
Abstract
Autosomal recessive ataxia is characterized primarily by gait and balance problems. Peripheral neuropathy, which affects peripheral nervous system is manifested as sensory loss, pain, numbness and/or burning sensation accompanied by distal muscle atrophy which can cause limb deformities. We recruited seven consanguineous families having symptoms of peripheral neuropathy or ataxia for the identification of possible genetic defects underlying their disease conditions. Exome sequencing was completed for multiple participants of each family and data were filtered to retain rare deleterious variants. We identified disease-causing variants for five out of seven families. Different variants of GDAP1 were delineated in members of two families; while those of AFG3L2, MFSD8 and SETX affected members in one family each. Interestingly, one patient was homozygous for both a GDAP1 and a pathogenic MMACHC variant. Genetic heterogeneity was observed in one family since a homozygous frameshift variant of ALS2 was found in a patient while it was absent in his two affected first cousins. No genetic cause was identified for these two patients and those in another family. Hence, exome sequencing pinpointed molecular causes of recessively inherited ataxia or peripheral neuropathy in five participating families. This research has broadened the clinical spectrum of some of these genetic disorders.
Similar content being viewed by others


Introduction
Autosomal recessive ataxia is characterized primarily by gait and balance problems, truncal swaying and frequent falls. Speech dysfunction and abnormal movements of eyes and limbs are other commonly observed phenotypes accompanying ataxia1. Ataxic disorders either present symptoms of ataxia as a sole disease condition or have other associated neurological manifestations including chorea, dystonia, spasticity, Parkinsonism or seizures, depending upon the regions of central nervous system (CNS) affected in the disease process. Peripheral neuropathies, on the other hand, are heterogeneous neurological disorders resulting from nerve damage in the peripheral nervous system2. Patients with pure neuropathies have symptoms of distal muscle weakness leading to hand and foot deformities, reduced or lost tendon reflexes, and sensory loss. Peripheral neuropathy can also be a primary disease feature or part of a multisystem disease such as inherited ataxias (Friedreich ataxia), lysosomal storage diseases (Tangier’s disease) and many mitochondrial disorders3.
The list of genes causing ataxic disorders has grown over the last decade4. Mechanisms that are commonly affected due to underlying genetic defects include DNA repair, protein folding, mitochondrial functions, oxidative stress, or cytoskeletal processes5,6,7,8. Molecular cascades carrying out functions of different brain regions such as the cerebellum, basal ganglia, and corticospinal tracts are vulnerable to some common pathophysiological processes and result in neurological manifestations with overlapping phenotypes. Examples of ataxic disorders that often present dystonia are ataxia telangiectasia due to variants of ATM and ataxia with vitamin E deficiency manifested because of TTPA variants9,10. Individuals with spastic ataxia of Charlevoix-Saguenay have phenotypes of ataxia with spasticity11. Similarly, ataxic disorders that are accompanied by neuropathy include NARP syndrome (neuropathy, ataxia, retinitis pigmentosa) and ataxia with oculomotor apraxia12,13. CANVAS (cerebellar ataxia, neuropathy, vestibular areflexia syndrome) is an adult-onset autosomal recessive disorder in which patients present symptoms of gait and limb ataxia, sensory neuropathy and impaired vestibular function14. A five nucleotide repeat expansion AAGGG in RFC1 was first identified as a cause of CANVAS in 25 patients from 11 different unrelated families15.
Neuropathies can be due to immune-mediated problems, genetic, infectious or idiopathic reasons16. Among the genetic neuropathies, Charcot-Marie-Tooth (CMT) disease is the most prevalent3. More than 100 genes are found to be associated with the pathogenesis of CMT17. Different neurological diseases have overlapping phenotypes18. However, there are variants of a few genes which are associated with the etiology of more than one disease, depending upon the nature of the mutation or the region of the protein affected19. For example, variants of SPG11; a well characterized gene associated with spastic paraplegia 11, autosomal recessive (OMIM 604360), also play a significant role in the pathogenicity of a neuropathy disorder termed CMT, axonal type 2X (OMIM 616668)20.
In this study, seven consanguineous Pakistani families were recruited that have multiple affected individuals presenting pure peripheral neuropathies or ataxia with or without peripheral neuropathy. Exome sequencing identified genetic causes of disorders for most of the patients.
Methods
Ethical approval
This study was approved by the institutional review board of School of Biological Sciences (IRB# 00005281, FWA 00010252), University of the Punjab, and Yale University, School of Medicine (HIC#1411014977). Written informed consents were taken from all participating individuals and from parents for individuals under 18 years of age.
Subjects
Seven consanguineous families with multiple affected individuals were recruited for the study from different areas of the Punjab province. Individuals presented phenotypes of pure ataxia or ataxia with associated neurological conditions including dystonia, peripheral neuropathy, or spasticity. Patients of some families had peripheral neuropathy as a sole disease condition. Families were interviewed to obtain clinical data and other relevant information. Patients were videotaped according to a standard videotaping protocol to record their abnormal gaits, body movements, and any speech dysfunction21. The videos were assessed by a neurologist at a local hospital in Lahore for clinical characterization of the phenotypes.
Molecular analysis
Five to ten ml of whole-blood samples were collected from all available affected members of the families, as well as patients’ biological parents and available unaffected siblings. DNA was extracted with a standard protocol involving sucrose lysis and salting out. Exome sequencing was performed with the DNA sample of one affected individual with the most severe phenotype or with the samples from additional affected individuals and some unaffected siblings or parents. Exome sequencing was carried out at Macrogen Inc. (Seoul, South Korea), Baylor-Hopkins Center for Mendelian Genomics, (Baltimore, MD, USA), Yale Center for Genome analysis, (New Haven, CT, USA) or at 3 billion Inc. (Seoul, South Korea). Different kits were used for exome capture at the centers: Agilent SureSelect All Exon V5, Agilent SureSelect Human All Exon V6, IDT xGen, xGen, or Exome Research Panel v2. Depending on the sequencing site, the constructed libraries were sequenced with Illumina HiSeq 2000, Illumina HiSeq2500, Illumina HiSeq 4000, or Illumina NovaSeq 6000. Alignment of the data to the GRCh37/hg19 reference human genome assembly, variant calling and annotation were completed.
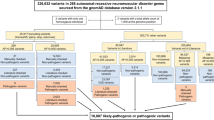
Data annotations were performed for all samples by using the Franklin tool (v.91) (https://franklin.genoox.com/clinical-db/home), the web-based server wANNOVAR (http://wannovar.wglab.org, accessed 2020)22, or the 3 billion automated prioritization system. Variants with frequencies less than 0.01 minor allele frequency in the public databases such as the genome aggregation database (gnomAD) (v2.1) (https://gnomad.broadinstitute.org/)23, exome aggregation consortium (ExAC) (v1.0) (https://exac.broadinstitute.org/)24, 1000 Genome (https://www.internationalgenome.org/, accessed 2020)25, and exon variant server (EVS) (https://bio.tools/exome_variant_server, accessed 2020)26 were selected. Variant frequencies were also observed in the in-house exome data of 300 unrelated ethnically matched individuals from Pakistan. Variants were further prioritized on the basis of prediction of pathogenicity by different algorithms and software such as CADD (v1.3) (https://cadd.gs.washington.edu)27, REVEL28, SIFT (https://sift.bii.a-star.edu.sg)29, PROVEAN (v1.1.3) (https://www.jcvi.org/research/provean)30, MutationTaster (2) (https://www.mutationtaster.org)31 and FATHMM (v2.3) (https://fathmm.biocompute.org.uk)32. Homozygous variants were analyzed first while compound heterozygous variants were considered later. In cases where exome data of more than one participant was available per family, shared predicted pathogenic variants were prioritized. If none existed, exome data analyses were repeated for each individual sample separately.
Copy number variants were assessed using CoNIFER or ExomeDepth as described33. The exome VCF data were also observed using AgileVCFMapper34 to determine the regions of homozygosity shared by the patients in each family. All samples of the participants from the families were analyzed by PCR amplification of the regions spanning the specific exons (Table S1) followed by Sanger sequencing to check the segregation of the prioritized variants. Tetra Primer ARMS PCR was done with 4 specific primers in each reaction (Table S1) to analyze the segregation of gene variants in MFSD8 and MMACHC with the disease as described35. Conservation of amino acids affected by missense variants were checked by comparison of orthologous proteins. The protein sequences of diverse vertebrate species were obtained from UniProt (https://www.uniprot.org, accessed 2024)36 and aligned with ClustalO (v.1.2.4) (https://www.ebi.ac.uk/Tools/msa/clustalo/)37.
Results
Multiple affected individuals in each of the recruited families presented one or more of the following phenotypes: ataxia, dystonia, spasticity, and neuropathies. Exome sequencing identified the causative genetic defects in the patients of most of the families.
Clinical description of the patients
Family RDFA05
Family RDFA05 had four patients (Fig. 1A) in their late forties and fifties affected with cerebellar ataxia. Disease onset was between the 15th to 18th year of age and had a slow progression. Two patients were available for videotaping and clinical characterization. Proband IV:1, a 55-year-old male and individual IV:5, a 45-year-old male, presented a wide-based ataxic gait, lateral body sway and suffered from frequent falls. Both patients had slow saccadic oculomotor disturbances and had severe difficulties in speech as well as impaired limb coordination. Muscle wasting was observed in the arm and the leg muscles of both patients and they had pes cavus feet. Though other patients participated in the study and their blood samples were collected for DNA extraction, they were not available for videotaping. They had similar phenotypes as were observed in their affected siblings (Table 1).

Pedigrees and electropherograms of the families. (A) RDFA05 (B) RDFA09 (C) RDFA11 (D) RDFA07 (E) RDFA08, (F) RDFA03 (Note the inheritance of two deleterious variants in individual V:1) (G) RDFA04. Asterisks indicate the individuals who participated in the study and double asterisks represent individuals for whom whole-exome sequencing was carried out. Genotypes of individuals participating in the study are mentioned below their respective symbols in the pedigrees. Electropherograms for (H) RDFA05 (I) RDFA09 (J) RDFA11 (K) RDFA03 (L) RDFA04. Electropherogram for families RDFA05 (H), RDFA09 (J) represents the reverse complement sequence. Arrows indicate the nucleotide change due to the variants.
Family RDFA09
Family RDFA09 was a consanguineous family with two affected individuals (Fig. 1B) presenting recessive spastic ataxia type 5. Proband (IV:3), a 12-year-old female was reported to have developed ataxia by the age of 1 year. She exhibited facial spasms and had difficulty in speaking. Minor symptoms of dystonia in her neck and arms were evident. She has pes planus feet, a condition in which feet are flat or low arched. Another affected individual was a 10-year-old male who developed disease symptoms in early infancy. He presented severe symptoms of generalized dystonia. Dystonic postures were prominent in the limbs. Generalized muscular atrophy was evident and he was unable to walk (Table 1).
Family RDFA11
Family RDFA11 had multiple affected individuals in two consecutive generations (Fig. 1C) presenting ataxia with oculomotor apraxia type 2 (AOA2). Disease onset was reported to be between 15 and 17 years of age. The proband V:2 developed symptoms of ataxia during 15th year of his life. Ataxia got worse with the passage of time, and he lost the ability to walk. At the time of examination, he was 50 years old and presented head titubation with constant jerks. He exhibited symptoms of oculomotor apraxia with inability to move the muscles of eyes horizontally. He had speech dysarthria and contractures of the hands. He developed symptoms of peripheral neuropathy and had distal limb muscle amyotrophy. His feet were high arched (pes cavus). An affected sibling V:3, showed phenotypes of similar severity (Table 1). The affected nephews and nieces of the proband were in their early thirties or teen years and showed milder disease symptoms (Table 1), probably due to the progressive nature of the disease.
Family RDFA07
Family RDFA07 was a consanguineous family having two affected male individuals in two branches (Fig. 1D) who presented with signs of ataxic cerebral palsy. Individual IV:10 a fifteen-year-old male was born after an uneventful pregnancy and had slow developmental milestones. He could not speak and had severe intellectual disability. His eyes exhibited esotropia that was more severe in the left eye. He had dysmorphic facial features and microcephaly. He exhibited a wide-based ataxic gait with frequent falls (Table 1). Individual V:4, a seven-year-old male presented less severe disease symptoms. He had esotropia of the eyes, more pronounced on the right side. He did not have dysmorphic facial features or microcephaly. He had normal muscle coordination. He had an ataxic gait and pes planus feet (Table 1).
Family RDFA08
Family RDFA08 is a consanguineous family having three male individuals in two branches (Fig. 1E) presenting ataxic cerebral palsy. Patients have symptoms of a complex movement disorder. The 7-year-old proband V:3, was born prematurely after eight months of gestation. He developed the disease symptoms at the age of 14 months. He had dysmorphic facial features with a bulbous nose and slight microcephaly. He exhibited esotropia of both eyes, unclear speech, and a wide-based ataxic gait that worsened with the passage of time, eventually losing the ability to walk. He now moves by dragging himself. He presented muscular atrophy of the distal limbs and minor hand and feet contractures. Individual V:1, a 13-year-old male, the sibling of patient V:3, was said to have developed disease symptoms at the age of 1 year. He exhibited slurred speech and had symptoms of ataxia with a narrow-based gait and lateral body sway. He had minor hand contractures and normal body coordination. The first cousin of the two affected individuals, patient V:7, was a six-year-old male diagnosed with juvenile onset amyotrophic lateral sclerosis with disease onset at the age of two years. He exhibited gross motor delay. His cognition was normal at the time of examination and his eye movements were also unremarkable. He had dysmorphic facial features with a bulbous nose, and he exhibited impaired speech. He had severe hand and foot contractures. He exhibited toe walking and could not walk independently. He also had weakness and hypertonia or spasticity of the lower limbs (Table 1).
Families RDFA03 and RDFA04
Family RDFA03 had two branches with four affected individuals (Fig. 1F) all of whom suffered from progressive Charcot-Marie-Tooth disease (CMT). In the first branch, individual V:1, a 17-year-old male at the time of examination, was affected by the most severe disease phenotype. He was reported to have been unaffected till the age of nine years and had attended school. He developed the disease symptoms that worsened with the passage of time. At the time of observation, his head was constantly having involuntary jerks. He presented dysmorphic facial features and had dysphonia. His hands had contractures with curved fingers. He exhibited valgus wrists and ankles. He had severe muscular atrophy of the distal limbs. Although he was reported to have been able to walk till the age of 12 years, he had lost the ability to ambulate by the time he was 15. Now, he moves by dragging himself and the legs are crisscrossed during the movement. He exhibited generalized areflexia in the distal limb muscles. His feet were high arched (pes cavus). Other affected individuals showed a less severe phenotype. Two other affected male individuals: V:3 and V:8, had hand contractures with restricted wrist and ankle movements. They also had mild ataxic gaits. Their voices were normal and the voluntary movements of the body were also unaffected. The female patient (V:6) had lost wrist and ankle movements. She had foot drop and was unable to walk. She had no voice abnormality or contractures of hands and feet, unlike the conditions in the affected male patients in her family (Table 1).
Family RDFA04 is a consanguineous family with two affected male individuals (Fig. 1G). Individual VI:1 is a 35-year-old male with normal facial features and speech. He moved by dragging himself with the support of his hands. He presented amyotrophy and areflexia of distal limbs. He exhibited valgus wrists and ankles. He had foot contractures with curved fingers. One foot was severely bent while the other foot was pes planus. Individual VI:3 was a 30-year-old male who showed similar phenotypes as exhibited by the proband (Table 1). It was not possible to obtain the electrophysiological data for patients in families RDFA03 and RDFA04 to specify the CMT type as demyelinating, axonal or intermediate.
Identification of deleterious homozygous variants in five families after exome sequencing
Exome sequencing yielded multiple variants for further consideration for each family (Tables S2-S9). Most of these were designated as variants of unknown significance while only a few were classified as pathogenic or likely pathogenic according to the ACMG criteria (PM2, PP3). Still, single variants could be prioritized for analyses based on their deleterious predictions by multiple software or previous description to cause a similar disorder (Table 2, Tables S2-S9). A missense variant of (NM_152778.3) MFSD8 c.935T > C, p.Ile312Thr (Fig. 1H) (Table 2) was linked to the disease phenotype in family RDFA05. For family RDFA09, molecular analyses identified a missense variant of (NM_006796.3) AFG3L2 c.2167G > A, p.Val723Met (Fig. 1I) (Table 2) as segregating with the disorder. Similarly, a frameshift variant of (NM_015046.7) SETX c.4633_4636delAGTG, p.Ser1545AlafsTer25 (Fig. 1J) (Table 2) was linked to the disease in family RDFA11. Exome sequencing identified a nonsense variant of (NM_018972.4) GDAP1 c.840delC, p.Tyr280Ter (Fig. 1K) (Table 2) in patients of family RDFA03 and a missense variant GDAP1 c.347T > G, p.Met116Arg (Fig. 1L) (Table 2) as the cause of the disorder in family RDFA04.
All affected individuals in the five families were homozygous for the identified variants while the unaffected parents were heterozygous. The unaffected siblings were either heterozygous for the variants or carried the wild-type alleles. The identified variants were absent or extremely rare in the public databases (Tables S2-S9) and were also absent from 600 chromosomes of an ethnically matched control population. In addition, the proband V: I in family RDFA03 (Fig. 1F) was also homozygous for a known ACMG designated pathogenic missense variant of (NM_001330540.2) MMACHC c.176T > C, p.Leu59Pro (Table 2). His parents (Fig. 1F) were heterozygous for the variant. However, this variant was not present in the other three patients of his family. We had exome data of a single patient. We mapped the regions of homozygosity using AgileVCFMapper. There were multiple regions of homozygosity on chromosomes 1, 8, 9, 11 and 18. The two variants were present in regions of homozygosity for the proband, suggesting that they are identical by descent (Supplementary Fig. S1).
Families for which no Bona Fide genetic cause was identified for all members
Exome sequencing filtering identified multiple variants in patients’ data, of which four were homozygous in both affected individuals of family RDFA07, but none were predicted deleterious (Table S7, Extended Tables 1 and 2). Sanger sequencing determined that one of these variants in (NM_001166415.2) EHHADH c.805T > G, p.Leu269Val segregated with the phenotype. However, the variant had low REVEL and CADD scores and the affected amino acid was not conserved in orthologous proteins (Supplementary Fig.S2). Additionally, various online software programs predicted that the variant was tolerated and is likely to be benign. None of the other variants segregated with the phenotype. Analyses of the exome VCF data using AgileVCFMapper identified four regions on chromosomes 2, 3, 6, and 18, at which both affected individuals were homozygous (Supplementary Fig.S3) which included the EHHADH region on chromosome 3, spanning ~2 Mb. In addition, no predicted deleterious compound heterozygous variants were identified which could explain the phenotype of the patients.
Exome sequencing of the DNA from two affected siblings V:1, V:3, and their parents in one branch of family RDFA08 identified no significant homozygous or compound heterozygous disease-causing gene variant that segregated with the disorder (Table S8, Extended Tables 3 and 4). A known homozygous nonsense variant of (NM_020919.3) ALS2 c.3520 A > T, p.Lys1174Ter38 was found associated with the disease of their cousin, patient V:7 (Fig. 1E; Table 2, Table S9). The affected individuals V:1 and V:3, had the wild type sequence for ALS2. AgileVCFMapper identified multiple regions of homozygosity shared by the two affected individuals V:1 and V:3, located on chromosomes 3, 11, 18 and 19 which were heterozygous in their parents (Supplementary Fig.S4).
Discussion
We identified homozygous variants of MFSD8, AFG2L3, SETX and GDAP1 associated with cerebellar ataxia, spastic ataxia, ataxia with oculomotor apraxia type 2 and CMT respectively. MFSD8 encodes a transmembrane protein, a member of major facilitator superfamily (MFS) (Fig. 2A) which is involved in the transport of solutes to the lysosome. The missense variant of MFSD8 c.935T > C, p.Ile312Thr identified in patients of family RDFA05 is associated with an adult-onset disease. However, the variant was classified to be of uncertain significance and affected an amino acid that is conserved in all vertebrates except the mouse in which it is leucine instead, a conservative change (Fig. 2B). Although the pathogenicity of this variant remains to be determined, we note that this p.Ile312Thr variant has been previously reported to cause adult onset ataxia and maculopathy in two unrelated patients of South Asian origin39.

Protein domains and conservation of amino acids affected by missense variants. (A) Diagrammatic structure of MFSD8. Transmembrane domains and the variants identified in the current study (boxed) and the previous studies are shown. (B) The conservation of the amino acid altered due to the MFSD8 variant identified in family RDFA05 is shown by the alignment of diverse vertebrate orthologues of the human protein. The affected amino acid is boxed. (C) Different domains of AFG3L2 are illustrated. Variants associated with the recessively inherited spastic ataxia syndrome are shown. The variant identified in the family RDFA09 is boxed. (D) The absolute conservation of the amino acid altered due to the identified AFG3L2 variant in family RDFA09 is evident after alignment of orthologues proteins from different vertebrate lineages (E) Domains of SETX are shown with the identified frameshift variants associated with ataxia with oculomotor apraxia. The variant identified in the family RDFA11 is boxed. (F) Domains of GDAP1 are shown with the variants known to cause CMT disease. Nonsense variant identified in the family RDFA03, and the missense variant associated with disease in family RDFA04 are boxed. (G) The amino acid altered due to the missense variant of GDAP1, identified in the family RDFA04 is conserved in all vertebrate groups, except fish.
In most previous studies, variants of MFSD8 have been described to cause variant late infantile neuronal ceroid lipofuscinosis (vLINCL) (OMIM 610951), that appears in early childhood and patients usually die in their first or second decade of life due to the severe and progressive symptoms of the disease40. Symptoms of vLNCL include developmental regression, mental and motor regression, seizures, impairment of speech, ataxia, myoclonus and visual failure. Patients in the current study exhibited neurological symptoms of worsening gait ataxia, and speech difficulties with symptoms of dysarthria like the patients in the previous study39. However, they had symptoms of slow saccades which is contrary to the patients in the previous study. This may be either the result of variable expressivity of MFSD8 variant in patients of RDFA05 or due to phenotypic heterogeneity of MFSD8. Similar reports of phenotypic heterogeneity associated with variants of MFSD8 have been observed in previous studies. For example in a study patients of an Egyptian family showed all the symptoms of vLNCL except visual problems41. Another study showed that some variants of MFSD8 caused only maculopathies and cone disorders in some patients with no signs of neurological phenotypes or other phenotypes of vLNCLs42. Research has also shown that variants of CLN6, another gene well characterized to be associated with childhood-onset vLNCLs, also play a role in an adult-onset NCL known as Kuf’s disease43,44. Kuf’s disease is an adult onset NCL without visual problems. Our research suggests a similar role of MFSD8 in childhood as well as adult onset NCLs. Functional studies using animal models in the future can further pave the way towards understanding the role of variants of MFSD8 in adult onset vLNCLs.
A homozygous missense variant of AFG3L2 c.2167G > A, p.Val723Met (Fig. 2C) was identified as a cause of ataxia, dystonia and speech difficulty for one patient of family RDFA09. The variant affected an amino acid that was fully conserved in diverse vertebrates (Fig. 2D). The protein encoded by AFG3L2 is highly conserved and a ubiquitously expressed metalloprotease of the mitochondria which degrades old mitochondrial proteins. The identified variant affects the proteolytic domain of the protein where most of the variants involved in dominantly inherited spinocerebellar ataxia or recessive spastic ataxia are clustered (Fig. 2C). Nearly 67 variants of AFG3L2 identified to date are associated with dominant form of spinocerebellar ataxia type 28 (OMIM 610246), dominant optical atrophy (OMIM 618977), or recessively inherited spastic ataxia (OMIM 614487) representing AFG3L2 pleiotropy45,46.
Acting as a mitochondrial quality control and a regulatory enzyme, AFG3L2 helps maintain the homeostasis of mitochondrial protein synthesis and mitochondrial protein turnover. AFG3L2 forms homo-oligomeric (only AFG3L2) or hetero-oligomeric (AFG3L2 oligomerized with SPG7) mitochondrial proteases. The homozygous variant AFG3L2 c.2167G > A, p.Val723Met identified in the proband of family RDFA09, was previously reported as a compound heterozygous variant, identified in one male patient in whom it caused progressive cerebellar ataxia, bilateral ptosis and dysarthria. The variant was detected in trans with c.1847 A > G, p.Tyr616Cys change47. In that study, mild cerebellar atrophy was observed in brain imaging of the 39 years old index patient. Abnormal mitochondrial morphology and fragmentation were observed in the patient fibroblasts. His mother being heterozygous, showed bilateral ptosis and difficulties performing tandem walk. However, medical history and clinical investigation revealed no phenotypes in the heterozygous parents in family RDFA09. Thus, the heterozygous effect of the AFG3L2 variant remains to be explored.
We also identified a SETX variant, which encodes a DNA/RNA helicase involved in the protection of cells against the damage to DNA. Affected individuals of family RDFA11 had ataxia, dysarthria and neuropathy due to a homozygous frameshift variant of c.4633_4636 delAGTG, p.Ser1545AlafsTer26. This identified variant has been reported earlier as a compound heterozygous allele in trans with a SETX deletion of 20.7 kb spanning exons 10–15 in a single German patient. The proband in the previous study had onset of ataxia at 12th year of age. He lost the ability to walk without support after a few years. He also showed symptoms of dysarthria, distal muscle wasting, and pes cavus48. These symptoms are similar to the disease course of the proband in the current study.
The frameshift variant of SETX c.4633_4636 delAGTG, p.Ser1545fsTer26, introduces a premature stop codon in mRNA and it will likely induce nonsense mediated decay (NMD) of the mRNA (Fig. 2E). However, if mRNA escapes NMD, a protein lacking the helicase domain may be formed. This variant caused a very severe phenotype in the affected members of the family RDFA11 as observed in most AOA2 patients reported in other studies. The presence of the same disease-causing variant in distant populations i.e. Pakistani and Germany, suggests that the mutation may have an independent occurrence, and the region could be a mutational hotspot.
Exome sequencing identified biallelic variants of GDAP1, (NM_018972.4) c.840delC, p.Tyr280Ter segregating in family RDFA03 and c.347T > G, p.Met116Arg in the members of family RDFA04 (Fig. 2F). GDAP1 variants cause different types of CMT disease (OMIM 214400, OMIM 607706, OMIM 607831, OMIM 608340). CMT is a heterogeneous group of well-studied inherited neuromuscular disorders, which has a prevalence of 1:2500 individuals49. It involves clinical manifestations of distal muscle wasting or weakness, loss of deep tendon reflexes, foot deformity and sensory loss. GDAP1 encodes a protein located in the outer mitochondrial membrane that is highly expressed in the central and the peripheral nervous system. It plays a role in the signal transduction pathway during neuronal development19. GDAP1 is also required for the maintenance of structure and function of the mitochondria50,51,52.
Symptoms of CMT are more severe due to truncating GDAP1 variants while missense variants result in a less severe disease course. The nonsense variant of GDAP1 c.840delC, p.Tyr280Ter identified in family RDFA03 caused severe symptoms and an early onset of the CMT4A (OMIM 214400). This variant introduces a premature stop codon in GDAP1. It is proposed to be a loss of function allele because of either a complete absence of the protein due to the nonsense mediated decay of mRNA or missing important catalytic and regulatory domains as a consequence of protein truncation. A previous study identified the same variant in a Pakistani family53. The affected individual examined at 9 years of age showed delayed developmental milestones, muscle atrophy of hands and distal legs and bilateral foot drop which is a phenotype similar to most patients in the current study. Thus, the c.840delC, p.Tyr280Ter variant may represent a founder mutation since it was detected in two independent families from Pakistan, In this regard, it is interesting to note that there are 4 South Asian individuals in gnomADv.4 who are heterozygous for the same variant and to date, individuals from none of the other world populations have this variant.
Interestingly, the proband in family RDFA03 was homozygous for a missense variant of MMACHC c.176T > C, p.Leu59Pro in addition to the variant in GDAP1. MMACHC encodes a protein named as “methylmalonic aciduria combined with homocystinuria type c (cbl)” which plays roles in the metabolism of cobalamin (vitamin B-12). Biallelic MMACHC pathogenic variants cause methylmalonic aciduria and homocystinuria, cblC type (OMIM 277400). Individuals with this disorder have elevated levels of homocysteine (homocystinuria) and methylmalonic acid (methylmalonic aciduria). Disease course due to MMACHC variants is varied and symptoms include hematological abnormalities, developmental delays, ataxia with associated neurological symptoms54 maculopathy55 and other systemic diseases56. The proband who was homozygous for both GDAP1 and MMACHC variants presented the most severe neurological symptoms as compared to the other affected patients in his family who lacked the variant in the latter gene. He had constant head titubation, a hoarse voice and highly severe muscular atrophy. We were unable to measure the levels of plasma and urine methylmalonic acid and homocysteine of patient. The variant MMACHC c.176T > C, p.Leu59Pro affects an amino acid which is conserved in diverse vertebrates and was reported first in 2 individuals having vitamin B-12 deficiency phenotypes57. In previous studies, patients harboring defects of cobalamin metabolism due to variants of MMACHC showed significant improvement in their disease course with parenteral administration of hydroxocobalamin, oral betaine and intravenous L-carnitine. Administration of folic acid and compound vitamin B also helped in the management of the disease58,59,60. The above treatment regimens can be recommended to clinicians for the management of the disease symptoms of proband in family RDFA03.
The variant GDAP1 c.347T > G, p.Met116Arg (NM_018972.4) linked to the pathogenesis of CMT (OMIM 214400) in affected individuals of family RDFA04, resulted in relatively less severe symptoms of the disease as compared to those of patients in family RDFA03 who were homozygous for the frameshift variant. This GDAP1 missense variant has been previously reported to cause recessively inherited CMT in four Italian individuals in three different families61. Their disease phenotypes including early onset distal muscle atrophy, foot deformities, loss of independent ambulation within the third decade of life, and absence of vocal cord paresis were similar to the disease features observed in the patients of family RDFA04. Sural nerve biopsies of the Italian patients indicated problems in neuron myelination as well as axonal impairment. This same missense variant was later reported as a cause of CMT4 in two Polish siblings as well62. Patients presented similar disease phenotypes as affected individuals of the family RDFA04. Hence, the GDAP1 c.347T > G, p.Met116Arg variant causes a similar disease in Italian, Polish and Pakistani individuals.
The missense variant GDAP1 c.347T > G, p.Met116Arg affects an amino acid that was found to be conserved in diverse vertebrates examined except fish (Fig. 2G). However, the latter have a threonine or an alanine residue and none have an arginine in this position. Usually, pathogenic variants affect amino acids, which are completely conserved in evolution, though there are exceptions in which a variant causing disorder in humans does not cause any phenotype in an animal model62,63. Some other pathogenic variants of GDAP1 have also been reported to affect amino acids which are not conserved in fish. For example, GDAP1 c.679 A > G, p.Asn227Asp is reported to cause recessively inherited CMT in two affected individuals in compound heterozygous form with c.715 C > T, p.Leu239Phe64. The residue GDAP1 asparagine 227 is not conserved in a few fish species where it is replaced either by valine, glycine or serine. This indicates that variation of some non-conserved amino acids of GDAP1 can be pathogenic.
For families RDFA07 and RDFA08, no deleterious variant explained the phenotype of most of the patients. In family RDFA08 very few features were common to the patients in the two branches (Table 1). The proband and his sibling were differently affected from their cousin. The identification of a homozygous nonsense variant of ALS2 c.3520 A > T, p.Lys1174Ter (OMIM 607225) for the latter patient only, further confirmed this heterogeneity at the genetic level. Re-evaluation of the clinical data showed that the phenotype of spasticity of lower limbs was only exhibited by the patient for whom the variant of ALS2 was identified.
ALS2 encodes alsin protein which has a role as GTPase regulator and affects the mitochondrial and endosomal trafficking65. Variants of ALS2 are associated with the recessively inherited motor neuron diseases including infantile onset ascending hereditary spastic paralysis (IAHSP) (OMIM #607225), juvenile primary lateral sclerosis (JPLS) (OMIM #606353) and juvenile onset amyotrophic lateral sclerosis (JALS) (OMIM #205100)66. The homozygous recessive variant ALS2 c.3520 A > T, p.Lys1174Ter was previously reported in two unrelated male individuals in a study from Saudi Arabia38. The symptoms associated with motor delay and hypertonia were similar to the patients in family RDFA08. Moreover, the symptoms of intellectual disability, developmental regression, dystonia, seizures and tremors observed in the Saudi Arabian report were not seen in the patient of the current study. Additionally, speech impairment observed in the affected individual of current study was not reported in one of the Saudi Arabian patients. These findings suggest that the disease phenomenology can vary due to the similar ALS2 variant in different individuals.
No homozygous or compound heterozygous variants could be correlated with the phenotype of the proband in family RDFA08 and his affected sibling, either individually or in comparison. Shared mapped chromosomal regions of homozygosities from the exome data suggest that a pathogenic variant may be located in any element which was not captured by exome sequencing. Similarly, members of family RDFA07 only segregated a predicted benign EHHADH c.805T > G, p.Leu269Val variant. EHHADH encodes an enzyme involved in the peroxisomal oxidation of fatty acids and it is expressed in the proximal tubule. Heterozygous variants of EHHADH are associated with Fanconi renotubular syndrome 3 (OMIM 615605), a cause of metabolic disorder involving growth impairment, rickets, proteinuria and metabolic acidosis67. A study classified this c.805T > G, p.Leu269Val variant as moderately associated with the already known dominantly inherited Fanconi renotubular syndrome 3 68. Further work is needed to see if this EHHADH c.805T > G, p.Leu269Val variant has any role, when homozygous, in a neurological phenotype, or whether it is a coincidental finding due to its being in linkage disequilibrium with a pathogenic variant in a different gene.
In this study, exome sequencing identified molecular causes of ataxic and neuropathic disorders in five participating families. The gene variant identified in the family RDFA05 is very rarely associated with ataxia while a severely affected individual was identified who had pathogenic variants in two different genes. In addition, variants of SETX and AFG3L2 which were previously reported as disease causing in the compound heterozygous form, caused similar severe disorders in the patients homozygous for the variants. Genome sequencing in the future may uncover likely variants located in the regions which were not detected by exome sequencing for patients in two families which are currently unresolved.
Data availability
The variants segregating with the phenotypes have been deposited in ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/, *MFSD8* SCV007328882, *AFG3L2* SCV007130105, *SETX* SCV007328881, *ALS2* SCV007130107, *GDAP1* SCV007328883, *GDAP1* SCV007328884 and *MMACHC* SCV007130106. Phenotypic and genotypic data are provided with the manuscript. Any additional data will be provided on reasonable request to the corresponding author.
References
Pavone, P. et al. Ataxia in children: early recognition and clinical evaluation. Ital. J. Pediatr. 43, 1–9. https://doi.org/10.1186/s13052-016-0325-9 (2017).
Pareyson, D., Saveri, P. & Pisciotta, C. New developments in Charcot–Marie–Tooth neuropathy and related diseases. Curr. Opin. Neurol. 30, 471–480. https://doi.org/10.1097/WCO.0000000000000474 (2017).
Morena, J., Gupta, A. & Hoyle, J. C. Charcot-Marie-Tooth: from molecules to therapy. Int. J. Mol. Sci. 20, 3419. https://doi.org/10.3390/ijms20143419 (2019).
Lopergolo, D. et al. Autosomal recessive cerebellar ataxias: a diagnostic classification approach according to ocular features. Front. Integr. Nuerosci. 17, 1275794. https://doi.org/10.3389/fnint.2023.1275794 (2024).
Gupta, S., You, P., SenGupta, T., Nilsen, H. & Sharma, K. Crosstalk between different DNA repair pathways contributes to neurodegenerative diseases. Biology 10, 163. https://doi.org/10.3390/biology10020163 (2021).
Bustamante-Barrientos, F. A. et al. Mitochondrial dysfunction in neurodegenerative disorders: potential therapeutic application of mitochondrial transfer to central nervous system-residing cells. J. Transl Med. 21, 613. https://doi.org/10.1186/s12967-023-04493-w (2023).
Zocchi, R. et al. Novel loss of function mutation in TUBA1A gene compromises tubulin stability and proteostasis causing spastic paraplegia and ataxia. Front. Cell. Neurosci. 17 https://doi.org/10.3389/fncel.2023.1162363 (2023).
Gomes, C. M. & Santos, R. Neurodegeneration in Friedreich’s ataxia: from defective frataxin to oxidative stress. Oxid Med Cell Longev 487534. (2013). https://doi.org/10.1155/2013/487534 (2013).
Huff, L. A., Yan, S. & Clemens, M. G. Mechanisms of ataxia telangiectasia mutated (ATM) control in the DNA damage response to oxidative stress, epigenetic regulation, and persistent innate immune suppression following sepsis. Antioxid 10, 1146. https://doi.org/10.3390/antiox10071146 (2021).
Maalej, M. et al. A first description of ataxia with vitamin E deficiency associated with MT-TG gene mutation. Acta Neurol. Belg. 121, 1733–1740. https://doi.org/10.1007/s13760-020-01490-4 (2021).
Gana, S. & Valente, E. M. Movement disorders in genetic pediatric ataxias. Mov. Disord Clin. Pract. 7, 383–393. https://doi.org/10.1002/mdc3.12937 (2020).
Del Dotto, V., Musiani, F., Baracca, A. & Solaini, G. Variants in human ATP synthase mitochondrial genes: biochemical Dysfunctions, associated Diseases, and therapies. Int. J. Mol. Sci. 25, 2239. https://doi.org/10.3390/ijms25042239 (2024).
Cohen, S. et al. Senataxin resolves RNA: DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 9, 533. https://doi.org/10.1038/s41467-018-02894-w (2018).
de Silva, R. N. et al. Diagnosis and management of progressive ataxia in adults. Pract. Neurol. 19, 196–207. https://doi.org/10.1136/practneurol-2018-002096 (2019).
Cortese, A. et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 51, 649–658. https://doi.org/10.1038/s41588-019-0422-y (2019).
Eggermann, K. et al. Hereditary neuropathies: clinical presentation and genetic panel diagnosis. Dtsch. Arztebl Int. 115, 91. https://doi.org/10.3238/arztebl.2018.0091 (2018).
Rudnik-Schöneborn, S., Auer-Grumbach, M. & Senderek, J. Hereditary neuropathies: update 2017. Neuropediatrics 48, 282–293. https://doi.org/10.1055/s-0037-1603518 (2017).
Pang, S. Y. Y. et al. The role of gene variants in the pathogenesis of neurodegenerative disorders as revealed by next generation sequencing studies: a review. Transl Neurodegener. 6, 1–11. https://doi.org/10.1186/s40035-017-0098-0 (2017).
Africk, D. Mitochondrial dysfunction in a patient with 8q21. 11 deletion and Charcot-Marie-Tooth disease type 2K due to GDAP1 haploinsufficiency. Mol. Syndromol. 6, 204–206. https://doi.org/10.1159/000440660 (2015).
Montecchiani, C. et al. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot–Marie–Tooth disease. Brain 139, 73–85. https://doi.org/10.1093/brain/awv320 (2016).
Miller, S. A., Dykes, D. D. & Polesky, H. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215. https://doi.org/10.1093/nar/16.3.1215 (1988).
Chang, X. & Wang, K. wANNOVAR: annotating genetic variants for personal genomes via the web. J. Med. Genet. 49, 433–436. https://doi.org/10.1136/jmedgenet-2012-100918 (2012).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. https://doi.org/10.1038/s41586-020-2308-7 (2020).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. https://doi.org/10.1038/nature19057 (2016).
Consortium, G. P. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56. https://doi.org/10.1038/nature11632 (2012).
Fu, W. et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220. https://doi.org/10.1038/nature11690 (2013).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. https://doi.org/10.1093/nar/gky1016 (2019).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885. https://doi.org/10.1016/j.ajhg.2016.08.016 (2016).
Sim, N. L. et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457. https://doi.org/10.1093/nar/gks539 (2012).
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. https://doi.org/10.1093/bioinformatics/btv195 (2015).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods. 11, 361–362. https://doi.org/10.1038/nmeth.2890 (2014).
Shihab, H. A. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65. https://doi.org/10.1002/humu.22225 (2013).
Krumm, N. et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 22, 1525–1532. https://doi.org/10.1101/gr.138115.112 (2012).
Carr, I. M. et al. Autozygosity mapping with exome sequence data. Hum. Mutat. 34, 50–56. https://doi.org/10.1002/humu.22220 (2013).
Ye, S., Dhillon, S., Ke, X., Collins, A. R. & Day I. N. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res. 29, e88–e88. https://doi.org/10.1093/nar/29.17.e88 (2001).
Consortium, U. The universal protein resource (UniProt). Nucleic Acids Res. 36, D190–D195. https://doi.org/10.1093/nar/gkl929 (2007).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal Omega. Mol. Syst. Biol. 7, 539. https://doi.org/10.1038/msb.2011.75 (2011).
Monies, D. et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum. Genet. 136, 921–939. https://doi.org/10.1007/s00439-017-1821-8 (2017).
Dobloug, S. et al. Maculopathy and adult-onset ataxia in patients with biallelic MFSD8 variants. Mol. Genet. Genomic Med. 12, e2505. https://doi.org/10.1002/mgg3.2505 (2024).
Siintola, E. et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 81, 136–146. https://doi.org/10.1086/518902 (2007).
Stogmann, E. et al. A novel mutation in the MFSD8 gene in late infantile neuronal ceroid lipofuscinosis. Neurogenetics 10, 73–77. https://doi.org/10.1007/s10048-008-0153-1 (2009).
Roosing, S. et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology 122, 170–179. https://doi.org/10.1016/j.ophtha.2014.07.040 (2015).
Gao, H. et al. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am. J. Hum. Genet. 70, 324–335. https://doi.org/10.1086/338190 (2002).
Arsov, T. et al. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am. J. Hum. Genet. 88, 566–573. https://doi.org/10.1016/j.ajhg.2011.04.004 (2011).
Di Bella, D. et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 42, 313–321. https://doi.org/10.1038/ng.544 (2010).
Patron, M., Sprenger, H. G. & Langer, T. m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell. Res. 28, 296–306. https://doi.org/10.1038/cr.2018.17 (2018).
Tunc, S. et al. Spinocerebellar ataxia type 28—Phenotypic and molecular characterization of a family with heterozygous and compound-heterozygous mutations in AFG3L2. Cerebellum 18, 817–822. https://doi.org/10.1007/s12311-019-01036-2 (2019).
Bernard, V. et al. Exon deletions and intragenic insertions are not rare in ataxia with oculomotor apraxia 2. BMC Med. Genet. 10, 1–9. https://doi.org/10.1186/1471-2350-10-87 (2009).
Laurá, M., Pipis, M., Rossor, A. M. & Reilly, M. M. Charcot–Marie–Tooth disease and related disorders: an evolving landscape. Curr. Opin. Neurol. 32, 641–650. https://doi.org/10.1097/WCO.0000000000000735 (2019).
Niemann, A., Wagner, K. M., Ruegg, M. & Suter, U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol. Dis. 36, 509–520. https://doi.org/10.1016/j.nbd.2009.09.011 (2009).
Huber, N., Guimaraes, S., Schrader, M., Suter, U. & Niemann, A. Charcot-Marie‐Tooth disease‐associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission. EMBO Rep. 14, 545–552. https://doi.org/10.1038/embor.2013.56 (2013).
González-Sánchez, P. et al. CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca2 + entry-stimulated respiration. Sci. Rep. 7, 42993. https://doi.org/10.1038/srep42993 (2017).
Zimoń, M. et al. Unraveling the genetic landscape of autosomal recessive Charcot-Marie-Tooth neuropathies using a homozygosity mapping approach. Neurogenetics 16, 33–42. https://doi.org/10.1007/s10048-014-0422-0 (2015).
Cui, J. et al. Isolated subacute combined degeneration in late-onset cobalamin C deficiency in children: two case reports and literature review. Medicine 98, e17334. https://doi.org/10.1097/MD.0000000000017334 (2019).
Collison, F. T. et al. Whole exome sequencing identifies an adult-onset case of methylmalonic aciduria and homocystinuria type C (cblC) with non-syndromic bull’s eye maculopathy. Ophthalmic Genet. 36, 270–275. https://doi.org/10.3109/13816810.2015.1010736 (2015).
Kömhoff, M. et al. Combined pulmonary hypertension and renal thrombotic microangiopathy in cobalamin C deficiency. Pediatrics 132, e540–e544. https://doi.org/10.1542/peds.2012-2581 (2013).
Lerner-Ellis, J. P. et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, CblC type. Nat. Genet. 38, 93–100. https://doi.org/10.1038/ng1683 (2006).
Carrillo-Carrasco, N., Chandler, R. J. & Venditti, C. P. Combined methylmalonic acidemia and homocystinuria, CblC type. I. Clinical presentations, diagnosis and management. J. Inherit. Metab. Dis. 35, 91–102. https://doi.org/10.1007/s10545-011-9364-y (2012).
Wang, S., Yan, C., Liu, Y. & Zhao, Y. Late-onset cobalamin C deficiency Chinese sibling patients with neuropsychiatric presentations. Metab. Brain Dis. 33, 829–835. https://doi.org/10.1007/s11011-018-0189-3 (2018).
Sun, M. & Dai, Y. Late-onset cobalamin C deficiency type in adult with cognitive and behavioral disturbances and significant cortical atrophy and cerebellar damage in the MRI: a case report. Front. Neurol. 14, 1308289. https://doi.org/10.3389/fneur.2023.1308289 (2023).
Di Maria, E. et al. A novel mutation of GDAP1 associated with Charcot-Marie-Tooth disease in three Italian families: evidence for a founder effect. J. Neurol. Neurosurg. Psychiatry. 75, 1495–1498. https://doi.org/10.1136/jnnp.2003.028100 (2004).
Kabzińska, D. et al. A novel Met116Thr mutation in the GDAP1 gene in a Polish family with the axonal recessive Charcot–Marie–Tooth type 4 disease. J. Neurol. Sci. 241, 7–11. https://doi.org/0.1016/j.jns.2005.10.002 (2006).
Xu, Q., Shenoy, S. & Li, C. Mouse models for LRRK2 parkinson’s disease. Parkinsonism Relat. Disord. 18, S186–S189. https://doi.org/10.1016/S1353-8020(11)70058-X (2012).
Kabzińska, D. et al. A new missense GDAP1 mutation disturbing targeting to the mitochondrial membrane causes a severe form of AR-CMT2C disease. Neurogenetics 12, 145–153. https://doi.org/10.1007/s10048-011-0276-7 (2011).
de Souza, P. V. S., de Rezende Pinto, W. B. V., de Rezende Batistella, G. N., Bortholin, T. & Oliveira, A. S. B. Hereditary spastic paraplegia: clinical and genetic hallmarks. Cerebellum 16, 525–551. https://doi.org/10.1007/s12311-016-0803-z (2017).
Helal, M. et al. Clinical presentation and natural history of infantile-onset ascending spastic paralysis from three families with an ALS2 founder variant. Neurol. Sci. 39, 1917–1925. https://doi.org/10.1007/s10072-018-3526-8 (2018).
Klootwijk, E. D. et al. Mistargeting of peroxisomal EHHADH and inherited renal fanconi’s syndrome. N Engl. J. Med. 370, 129–138. https://doi.org/10.1056/NEJMoa1307581 (2014).
Bastarache, L. et al. Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science 359, 1233–1239. https://doi.org/10.1126/science.aal4043 (2018).
Acknowledgements
We express our thanks to Lauren Jefferies from Yale school of Medicine for evaluation of the family RDFA11 videos and to Monica Konstantino from Yale school of Medicine for technical assistance.
Author information
Authors and Affiliations
Contributions
Conceptualization, SN; Investigation, FA, MW, AIB, EW, GHS, WJ, SAL, NS, SN; Writing- original draft, FA, SN; Writing-review and editing, MW, AIB, EW, GHS, WJ, SAL, NS; Visualization FA. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was conducted in accordance with the Declaration of Helsinki. The Institutional Review Boards of the School of Biological Sciences, University of the Punjab, Lahore, Pakistan, and Yale University, School of Medicine, New Haven, CT, USA approved the study.
Consent to participate and to publish
Written informed consents were obtained from all the participants. Parents provided consent for their minor children.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-37808-0
