapy. J. Clin. Med. 11 https://doi.org/10.3390/jcm11216498 (2022).
Thongchot, S. et al. Cancer-Associated Fibroblast-Derived IL-6 determines unfavorable prognosis in cholangiocarcinoma by affecting Autophagy-Associated chemoresponse. Cancers (Basel). 13. https://doi.org/10.3390/cancers13092134 (2021).
Kittirat, Y. et al. Interleukin-6-derived cancer-associated fibroblasts activate STAT3 pathway contributing to gemcitabine resistance in cholangiocarcinoma. Front. Pharmacol. 13, 897368. https://doi.org/10.3389/fphar.2022.897368 (2022).
Chen, S. et al. Magnolol suppresses pancreatic cancer development in vivo and in vitro via negatively regulating TGF-β/Smad signaling. Front. Oncol. 10, 597672. https://doi.org/10.3389/fonc.2020.597672 (2020).
Penning, T. M. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): roles in malignancy and endocrine disorders. Mol. Cell. Endocrinol. 489, 82–91. https://doi.org/10.1016/j.mce.2018.07.002 (2019).
Xiao, X. Z., Lin, L. Y., Zhuang, M. K., Zhong, C. M. & Chen, F. L. Roles of AKR1C3 in malignancy. Chin. Med. J. (Engl). 134, 1052–1054. https://doi.org/10.1097/cm9.0000000000001379 (2021).
Pan, D. et al. AKR1C3 regulated by NRF2/MAFG complex promotes proliferation via stabilizing PARP1 in hepatocellular carcinoma. Oncogene 41, 3846–3858. https://doi.org/10.1038/s41388-022-02379-7 (2022).
Zhu, P. et al. Diagnostic and prognostic values of AKR1C3 and AKR1D1 in hepatocellular carcinoma. Aging (Albany NY). 13, 4138–4156. https://doi.org/10.18632/aging.202380 (2021).
Peraldo-Neia, C. et al. AKR1C3 is a biomarker and druggable target for oropharyngeal tumors. Cell. Oncol. (Dordr). 44, 357–372. https://doi.org/10.1007/s13402-020-00571-z (2021).
Pan, D. et al. AKR1C3 decreased CML sensitivity to Imatinib in bone marrow microenvironment via dysregulation of miR-379-5p. Cell. Signal. 84, 110038. https://doi.org/10.1016/j.cellsig.2021.110038 (2021).
Adeniji, A. O., Chen, M. & Penning, T. M. AKR1C3 as a target in castrate resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 137, 136–149. https://doi.org/10.1016/j.jsbmb.2013.05.012 (2013).
Zhao, J. et al. AKR1C3 overexpression mediates methotrexate resistance in choriocarcinoma cells. Int. J. Med. Sci. 11, 1089–1097. https://doi.org/10.7150/ijms.9239 (2014).
Zheng, J., Yang, Z., Li, Y., Yang, L. & Yao, R. Knockdown of AKR1C3 promoted Sorafenib sensitivity through inhibiting the phosphorylation of AKT in hepatocellular carcinoma. Front. Oncol. 12, 823491. https://doi.org/10.3389/fonc.2022.823491 (2022).
Pant, K., Richard, S., Peixoto, E. & Gradilone, S. A. Role of glucose metabolism reprogramming in the pathogenesis of cholangiocarcinoma. Front. Med. (Lausanne). 7, 113. https://doi.org/10.3389/fmed.2020.00113 (2020).
Raggi, C., Taddei, M. L., Rae, C., Braconi, C. & Marra, F. Metabolic reprogramming in cholangiocarcinoma. J. Hepatol. 77, 849–864. https://doi.org/10.1016/j.jhep.2022.04.038 (2022).
Wang, S., Yang, Q., Fung, K. M. & Lin, H. K. AKR1C2 and AKR1C3 mediated prostaglandin D2 metabolism augments the PI3K/Akt proliferative signaling pathway in human prostate cancer cells. Mol. Cell. Endocrinol. 289, 60–66. https://doi.org/10.1016/j.mce.2008.04.004 (2008).
Kitamura, H. et al. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 108, 1947–1952. https://doi.org/10.1111/cas.13332 (2017).
Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 33, 127–148. https://doi.org/10.1093/intimm/dxaa078 (2021).
Xu, J., Lin, H., Wu, G., Zhu, M. & Li, M. IL-6/STAT3 is a promising therapeutic target for hepatocellular carcinoma. Front. Oncol. 11, 760971. https://doi.org/10.3389/fonc.2021.760971 (2021).
Isomoto, H. Epigenetic alterations in cholangiocarcinoma-sustained IL-6/STAT3 signaling in cholangio- carcinoma due to SOCS3 epigenetic Silencing. Digestion 79 (Suppl 1), 2–8. https://doi.org/10.1159/000167859 (2009).
Leng, K. et al. Akirin2 is modulated by miR-490-3p and facilitates angiogenesis in cholangiocarcinoma through the IL-6/STAT3/VEGFA signaling pathway. Cell. Death Dis. 10, 262. https://doi.org/10.1038/s41419-019-1506-4 (2019).
Zheng, T. et al. Gankyrin promotes tumor growth and metastasis through activation of IL-6/STAT3 signaling in human cholangiocarcinoma. Hepatology 59, 935–946. https://doi.org/10.1002/hep.26705 (2014).
Mao, X. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer. 20, 131. https://doi.org/10.1186/s12943-021-01428-1 (2021).
Chen, X. & Song, E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 18, 99–115. https://doi.org/10.1038/s41573-018-0004-1 (2019).
Cantallops Vilà, P., Ravichandra, A., Agirre Lizaso, A., Perugorria, M. J. & Affò, S. Heterogeneity, crosstalk, and targeting of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 79, 941–958. https://doi.org/10.1097/hep.0000000000000206 (2024).
Acknowledgements
This work was supported by the Natural Science Foundation of Fujian Province (Grant Number: 2022J01776).
Author information
Authors and Affiliations
Contributions
Tian-cong Huang and Wen-du Feng conceived the study; Guo-xu Fang, Qing-hua Zhang, Guang-ya Wei and Cheng-zong Li collected the report; Tian-cong Huang, Jian-min Wang and Jing-Feng Liu wrote the manuscript and edited the manuscript. All authors have approved publishment of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The author (s) declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Tc., Feng, Wd., Fang, Gx. et al. Cancer-Associated fibroblasts regulate the development of cholangiocarcinoma through IL-6/STAT3/AKR1C3 signaling axis. Sci Rep 16, 10467 (2026). https://doi.org/10.1038/s41598-026-37583-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-37583-y
