
Abstract
An innovative, sustainable analytical approach was developed by coupling an Epsom salt, E518 (magnesium sulfate)–facilitated salting-out assisted liquid–liquid extraction (SALLE) system with a safe molecular fluorescent probe, Celfia Pink B (CPB), employing a fluorescence turn-off sensing strategy. MgSO₄ acts as a green salting-out agent, enhancing phase separation efficiency and producing cleaner extracts with higher analyte recovery. CPB, a safe, food-grade dye, serves as a selective fluorogenic probe whose emission at 553 nm is quenched upon electrostatic ion-pair complex formation with Cinacalcet HCl under mildly acidic conditions. The method exhibited excellent linearity (0.08–1.3 µg/mL) with high sensitivity (LOD = 0.024 µg/mL; LOQ = 0.075 µg/mL). Validation according to ICH guidelines demonstrated robustness, precision, and accuracy across pharmaceutical formulations and biological matrices. Sustainability assessment using the WAC framework categorized the platform as “white,” highlighting low environmental impact, safe operation, and simplicity. By minimizing organic solvent use and eliminating hazardous reagents, this MgSO₄-assisted SALLE/fluorescence turn-off approach provides a green, highly sensitive, and versatile tool for trace-level determination of Cinacalcet HCl in pharmaceutical and biological matrices.
Similar content being viewed by others


Introduction
Magnesium sulfate (E518, commonly known as Epsom salt) is a highly versatile and safe compound with wide applications in food, pharmaceutical, and analytical sciences. In salting-out assisted liquid–liquid extraction (SALLE), it plays a dual role as an effective phase-separation agent and drying reagent, efficiently removing residual water from organic phases such as acetonitrile to ensure cleaner extracts and enhanced analyte recovery. Comparative studies have shown its superiority to sodium sulfate, making it especially valuable in trace-level pharmaceutical assays. As a non-toxic, registered food additive (E518) with high water solubility, environmental compatibility, and lack of bioaccumulation, magnesium sulfate represents a cost-effective and sustainable option that aligns with green and sustainable chemistry principles1,2,3,4,5,6.
However, there is a continuous demand for more efficient, versatile, rapid, and environmentally friendly extraction systems in modern pharmaceutical and bioanalytical chemistry. Salting-out assisted liquid–liquid extraction (SALLE) has gained increasing popularity due to its simplicity, cost-effectiveness, and alignment with green chemistry principles. In SALLE, the addition of an appropriate salt induces phase separation between an aqueous sample and a water-miscible organic solvent, resulting in selective partitioning of target analytes into the organic phase7. The method is remarkably fast, safe, and economical, and the obtained extracts can be directly integrated into subsequent spectrofluorimetric measurements. Various organic solvents (acetonitrile, acetone, ethyl acetate, isopropanol) and salting-out agents (magnesium sulfate, ammonium sulfate, calcium chloride, potassium carbonate, calcium sulfate) have been successfully employed8.
In the present work, magnesium sulfate (E518) was employed as the salting-out agent, providing efficient phase separation and enhanced extraction performance. To the best of our knowledge, this is the first attempt to couple SALLE with a fluorimetric “switch-off” strategy using Celfia Pink B (CPB), a safe food dye repurposed as a fluorescent probe for the determination of cinacalcet HCl. The quenching of CPB’s intrinsic fluorescence occurs through electrostatic or ion-pair complexation with the target analyte, offering a highly selective and sensitive platform. Such a hybrid methodology not only extends the application of SALLE in pharmaceutical analysis but also establishes a novel, sustainable, and cross-disciplinary analytical approach for ultra-trace determination of the target analyte, cinacalcet HCl.
Cinacalcet (CCT.HCl, Fig. 1a), chemically designated as N-[1®(−)-(1-naphthyl)ethyl]-3-[3-(trifluoromethyl)phenyl]-1-aminopropane, functions as a calcimimetic moderator by selectively modulating calcium-sensing receptors in parathyroid tissues9. This mechanism enhances receptor sensitivity to extracellular calcium, effectively suppressing parathyroid hormone secretion10,11. CCT.HCl is clinically approved for managing secondary hyperparathyroidism in chronic kidney syndrome patients undergoing dialysis12 and hypercalcemia associated with diagnosed parathyroid carcinoma13.
Fluorometric quantification of CCT.HCl has employed aromatic fluorogenic reagents such as 5-nitrobenzofuran derivatives14,15, 1,2,3-indantrione16, aminonaphthalene analogs17, fluorescamine18, diformylbenzene19, and o-phthalaldehyde/2-mercaptoethanol20. These reagents form stable, fluorescent covalent adducts upon reaction with primary amine groups of the analyte.
Prior methodologies for CCT.HCl quantification in pharmaceutical and biological matrices encompasses spectrophotometric21,22,23, spectrofluorometric24,25, HPTLC26, HPLC27,28,29, and LC-MS30,31 techniques. However, these approaches have been associated with operational limitations, including reliance on sophisticated instrumentation, labor-intensive protocols, and high costs.
To date, few spectrofluorometric derivatization strategies have been reported for CCT.HCl analysis24. While demonstrating superior sensitivity to the current method, this methodology demands a harsh derivatization procedure with 4-chloro-7-nitro-1,2,3-benzoxadiazole, involving 20-minute heating at 60 °C followed by acidic quenching with HCl. The procedure is complicated by the requirement to control multiple variables, such as NBD-Cl reagent concentration, pH, buffer composition, temperature, reaction duration, cooling conditions, HCl strength, and solvent effects. Furthermore, NBD-Cl degradation under heating releases corrosive vapors, posing risks of respiratory and dermal irritation. In contrast, the proposed methodology eliminates hazardous reagents, streamlines workflow through a single-step reaction, and aligns with green chemistry principles while retaining sufficient sensitivity to quantify CCT.HCl within therapeutic plasma concentrations.
The predominant methodologies for cinacalcet (CCT.HCl) analysis have historically depended on high-performance liquid chromatography (HPLC), which is often constrained by prolonged analysis durations, substantial consumption of high-purity organic solvents during sample purification, elevated operational costs, and adverse environmental implications32,33,34,35. While spectrophotometric techniques offer simplicity, their limited sensitivity restricts their utility in biological fluid analysis. In contrast, spectrofluorimetry emerges as a superior alternative owing to its rapidity, cost efficiency, operational simplicity, minimal sample preparation, and inherent methodological straightforwardness.
celfia pink B (CPB, Fig. 1b), a disodium salt of 2,4,5,7-tetraiodofluorescein, functions as both a biological stain and an ion-association reagent, enabling its application as a fluorogenic probe. The protonatable amino group within CCT.HCl facilitates electrostatic interaction with CPB’s anionic moieties under acidic conditions, forming a stable ion-paired complex. Although alternative dyes such as merbromin and Rhodamine 6G have been utilized in spectrofluorimetric assays, their applicability is limited: merbromin’s mercury content raises toxicity worries, while Rhodamine 6G exhibits structural incompatibility with CCT.HCl, hindering effective electrostatic interactions.
CPB was selected for its exceptional sensitivity, robust negative charge (promoting stable ion-pair formation with cationic analytes), aqueous solubility, cost-effectiveness, and alignment with green chemistry ethics. These attributes render it ideal for quantifying amine-containing pharmaceuticals, including antidepressants, antihypertensives, and anticancer agents36,37, without necessitating hazardous reagents or derivatization.
Prior fluorometric method24 for CCT.HCl quantification relied on a harsh derivatization procedure involving heating and using hydrochloric acid, a corrosive and hazardous medium, underscoring the novelty of the current approach, which prioritizes safety and sustainability. This study introduces an innovative, eco-friendly spectrofluorimetric strategy for CCT.HCl determination in bulk materials and dosage forms, validated through comprehensive green metrics. The method’s simplicity further permits its adaptation to content uniformity testing in tablet formulations, ensuring batch-to-batch consistency.
By leveraging CPB’s intrinsic fluorescence quenching upon interaction with CCT.HCl in a mildly acidic aqueous buffer, this methodology achieves high sensitivity (detection limits surpassing conventional techniques), accuracy, and operational safety. The elimination of toxic solvents and complex instrumentation positions this approach as a paradigm shift in pharmaceutical analysis, combining analytical rigor with environmental stewardship.
Analytical methods for trace-level quantification of pharmaceuticals in complex matrices demand high sensitivity, selectivity, and minimal environmental impact.
Conventional chromatographic techniques (e.g., HPLC, GC–MS) demonstrate robust performance but typically involve expensive instrumentation, extensive purification protocols, and substantial organic solvent volumes.
Salting-out assisted liquid–liquid extraction (SALLE) has emerged as a facile and efficient sample-cleanup technique in pharmaceutical analysis, where an inorganic salt—such as magnesium sulfate—induces rapid phase separation of a water-miscible organic solvent from an aqueous matrix, enriching the target analyte while reducing matrix interferences38,39. This approach offers significant advantages over conventional liquid–liquid extraction, including reduced consumption of hazardous organic solvents and simplified operational procedures. Fluorescence “switch-off” tagging strategies complement SALLE by converting target drugs into non-fluorescent ion-pair complexes with food-grade dyes (e.g., Celfia Pink B), enabling ultra-trace detection via quenching measurements at specific wavelengths with exceptional sensitivity.
The present work introduces a novel hybrid analytical platform for the ultra-trace quantification of cinacalcet HCl, a clinically significant calcimimetic agent used in the management of parathyroid disorders. The novelty lies in the synergistic integration of magnesium sulfate-based SALLE with a fluorescence quenching detection strategy, providing both efficient sample clean-up and highly selective signal transduction. Unlike conventional extraction methods, magnesium sulfate ensures superior phase separation, enhanced recovery of cinacalcet, and minimal interference from matrix components, representing a greener and safer alternative that aligns with sustainable analytical chemistry principles.
The second innovative dimension is the strategic repurposing of Celfia Pink B (CPB), a food-grade dye, as a selective fluorescent probe. While CPB is typically recognized as a food colorant, its intrinsic fluorescence properties are exploited here for sensitive detection of cinacalcet through electrostatic or ion-pair complex formation. The resulting fluorescence quenching (“switch-off” signal) offers a unique analytical response with high sensitivity and selectivity. This dual-function platform combines the sample purification advantages of SALLE with the analytical precision of fluorescence quenching, utilizing safe reagents (MgSO₄ and CPB), reduced solvent consumption, and an eco-friendly workflow as a sustainable, cost-effective, and innovative tool for pharmaceutical quality control and clinical sample analysis.
Experimental section
Instrumentation
Fluorimetric measurements were conducted using an FS-2-SCINCO spectrofluorometer (Korea) fitted with a xenon arc lamp (150 W). The spectra of emission and excitation were recorded at a scan rate of 571 nm/min. Sample sonication was performed using an SC-101TH SONICOR ultrasonic bath (USA) to enhance solubility. pH adjustments were carried out with an AD11P pH measuring equipment (ADwa, Romania).
Materials and reagents
Cinacalcet (CCT.HCl) reference standard was generously provided by the Egyptian Group for Pharmaceutical Industries (EGPI). Cinacalcet® F.C. Tablets (30, 60, and 90 mg/tab) were procured from local pharmacies. Celfia pink B (CPB; 2,4,5,7-tetraiodofluorescein disodium salt, 4.34 × 10− 2 mM) was prepared by dissolving 15 mg in 50 mL deionized water. Analytical-grade reagents—phosphoric acid, sodium hydroxide, hydrochloric acid, anhydrous magnesium sulphate, acetic acid, methanol, ethanol, acetonitrile, dimethylformamide (DMF), and dimethyl sulfoxide (DMSO)—were sourced from El-Naser Co. (Egypt). Buffers (Britton-Robinson40, Teorell-Stenhagen41,42, McIlvaine43, acetate44 were employed for pH optimization.
Standard solution preparation
A primary CCT.HCl stock solution (100.0 µg mL− 1) was prepared by dissolving 10.0 mg CCT.HCl in distilled water and diluting to 100 mL. Working standards (80–1300 ng mL− 1) were derived via serial dilution. Anhydrous magnesium sulphate (2.0 M) solution was prepared by dissolving about 24.05 g dissolved in 100 mL water. Solutions were stored at 4 °C to ensure stability.
Calibration protocol
Aliquots of CCT.HCl working standards (800–13000 ng mL− 1) were taken into 10 mL standard volumetric flasks. Teorell-Stenhagen (1.80 mL, pH 4.2) and CPB solution (1.4 mL, 3.41 × 102 µM) were added, followed by dilution to volume with deionized water. Fluorescence production intensity (λex = 526 nm; λem = 553 nm) was measured. A calibration graph was created by plotting ΔRFI (Fo − F) against CCT.HCl concentration, where Fo and F represent the fluorescence intensities of free CPB and the CCT.HCl -CPB complex, respectively.
Analytical applications
Pharmaceutical applications
Tablet assay
Ten Cinacalcet® tablets (90 and 60 mg) were pulverized. A portion equivalent to 10 mg CCT.HCl was sonicated in water, filtered, and diluted. CCT.HCl content was quantified using the calibration curve, with triplicate measurements per concentration.
Content uniformity testing
Compliance with USP guidelines45 was verified by individually analyzing ten Cinacalcet® tablets (30 mg). Each tablet was processed as per Section 2.5.1 and CCT.HCl content was assessed against label claims.
Biological sample investigation
Plasma sample preparation
Ethical approval for human plasma collection was secured from the institutional review board (Al-Azhar University blood store, Assiut branch), and informed consent was obtained from participants prior to sample acquisition. Venous blood (5.0 mL) was drawn from a fasting healthy volunteer into heparinized tubes to prevent coagulation. Plasma separation was achieved via centrifugation at 5000 rpm for 5 min. Aliquots of the supernatant were spiked with known CCT.HCL concentrations and subjected to Salting-out MgSO4-assisted liquid-liquid extraction (SALLE) using acetonitrile for plasma purification and analyte isolation.
This involved sequential addition of 500 µL plasma, 1000 µL acetonitrile (ACN), and 250 µL saturated MgSO₄ solution (2.0 M) to the sample in 10 mL working volumetric flask. The mixture underwent vortex-mixing (5 min) followed by centrifugation (5000 rpm, 5 min). The cinacalcet HCl-enriched acetonitrile layer was retrieved and reconstituted in working flasks for fluorimetric analysis. The validated methodology was subsequently applied to pretreated plasma specimens. A blank measurement was acquired by executing identical processing on plasma matrices lacking cinacalcet.
Urine sample preparation
Fresh urine specimens were collected from a fasting healthy donor and filtered twice over a 0.45 μm membrane to retain particulate matter. Filtrate aliquots were conveyed to 10 mL standard flasks and analyzed by the same spectrofluorimetric protocol applied to plasma. Method specificity was confirmed by comparing spiked urine samples with unspiked controls.
Results and discussion
Spectral characteristics of the CCT.HCl-CPB complex system
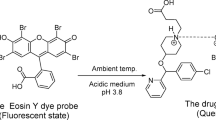
Amino-containing analytes, such as cinacalcet (CCTs.HCL), can be quantified through spectrofluorometric analysis by forming binary ion-pair complexes with acidic fluorophores like (CPB). In this study, electrostatic interactions between the protonated amino group of CCT.HCl and the anionic species of CPB (HL⁻) facilitated complex generation in the Teorell-Stenhagen buffer (pH 4.2). This interaction induced structural modifications in CPB’s conjugated π-electron system, redistributing electron density and leading to fluorescence quenching. The resultant “turn-off” fluorescence mechanism enabled precise CCT.HCl detection, as evidenced by the linear reduction in CPB’s intrinsic emission intensity at 553 nm (λex = 526 nm) with increasing analyte concentration (Fig. 2).

The drug (A) and dye (B) chemical structures.

The fluorescence spectra (excitation and emission) of the developed complex between the dye (CPB) and the target drug (CCT.HCl).
Mechanistic insights into complex formation
pH-dependent ionization of CPB
The ionization state of CPB, critically influenced by pH, governs its interaction with CCT.HCl. With dissociation constants (pKa1 = 3.9 and pKa2 = 5.046, CPB predominantly exists as the monovalent anion at pH 4.2, as confirmed by maximal fluorescence quenching within the pH range of 3.8–4.6. Iodine substituents in the xanthene structure reduced electron density at the hydroxyl group, enhancing its reactivity relative to the carboxyl group37,47. Quantum chemical computations (AM1 method) revealed hydroxyl ionization released 120.3 kJ/mol more energy (− 285.8 kJ/mol) than carboxyl dissociation (− 165.5 kJ/mol), favoring HL⁻ stability48.
Protonation behavior of CCT.HCl
CCT.HCl compound’s chemical structure contains only one nitrogen atom, and exhibits sufficient basicity for protonation under acidic conditions. Protonation generated a cationic species (CCT.HClH⁺), which interacted electrostatically with CPB⁻ to form a stable 1:1 ion-pair complex (Fig. 3). Hydrophobic interactions further stabilized the complex, underscoring its analytical robustness.

The suggested complex formation pathway between the dye (CPB) and the studied drug (CCT.HCl).
Mechanistic analysis of fluorescence quenching
The fluorescence attenuation of CPB upon interaction with cinacalcet (CCT.HCl) was investigated to elucidate the underlying quenching mechanism. Potential pathways, including dynamic collision, energy transfer, and static complexation, were evaluated using the Stern-Volmer relationship:
Here, Fo and F denote the fluorescence powers of CPB in the absence and presence of CCT.HCl (quencher, [Q]), respectively, while KSV represents the Stern-Volmer constant. A linear correlation between Fo/F and [Q] (See Supplementary Materials, Fig. S1a) confirmed quenching dependence on CCT.HCl concentration. The Stern-Volmer constant (Ksv) as calculated from the plot was 775.76 M− 1. The bimolecular quenching constant (Kq) was derived using KSV = Kq τ0, where τo (CPB’s intrinsic fluorescence lifetime (τ₀) = 89 ps49 yielding Kq = 8.72 × 1012 M− 1 s− 1. This value significantly exceeds both the theoretical maximum for dynamic quenching (2 × 1010 M− 1 S− 1)50 and thediffusion-controlled limit (~ 1010 M⁻¹ s⁻¹), conclusively strongly supporting a static quenching mechanism in which a stable ground-state ion-pair complex forms between CPB and cinacalcet HCl, rather than quenching occurring solely through dynamic collisions.
Thermodynamic and binding affinity studies
The binding stoichiometry (n) and association constant (Ka) were determined via the modified Stern-Volmer equation (double-log Stern–Volmer plot)51:
In applying the modified Stern–Volmer (double-log) plot for erythrosine B fluorescence quenching by cinacalcet HCl, the linear equation obtained was (Fig. S1b):
From this, the slope (n) is 1.0889, which signifies approximately one binding site per CPB molecule, indicative of a 1:1 stoichiometry, consistent with Job’s method. The intercept, representing logKa, is 6.4072, which corresponds to a binding constant Ka ≈ 2.55 × 106 M−1, reflecting strong affinity between the dye and cinacalcet HCl.
These findings support the development of a static quenching mechanism, where a stable non-fluorescent ion-pair complex forms in the ground state. The value of n ≈ 1 confirms one site involved in complexation, and the high Ka highlights strong binding. Collectively, this confirms that cinacalcet HCl effectively quenches CPB via complex formation, validating the use of this binding model in the analytical platform.
Optimization of methodological parameters
The experimental variables influencing the fluorescence quenching efficiency of the CCT.HCl-CPB system were systematically investigated to refine the spectrofluorimetric protocol. Each parameter was meticulously adjusted to maximize analytical performance while adhering to sustainability principles.
Influence of pH
The pH of the system medium was evaluated using Teorell-Stenhagen buffer across a 2.2–6.5 range. Optimal fluorescence attenuation was observed at pH 4.2 (Fig. 4), where CCT.HCl undergoes protonation to form a cationic species (HCCT.HCl⁺), while CPB predominantly exists in its monovalent anionic state (CPB⁻). This pH facilitates robust electrostatic interactions and hydrophobic stabilization between the drug and dye, promoting the construction of a stable electrostatic-based complex. Deviations from this range resulted in reduced quenching efficiency due to suboptimal ionization states of the reactants.
Selection of buffer system
Four buffer systems—Britton-Robinson, McIlvaine, Toerell-Stenhagen, and acetate—were compared at a fixed volume (1.8 mL). The Teorell-Stenhagen buffer demonstrated superior performance (Fig. 4), attributed to its broad buffering capacity and compatibility with the reaction milieu, ensuring stable complex formation and maximal signal attenuation.
Effect of CPB volume
Volumes of CPB reagent ranging from 0.1 to 2.8 mL were tested to fix the optimal quantity for maximal fluorescence suppression. A volume of 1.4 mL (Fig. S2) was identified as ideal, yielding consistent quenching without signal saturation. Volumes below 1.2 mL provided insufficient reagent for complete complexation, while quantities exceeding 1.8 mL induced self-quenching due to dye aggregation, diminishing the analytical response.
Buffer volume optimization
The Teorell-Stenhagen buffer volume ( in a 0.2–3.0 mL range) impact on signal intensity was assessed. A volume of 1.8 mL was found to balance pH stabilization and minimize competitive anion interference (e.g., phosphate). Excessive buffer (> 2.4 mL) introduced competing ions that disrupted CPB-CCT.HCl interactions, whereas insufficient volumes (< 1.2 mL) failed to maintain consistent pH, leading to variability in quenching efficiency (Fig. S2).
Solvent compatibility assessment
Aqueous and organic solvents (ethanol, acetonitrile, methanol, and acetone) were evaluated as dispersion media. Aqueous solutions achieved maximal quenching due to water’s high polarity of value = 9.0, dielectric constant = 80.2)52 and hydrogen-bonding capacity, which stabilized the excited-state orientation of the complex (Fig. S3). Organic solvents, particularly short-chain alcohols, disrupted hydrophobic interactions and complex assembly at raised concentrations53. Water’s dual role as a hydrogen bond donor and acceptor further enhanced proton transfer, ensuring amino group protonation and complex stability54. Additionally, the elevated polarity of aqueous systems suppressed π-π* electronic transitions while promoting n-π* interactions, stabilizing the excited state, and amplifying the quenching effect55,56.
Temporal stability of the complex
The CCT.HCl-CPB complex exhibited rapid formation at ambient temperature, with signal stabilization achieved within 4 min post-reaction (Fig. 4). Prolonged incubation beyond this period showed no significant variation, confirming the method’s suitability for high-throughput analysis.

The study of the medium pH, buffer system type used, and the reaction required for the complex formation between the dye (CPB) and the investigated drug (CCT.HCl).
Stoichiometric determination via Job’s method
Job’s continuous variation analysis57 was used to establish the binding ratio between CPB and CCT.HCl. Fluorescence intensities were measured for solutions containing fixed total molar concentrations of CPB and CCT.HCl, with mole fractions varying between 0.1 and 0.9. Corrected fluorescence values (ΔF = F₀ − F) were plotted counter to the drug’s mole fraction, yielding a peak value at 0.5 (Fig. S4), confirming a 1:1 stoichiometry for the CPB: CCT.HCl complex. This finding aligned with prior mechanistic and thermodynamic analyses.
Application of Scatchard model for binding site(s) and association affinity estimations
The Scatchard model (Fig. 5) is a graphical technique used in analytical chemistry to study binding interactions between molecules, such as dyes and drugs, by determining the binding affinity constant (K) and the number of binding sites (n)58.

Application of the Scatchard model for the binding site(s) and association affinity estimations.
It applies the equation: \(\varvec{r}/\left[\varvec{L}\right]=\varvec{n}\varvec{K}\varvec{a}-\varvec{r}\varvec{K}\varvec{a}\)
Here, ( r ) is the number of bound ligands, ( [L] ) is the concentration of free ligands, ( n ) is the number of binding sites, and ( Ka ) is the association constant59.
In the investigation of the interaction between CPB dye and Cinacalcet, Scatchard plot analysis yielded a linear relationship. The slope represents -Ka, thus Ka = 7.58 × 105 M-1, reflecting a very strong binding affinity. The y-intercept represents nKa, yielding n ≈ 0.94 (approximately 1), which confirms that each molecule of the dye binds to one molecule of the drug in a stable 1:1 stoichiometric interaction60,61. This strong binding constant (Ka) indicates a robust interaction between CPB and Cinacalcet HCl, reducing the amount of free dye available and altering its fluorescence properties through ground-state complex formation. This phenomenon supports the system’s efficiency as a fluorescence quenching platform and can be leveraged to develop precise analytical methods for drug detection or to study interaction mechanisms61.
Validation of the developed approach
Following comprehensive optimization of all experimental variables, the proposed spectrofluorimetric method was rigorously validated in compliance with International Council for Harmonization (ICH) strategies62. Critical validation considerations, including linearity, sensitivity, accuracy, precision, robustness, selectivity, and applicability to pharmaceutical formulations, were systematically evaluated.
Linearity and range
The linear relationship between CCT.HCl concentration and fluorescence quenching response (ΔRFI) was established under standardized experimental conditions. Serial dilutions of CCT.HCl standard solutions spanning 80–1300 ng mL⁻¹ were analyzed, with ΔRFI values demonstrating proportional attenuation relative to analyte concentration. Regression analysis yielded a correlation coefficient (r) approaching unity (Table 1), confirming exceptional linearity across the specified range.
Sensitivity limits (LOD and LOQ)
Method sensitivity was quantified via statistical determination of the limit of detection (LOD) and limit of quantification (LOQ). These parameters were derived via the formulas LOD = 3.3σ/S and LOQ = 10σ/S, where σ symbolizes the standard deviation of the intercept and S signifies the calibration curve slope. Calculated values of 24.8 ng mL− 1 (LOD) and 75.2 ng mL− 1 (LOQ) (Table 1) underscored the method’s capability for trace-level CCT.HCl detection.
Accuracy assessment
Accuracy was assessed through recovery readings at four CCT.HCl concentrations (200, 700, 1000, 1200 ng mL− 1), with triplicate measurements per level. Percent recoveries ranged from 99.78% to 101.98%, accompanied by %RSD (relative standard deviation) values below 2.0% (Table 2). The proximity of recoveries to 100% validated the method’s accuracy in quantifying CCT.HCl across its linear range.
Precision evaluation
Intra-day and inter-day precision were assessed to determine method reproducibility. Intra-day precision was evaluated via triplicate analyses of three CCT.HCl concentrations (300, 700, 1000 ng mL− 1) within a single analytical run. Inter-day precision involved replicate measurements across three consecutive days. %RSD values remained below 1.75% for both precision tiers (Table 2), confirming minimal variability under repeatability and intermediate precision conditions.
Robustness testing
Method resilience was examined by introducing minor modifications to three critical parameters: buffer pH (± 0.2), buffer solution volume (± 0.2 mL), and CPB solution volume (± 0.2 mL). Recovery percentages (99.91–101.93%) and %RSD values (< 1.85%) remained consistent across all variations (Table 3), demonstrating insensitivity to controlled procedural deviations.
Selectivity and interference analysis
Selectivity was verified by analyzing CCT.HCl in the presence of common pharmaceutical excipients and structurally analogous compounds. Recovery rates exceeding 98% with %RSD < 2% (Table S1) confirmed negligible matrix interference. The absence of reactive aliphatic amine groups in excipients prevented competitive complexation with CPB, ensuring specificity for CCT.HCl.
Applications of the approach
Pharmaceutical application
The validated approach was used to quantify CCT.HCl in Cinacalcet® tablets (60 and 90 mg/tablet). Results were statistically compared with a reference method24 using Student’s t-test and F-test. Calculated t- and F-values fell below critical thresholds (2.78 and 6.39 at 95% confidence), confirming equivalence in accuracy and precision (Table 4).
Content uniformity evaluation
Content uniformity testing was performed on ten Cinacalcet® tablets (30 mg) per USP criteria63. The AV, acceptance value, calculated as AV = |R − Ā| + kS (where k = 2.4, S = standard deviation, R = reference value, Ā = mean content), yielded values below the maximum allowable limit (L1 = 15) (Table 5). This confirmed homogeneous drug distribution within the tested batch.
Application to spiked human plasma and urine specimens
Salting-out assisted liquid–liquid extraction (SALLE)—a streamlined sample preparation approach—employs water-miscible organic solvents (e.g., acetonitrile) and salting-out agents (e.g., MgSO4) to induce phase separation. This technique yields purified extracts while diminishing matrix interference relative to classical liquid–liquid extraction or protein precipitation methods38,39.
The enhanced sensitivity of the developed methodology enabled its successful implementation for quantifying CCT.HCl spiked in biological matrices (human plasma and urine) within the validated calibration range. For plasma samples fortified with CCT.HCl, an average recovery of 99.60-101.98% ± 1.43% was achieved (Table 6), demonstrating high accuracy and minimal matrix interference. Similarly, urine samples spiked with CCT.HCl yielded a mean recovery of 99.38-101.86% ± 1.76%, confirming the method’s reliability and suitability for biofluid analysis.
Sustainability evaluation of the method
An environmental sustainability assessment is characterized by minimal or zero utilization of hazardous reagents, complete waste mitigation, and low energy demands. The ecological compatibility of the planned spectrofluorometric technique was systematically examined through multiple modern green chemistry metrics.
Analytical professionals in chemical and pharmaceutical disciplines bear significant responsibility for safeguarding human and environmental health from harmful substances16,19,64,65,66,67,68,69. Continuous emphasis on advancements in green chemistry principles remains essential. The current spectrofluorimetric methodology was rigorously evaluated using Green Analytical Chemistry (GAC) rating criteria.
As environmentally conscious methodologies gain prominence, diverse qualitative and quantitative metrics have been established by researchers to assess compliance with GAC standards70,71,72. Notably, theoretical indicators of ecological sustainability have been widely implemented. These include the Eco-Scale73, AGREE74, GAPI75, NEMI76, the AGREEprep tool for material preparation77, the RGB12 platform, and the BAGI tool (Table S2).
Analytical eco-scale assessment
The Eco-Scale quantifies ecological impacts by measuring penalty scores built on chemical quantities, occupational hazards, waste output, and energy consumption. The total penalty points are subtracted from 100 to yield the Eco-Scale score73. Methods scoring above 75 qualify as sustainable. This fluorometric method achieved a score of 95 (compared to 74for conventional methods), confirming its environmental safety (Table S2).
National environmental methods index (NEMI)
The NEMI framework was employed to evaluate the procedural sustainability76. This metric evaluates solvent persistence, bioaccumulation potential, and toxicity. Methanol, employed in this spectrofluorometric procedure, is classified as a non-PBT (persistent, bioaccumulative, toxic) solvent. The Teorell-Stenhagen buffer system (pH 4.2) used in the procedure was non-caustic, and overall waste generation was kept less than 50 mL. Findings confirmed minimal solvent consumption and waste production, fulfilling all four NEMI criteria (Table S2), thereby designating the method as eco-friendly.
Application of the complexGAPI tool
In analytical chemistry, the complexGreen Analytical Procedure Index (complexGAPI) is a widely used tool for evaluating the environmental impact of analytical techniques based on green chemistry principles. The complex GAPI, an advanced version, provides a detailed assessment by breaking down the evaluation into multiple criteria, often represented visually as a pentagonal or hexagonal diagram with color-coded sections. Each color—green (little impact), yellow (reasonable impact), red (great impact), and white (unevaluated)—indicates the level of environmental friendliness for specific criteria, such as the usage of renewable resources, energy efficiency, waste generation, and toxicity of reagents78,79.
The analysis involved examining the color distribution in each image to assess the green chemistry criteria. The complex GAPI tool typically evaluates 15 sub-indicators grouped into categories like sample treatment, reagent use, energy consumption, and waste management75. Colors are interpreted as follows: green for little impact, yellow for reasonable impact, red for great impact, and white for unevaluated criteria. The images were compared based on the percentage of each color to determine which method is greener69,79,80,81.
The GAPI pictogram dissects the analytical lifecycle into 15 stages, color-coded as follows:
-
Green (10 stages): sample preparation (aqueous dilution), detection (non-destructive fluorescence), and waste management (low-volume disposal).
-
Yellow (4 stages): offline analysis (manual data collection) and derivatization (CBP complexation).
-
Red (1 stage): limited automation and use of organic solvents (It’s the small amount of acetonitrile in SALLE for biological samples). The predominance of green zones (Table S2) underscored the method’s alignment with GAC principles.
AGREE framework analysis
The AGREE metric, a software-driven GAC tool, was implemented to assess the methodology’s sustainability74. Twelve GAC-aligned criteria are graded on a 0.0 (red) to 1.0 (green) scale, generating a radial chart with a central color indicator (aggregate score: 0.78)82,83,84. Key evaluations included:
Criterion 1 (sample handling): yellow (offline analysis requirement).
Criterion 2 (sample volume): light green (minimal consumption).
Criterion 3 (in situ analysis): red (offline instrumentation).
Criterion 4 (preparation steps): green (simplified workflow).
Criterion 5 (automation): yellow (manual operation).
Criterion 6 (derivatization): light green.
Criterion 7 (waste): green (low volume).
Criterion 8 (throughput): green (high hourly analyte capacity).
Criterion 9 (energy): green (low-energy detection).
Criterion 10 (renewables): green (water-based solvents).
Criterion 11 (solvent safety): green (low toxicity).
Criterion 12 (operator safety): green (safe practices).
The high summative score (0.78) reflects distilled water implementation, reducing processing time and ecological burden (Table S2).
AGREEprep tool implementation
AGREEprep evaluates the sustainability of the sample preparation via a circular diagram with 10 segmented areas signifying Green Sample Preparation (GSP) principles77. The proposed method scored 0.80, attributed to streamlined workflows, elimination of thermal cycling steps (reducing energy use), and enhanced safety via aqueous solvents (Table 6).
The following analysis details how the method aligns with each principle, supported by the AGREEprep.
Here’s a concise summary of how the CCT.HCl spectrofluorimetric method aligns with the 10 Green Sample Preparation (GSP) principles:
-
1.
Favor in situ sample preparation.
-
Lab-based, not in situ (yellow AGREE rating).
-
Not met.
-
2.
Use safer solvents and reagents.
-
1 mL methanol (minimal, not PBT), 9 mL water, safe CPB.
-
Met.
-
3.
Target sustainable, renewable, reusable materials.
-
Water is renewable, no organic solvents, and low waste (< 50 mL).
-
Met.
-
4.
Minimize waste.
-
< 50 mL waste (green AGREE score).
-
Fully met.
-
5.
Minimize sample, chemical, and material amounts.
-
Small volumes (0 mL organic solvent, 10 mL water).
-
Met.
-
6.
Maximize sample throughput.
-
High hourly rates (green AGREE rating).
-
Fully met.
-
7.
Integrate steps, promote automation.
-
Manual, not automated (yellow AGREE score).
-
Not fully met.
-
8.
Minimize energy consumption.
-
Ambient temperature, no heating/cooling (green AGREE rating).
-
Fully met.
-
9.
Select the greenest post-sample preparation.
-
Low-energy spectrofluorimetry.
-
Met.
-
10.
Confirm safe procedures for the worker.
-
Safe solvents/reagents (green AGREE rating).
-
Fully met.
Therefore, the planned CCT.HCl method excels in safety, waste reduction, and energy efficiency (AGREEprep 0.80, Eco-Scale 95), but falls short in in situ preparation and automation, suggesting areas for future enhancement (Table S2).
Green solvent selection tool (GSST)
GSST identifies sustainable solvents using Hansen Solubility Parameters (HSP) to assess dispersion, polarity, and hydrogen bonding interactions85,86. Solvents are graded (G-score: 1–10), with higher values indicating sustainability. Methanol (5.8) and water (7.3) in the method outperformed prior spectrophotometric solvents (Figure S2). GSST’s rapid online evaluation facilitates solvent comparisons87.
GSST prioritized solvents with optimal Hansen solubility parameters (HSPs):
-
Dispersion forces (δD): water (15.5 MPa1/2), methanol (15.1 MPa1/2).
-
Polarity (δP): water (16.0 MPa1/2), methanol (12.3 MPa1/2).
-
Hydrogen bonding (δH): water (42.3 MPa1/2), methanol (22.3 MPa1/2).
-
High G-scores for water (7.3) and methanol (5.8) confirmed their environmental suitability.
Whiteness evaluation via RGB12
The RGB12 framework integrates red (analytical efficiency), green (ecological compatibility), and blue (economic feasibility) metrics to compute a whiteness score88. The method demonstrated:
Red (performance): broad linear range (0.08–1.3 µg/mL), low LOD (24.8 ng/mL), precision (RSD < 1.75%), accuracy (99.78–101.98%).
Green (ecology): water-dominated reagents, no organic solvent, low energy, Eco-Scale 95.
Blue (economics): cost-effective, simple, solvent-efficient.
The composite whiteness score (Table S3) confirmed superior sustainability, efficiency, and practicality.
Blueness assessment via BAGI
The Blue Applicability Grading Index (BAGI) is a relatively new tool designed to grade the applicability of analytical methods in practical settings (25–100 scale). Research suggests it provides a scoring system to evaluate how well a method can be implemented, considering aspects like safety, cost-effectiveness, and operational simplicity89,90. It seems likely that BAGI uses a color-coded or numerical scoring system, with higher scores indicating better applicability, making it useful for quick assessments in laboratories. The evidence leans toward BAGI being particularly valuable in resource-limited environments, where cost and safety are critical, but it may lack depth in environmental impact assessment compared to other tools91.
BAGI focuses on applicability, with criteria likely including safety (e.g., handling hazards), cost (e.g., reagent and equipment expenses), and ease of use (e.g., training requirements). These criteria indirectly relate to green chemistry, as lower costs and safer methods often align with reduced environmental impact, but BAGI does not explicitly evaluate detailed environmental factors like waste or energy use. The method scored 72.5, reflecting high applicability and ease of comparative analysis (Table S2).
Conclusion
A green hybrid analytical platform combining MgSO₄-SALLE with “switch-off” fluorescence detection has been successfully developed for the ultra-trace quantification of Cinacalcet HCl in pharmaceutical and biological matrices. The method relies on charge-mediated ion-pair formation between Cinacalcet HCl and Celfia Pink B (CPB), a food-grade fluorescent dye, in a weakly acidic environment, with quantification achieved through spectrofluorimetric monitoring of CPB fluorescence quenching. Excellent analytical performance was demonstrated, including linearity over 80–1300 ng mL-1, with detection and quantification limits of 24.8 ng mL-1 and 75.2 ng mL-1, respectively. The method was successfully applied to tablet homogeneity assessments and dosage-form analysis, confirming its practical utility in pharmaceutical quality control.
Key advantages of this approach include simplified implementation through aqueous-based sample preparation, direct analysis without chromatographic separation, elimination of volatile organic solvents, and reduced procedural complexity, resulting in faster analysis and enhanced cost efficiency. The methodology’s environmental sustainability was rigorously evaluated using multiple contemporary metrics, yielding an exceptional holistic sustainability profile across integrated greenness, whiteness, and blueness frameworks.
Beyond immediate analytical performance, this work presents several broader implications for pharmaceutical analysis and green analytical chemistry. The hybrid SALLE-fluorescence platform represents a versatile framework that can be extended to other basic or cationic drugs capable of forming ion-pair complexes with anionic fluorescent dyes. The successful use of food-grade CPB as a fluorescent probe establishes a precedent for developing safe, sustainable sensor systems applicable across pharmaceutical quality control, bioanalytical testing, and environmental monitoring. By demonstrating that high sensitivity and selectivity need not compromise environmental responsibility, this methodology contributes to advancing eco-conscious analytical practices and addresses critical sustainability challenges in routine pharmaceutical laboratories.
The compatibility with both pharmaceutical and biological matrices, combined with minimal reliance on sophisticated instrumentation, underscores the practical scalability of this approach. Future applications may include adaptation for multi-analyte detection, integration with portable spectrofluorimeters for point-of-care testing, and expansion to other therapeutic classes where ion-pair formation mechanisms are feasible. The simplicity, cost-efficiency, and minimal ecological footprint make this hybrid methodology particularly suitable for resource-limited settings and large-scale quality control operations. Ultimately, this work supports the global transition toward sustainable analytical practices in pharmaceutical sciences, positioning the SALLE-fluorescence platform as a model for future method development that harmonizes analytical excellence with environmental stewardship.
Data availability
All data are available in the submitted manuscript and its supplementary file.
References
Schenck, F. J., Callery, P., Gannett, P. M., Daft, J. R. & Lehotay, S. J. Comparison of magnesium sulfate and sodium sulfate for removal of water from pesticide extracts of foods. J. AOAC Int. 85, 1177–1180 (2002).
Eller, K. I. & Lehotay, S. J. Evaluation of hydromatrix and magnesium sulfate drying agents for supercritical fluid extraction of multiple pesticides in produce. Analyst 122, 429–435 (1997).
Jayaganesh, S. & Venkatesan, S. Impact of magnesium sulphate on biochemical and quality. Am. J. Food Techn. 5, 31–39 (2010).
Arroyo-Manzanares, N., Huertas-Pérez, J. F., Lombardo-Agüí, M., Gámiz-Gracia, L. & García-Campaña, A. M. A high-throughput method for the determination of quinolones in different matrices by ultra-high performance liquid chromatography with fluorescence detection. Anal. Methods. 7, 253–259 (2015).
Mariño-Repizo, L., Goicoechea, H., Raba, J. & Cerutti, S. A simple, rapid and novel method based on salting-out assisted liquid–liquid extraction for ochratoxin A determination in beer samples prior to ultra-high-performance liquid chromatography coupled to tandem mass spectrometry. Food Addit. Contaminants: Part. A. 35, 1622–1632 (2018).
Legesse, A., Megersa, N. & Chandravanshi, B. S. Development of sensitive and efficient effervescence tablet-assisted salting-out liquid-liquid extraction for simultaneous enrichment of multiclass pesticide residues in water, fruits and vegetables. J. Food Compos. Anal. 108238 (2025).
Noche, G. G., Laespada, M. E. F., Pavón, J. L. P., Cordero, B. M. & Lorenzo, S. M. In situ aqueous derivatization and determination of non-steroidal anti-inflammatory drugs by salting-out-assisted liquid–liquid extraction and gas chromatography–mass spectrometry. J. Chromatogr. A. 1218, 6240–6247 (2011).
Liu, J., Jiang, M., Li, G., Xu, L. & Xie, M. Miniaturized salting-out liquid–liquid extraction of sulfonamides from different matrices. Anal. Chim. Acta. 679, 74–80 (2010).
Torres, P. U. Cinacalcet HCl: A novel treatment for secondary hyperparathyroidism caused by chronic kidney disease. J. Ren. Nutr. 16, 253–258 (2006).
Dru, T. B. & Ritz, E. Treatment of secondary hyperparathyroidism in CKD patients with cinacalcet and/or vitamin D derivatives. Clin. J. Am. Soc. Nephrol. 4, 234–241 (2009).
Levi, R. et al. Increased parathyroid hormone gene expression in secondary hyperparathyroidism of experimental uremia is reversed by calcimimetics: Correlation with posttranslational modification of the: trans: acting factor AUF1. J. Am. Soc. Nephrol. 17, 107–112 (2006).
Geibel, J. et al. Calcium-sensing receptor abrogates secretagogue-induced increases in intestinal net fluid secretion by enhancing cyclic nucleotide destruction. Proc. Natl. Acad. Sci. 103, 9390–9397 (2006).
Brown, E. The Calcium-Sensing Receptor: Physiology, Pathophysiology and Car-Based Therapeutics: Physiology and Pathophysiology of CaR, Calcium Signalling and Disease. 139–167 (Molecular Pathology of Calcium, 2007).
Elbashir, A. A., Suliman, F. E. O. & Aboul-Enein, H. Y. The application of 7-chloro-4-nitrobenzoxadiazole (NBD-Cl) for the analysis of pharmaceutical-bearing amine group using spectrophotometry and spectrofluorimetry techniques. Appl. Spectrosc. Rev. 46, 222–241 (2011).
El-Yazbi, A., Wagih, M., Ibrahim, F. & Barary, M. Spectrofluorimetric determination of topiramate and levetiracetam as single components in tablet formulations and in human plasma and simultaneous fourth derivative synchronous fluorescence determination of their co-adminstered mixture in human plasma. J. Fluoresc. 26, 1225–1238 (2016).
Hamad, A. A. Utility of a fluorescent probing strategy for designing a distinctive chemically mutagenized reaction for the determination of an antiepileptic agent; Topiramate. Talanta Open. 7, 100179 (2023).
Bartzatt, R. Fluorescent labeling of drugs and simple organic compounds containing amine functional groups, utilizing dansyl chloride in Na2CO3 buffer. J. Pharmacol. Toxicol. Methods. 45, 247–253 (2001).
Derayea, S. M., Madian, H., Samir, E., Hamad, A. A. & Badr El-Din, K. M. A feasible fluorimetric approach anchored in diaryl pyrrolone derivative for the facile analysis of milnacipran in tablets; Evaluation of the method greenness. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 273, 121024 (2022).
Hamad, A. A. & Derayea, S. M. A novel and unusual utility of the cardiosintol drug as a fluoro-prober in the amendment of a highly fluorescent module for determining the non-fluorescent N-acetylcysteine drug. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 293, 122460 (2023).
El Hamd, M. A. et al. Roth’s switch-on fluoremetric probe for green tracking and quantifying of 1.4-dihydropyridine medication: Evaluation of greenness, whiteness, and blueness. Sustainable Chem. Pharm. 36, 101294 (2023).
Loni, A. B., Ghante, M. R. & Sawant, S. Spectrophotometric estimation of cinacalcet hydrochloride in bulk and tablet dosage form. Int. J. Pharm. Pharm. Sci. 4, 513–515 (2012).
Darwish, I. A., Al-Shehri, M. M. & El-Gendy, M. A. Novel spectrophotometric method for determination of cinacalcet hydrochloride in its tablets via derivatization with 1, 2-naphthoquinone-4-sulphonate. Chem. Cent. J. 6, 1–8 (2012).
Mallikarjuna Rao, N. & Gowrisankar, D. Development and validation of novel stability indicating UV spectrophotometric method for the estimation of cinacalcet hydrochloride in bulk and tablet dosage forms. Indian Drugs. 53, 31 (2016).
Darwish, I. A., AlShehri, M. M. & Al-Gendy, M. A. A highly sensitive fluorimetric method for determination of cinacalcet hydrochloride in tablets and plasma via derivatization with 7-chloro-4-nitrobenzoxadiazole. Curr. Anal. Chem. 9, 504–512 (2013).
Zaid, M. H. A., El-Enany, N., Mostafa, A. E., Hadad, G. M. & Belal, F. Spectrofluorometric determination of cinacalcet hydrochloride: Greenness assessment and application to biological fluids and in-vitro dissolution testing. Methods Appl. Fluoresc. 11, 045005 (2023).
Kamatham, S. & Veeresham, C. Determination and validation of HPTLC method for cinacalcet hydrochloride. Am. J. Anal. Chem. 10, 55 (2019).
Panigrahi, D., Mishra, A. & Sahu, S. K. Development and validation of a RP-HPLC method for quantitative estimation of cinacalcet in tablet dosage form. World J. Pharm. Res. 7, 1016–1025 (2018).
Ganesh, N. S. et al. Development and validation RP-HPLC method for estimation of cinacalcet in bulk and tablet dosage form. Am. J. Pharm. Tech. Res. 5, 454–463 (2015).
Rao, N. M. & Sankar, D. G. Development and validation of stability indicating RP-HPLC method for the estimation of cinacalcet hydrochloride in bulk and their formulations. Biointerface Res. Appl. Chem. 10, 6610–6618 (2020).
Li, L. et al. An improved LC–MS/MS method for determination of cinacalcet in human plasma and its application to the evaluation of food intake effect on the pharmacokinetics of cinacalcet in healthy volunteers. Biomed. Chromatogr. 33, e4631 (2019).
Ramisetti, N. R. & Bompelli, S. LC-MS/MS determination of cinacalcet enantiomers in rat plasma on chirobiotic V column in polar ionic mode: application to a pharmacokinetic study. Biomed. Chromatogr. 28, 1846–1853 (2014).
Muhammad, N. et al. Online clean-up setup for the determination of non-fluorescent acidic pharmaceutical drugs in complex biological samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 121708, 1126–1127 (2019).
Muhammad, N. et al. Dual application of synthesized SnO 2 nanoparticles in ion chromatography for sensitive fluorescence determination of ketoprofen in human serum, urine, and canal water samples. New J. Chem. 41, 9321–9329 (2017).
Muhammad, N. et al. A comprehensive review of liquid chromatography hyphenated to post-column photoinduced fluorescence detection system for determination of analytes. Arab. J. Chem. 15, 104091 (2022).
Muhammad, N. et al. Ion chromatography coupled with fluorescence/UV detector: A comprehensive review of its applications in pesticides and pharmaceutical drug analysis. Arab. J. Chem. 14, 102972 (2021).
Al-Farhan, B. S., Ibrahim, A. M. M., Hassan, D. A., Saleh, S. F. & Hamad, A. A. Facile and sustainable electrostatic integration of eosin Y dye for the assay of the atomoxetine drug through the enhancement of the RRS phenomenon. J. Fluoresc. (2024).
Alqarni, A. M. et al. Application of a white and green spectrofluorimetric approach for facile quantification of amlodipine, a hypotensive drug, in batch materials, dosage forms, and biological fluids; Content homogeneity testing. Luminescence 39, e4661 (2024).
Majors, R. Salting-Out Liquid-Liquid Extraction (SALLE) (2009).
Tang, Y. Q. & Weng, N. Salting-Out Assisted Liquid–Liquid Extraction for Bioanalysis. Vol. 5. 1583–1598 (Bioanalysis, 2013).
Britton, H. T. S. & Robinson, R. A. CXCVIII.—Universal buffer solutions and the dissociation constant of veronal. J. Chem. Soc. (Resumed) 1456–1462 (1931).
Pesez, M. & Bartos, J. Colorimetric and Fluorimetrlc Analysis of Organic Compounds (Drugs Marcel Dekker, 1974).
Derayea, S. M., Hamad, A. A., Ali, R. & Ali, H. R. H. Investigating erythrosine B as a fluorimetric probe for the determination of benzimidazole drugs via facile complexation reaction. Microchem. J. 149, 104024 (2019).
McIlvaine, T. A buffer solution for colorimetric comparison. J. Biol. Chem. 49, 183–186 (1921).
Derayea, S. M., Abdel-Lateef, M. A., Mohammed, B. S., Hamad, A. A. & Samir, E. A comprehensive review on the utility of resonance Rayleigh scattering in pharmaceutical analysis; With emphasis on nanostructure role in analyte probing. Microchem. J. 191, 108774 (2023).
Rockville, M. The United States Pharmacopoeia 30, The National Formulary 25 US Pharmacopeial Convention. 2287–2288 (Electronic Version, 2007).
Snigur, D., Fizer, M., Chebotarev, A., Lukianova, O. & Zhukovetska, O. Spectroscopic and computational studies of erythrosine food dye protonation in aqueous solution. Dyes Pigm. 198, 110028 (2022).
Wang, J., Liu, Z., Liu, J., Liu, S. & Shen, W. Study on the interaction between fluoroquinolones and erythrosine by absorption, fluorescence and resonance Rayleigh scattering spectra and their application. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 69, 956–963 (2008).
Zang, X., Peng, J., Zhou, M. & Peng, H. A high selective and sensitive method for the detection of six psychotropic drugs in human urine by high performance liquid chromatography combined with resonance Rayleigh scattering spectra. RSC Adv. 5, 73091–73098 (2015).
Blatt, E., Chatelier, R. & Sawyer, W. Effects of quenching mechanism and type of quencher association on Stern-Volmer plots in compartmentalized systems. Biophys. J. 50, 349–356 (1986).
Lakowicz, J. R. & Lakowicz, J. R. Instrumentation for fluorescence spectroscopy. In Principles of Fluorescence Spectroscopy. 25–61 (1999).
Bi, S. et al. Investigation of the interaction between flavonoids and human serum albumin. J. Mol. Struct. 703, 37–45 (2004).
Kier, L. B. Quantitation of solvent polarity based on molecular structure. J. Pharm. Sci. 70, 930–933 (1981).
Shiao, S. et al. Chain length compatibility effects in mixed surfactant systems for technological applications. Adv. Colloid Interface Sci. 74, 1–29 (1998).
Hamad, A. A., Ali, R. & Derayea, S. M. A simple single jar on–off fluorescence designed system for the determination of mitoxantrone using an eosin Y dye in raw powder, vial, and human biofluids. RSC Adv. 12, 7413–7421 (2022).
Nigam, S. & Rutan, S. Principles and applications of solvatochromism. Appl. Spectrosc. 55, 362A–370A (2001).
Nasr, M. S., Kaddah, M. M. Y., Morshedy, S., Omran, G. & Talaat, W. Determination of baloxavir marboxil in pharmaceutical preparations and spiked human plasma using its quenching action on acetoxymercuric fluorescein reagent: Assessment of greenness and whiteness. Heliyon 10, e32120 (2024).
Coleman, J. S., Varga, L. P. & Mastin, S. H. Graphical methods for determining the number of species in solution from spectrophotometric data. Inorg. Chem. 9, 1015–1020 (1970).
Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 51, 660–672 (1949).
Cantor, C. R. & Schimmel, P. R. Biophysical Chemistry: Part II: Techniques for the Study of Biological Structure and Function (Macmillan, 1980).
Klotz, I. M. Numbers of receptor sites from Scatchard graphs: Facts and fantasies. Science 217, 1247–1249 (1982).
Lakowicz, J. R. Principles of Fluorescence Spectroscopy (Springer, 2006).
Guideline, I. H. T. Validation of analytical procedures: Text and methodology. Q2 (R1) 1, 05 (2005).
Oliva, A., Fariña, J. B. & Llabrés, M. Development and validation of an UPLC method for determination of content uniformity in low-dose solid drugs products using the design space approach. Talanta 115, 490–499 (2013).
Hamad, A. A. et al. An eco-friendly matrix-augmented fluorescence spectroscopic approach for the analysis of mitoxantrone, An oncogenic therapy; Application to the dosage form and biological matrices. Luminescence 38, 166–175 (2023).
Hamad, A. A. & Batubara, A. S. Planning and projecting of a green isoindole-based fluoro-probe for feasible tagging and tracking of topiramate, a non-fluorescent drug in bulk powder and prescribed commercial products. Talanta Open. 7, 100205 (2023).
Hamad, A. A. & Haredy, A. M. Designing a unique molecular size-dependent approach for determining levamisole via synergizing Rayleigh Scattering response of Cilefa Pink B dye; Application to bulk, dosage forms, and biofluids; Method greenness evaluation. Talanta Open 7, 100196 (2023).
Abdulhafez Hamad, A. Utility of Cilefa Pink B, a food dye in a facile decoration of the first green molecular-size-based fluorescence probe (MSBFP) for determining trimebutine; Application to bulk, dosage forms, and real plasma. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 288, 122187 (2023).
Hussein, O. G., Ahmed, D. A., Abdelkawy, M., Rezk, M. R. & Rostom, Y. A novel green spectrofluorimetric method for simultaneous determination of antazoline and tetryzoline in their ophthalmic formulation. Luminescence 39, e4728 (2024).
Rostom, Y., Hussein, O. G., Abdelkawy, M., Rezk, M. R. & Ahmed, D. A. A novel spectrofluorimetric determination of antazoline and xylometazoline in their ophthalmic formulation; Green approach and evaluation. J. Fluoresc. (2024).
Tobiszewski, M. Metrics for green analytical chemistry. Anal. Methods. 8, 2993–2999 (2016).
Tobiszewski, M., Marć, M., Gałuszka, A. & Namieśnik, J. Green chemistry metrics with special reference to green analytical chemistry. Molecules 20, 10928–10946 (2015).
Phan, T. T., Gallardo, C. & Mane, J. Green motion: A new and easy to use green chemistry metric from laboratories to industry. Green Chem. 17, 2846–2852 (2015).
Gałuszka, A., Migaszewski, Z. M., Konieczka, P. & Namieśnik, J. Analytical eco-scale for assessing the greenness of analytical procedures. TRAC Trends Anal. Chem. 37, 61–72 (2012).
Pena-Pereira, F., Wojnowski, W. & Tobiszewski, M. AGREE—Analytical GREEnness metric approach and software. Anal. Chem. 92, 10076–10082 (2020).
Płotka-Wasylka, J. A new tool for the evaluation of the analytical procedure: Green analytical procedure index. Talanta 181, 204–209 (2018).
Keith, L. H., Gron, L. U. & Young, J. L. Green analytical methodologies. Chem. Rev. 107, 2695–2708 (2007).
Wojnowski, W., Tobiszewski, M., Pena-Pereira, F. & Psillakis, E. AGREEprep – Analytical greenness metric for sample preparation. TRAC Trends Anal. Chem. 149, 116553 (2022).
Mansour, F. R., Płotka-Wasylka, J. & Locatelli, M. Modified GAPI (MoGAPI) tool and software for the assessment of method greenness: Case studies and applications. Analytica 5, 451–457 (2024).
Płotka-Wasylka, J. & Wojnowski, W. Complementary green analytical procedure index (ComplexGAPI) and software. Green Chem. 23, 8657–8665 (2021).
Sajid, M. & Płotka-Wasylka, J. Green analytical chemistry metrics: A review. Talanta 238, 123046 (2022).
Hamad, A. A., Saleh, S. F., Mahdi, W. A., Alshehri, S. & Hamd, M. A. E. Facile integration of hanztsch’s switch-off/on modeled fluorogenic probe for feasible tagging and tracking of the midodrine drug in different matrices; First evaluation of the method’s greenness, whiteness, blueness, quantum yield, and tablets’ content homogeneity. J. Fluoresc. (2024).
Morgan, E. M. et al. ChlorTox scale assessment, greenness, and whiteness evaluation of selective spectrophotometric analysis of dimenhydrinate and cinnarizine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 321, 124740 (2024).
Amin, K. F. M., Obaydo, R. H. & Lotfy, H. M. Chemometric-assisted spectrophotometric methods for simultaneous drug determination in new Helicobacter pylori treatment regimens - Environmental sustainability assessment. Sustainable Chem. Pharm. 42, 101849 (2024).
Amin, K. F. M., Obaydo, R. H. & Abdullah, A. M. Eco-friendly chemometric analysis: Sustainable quantification of five pharmaceutical compounds in bulk, tablets, and spiked human plasma. Results Chem. 11, 101761 (2024).
Larsen, C. et al. A tool for identifying green solvents for printed electronics. Nat. Commun. 12, 4510 (2021).
Chang, Y. M. et al. All solution and ambient processable organic photovoltaic modules fabricated by slot-die coating and achieved a certified 7.56% power conversion efficiency. Sol. Energy Mater. Sol. Cells. 202, 110064 (2019).
Saleh, S. S. et al. Guidelines for accurate application of green and white analytical concepts: Merits versus demerits with insights of significant milestones of assessment tools applied for antiviral drugs. Microchem. J. 199, 109917 (2024).
Nowak, P. M., Wietecha-Posłuszny, R. & Pawliszyn, J. White analytical chemistry: An approach to reconcile the principles of green analytical chemistry and functionality. TRAC Trends Anal. Chem. 138, 116223 (2021).
Manousi, N., Wojnowski, W., Płotka-Wasylka, J. & Samanidou, V. Blue applicability grade index (BAGI) and software: A new tool for the evaluation of method practicality. Green Chem. 25, 7598–7604 (2023).
Abdulhafez Hamad, A. et al. Utility of Cilefa Pink B, a foodstuff dye as a fluoro-substrate in the devising of the first facile green molecular-mass-related fluorescence sensor for quantifying amlodipine in batched material and dosage forms; Content uniformity evaluation. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 308, 123744 (2024).
Elagamy, S. H., Fuente-Ballesteros, A., Chanduluru, H. K. & Obaydo, R. H. Systematic review of recent metrics (2020–2025) for greenness, applicability, and analytical performance with guidelines for practical use. Results Chem. 103048. https://doi.org/10.1016/j.rechem.2025.103048 (2026).
Funding
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-DDRSP2601).
Author information
Authors and Affiliations
Contributions
Ahmed Abdulhafez Hamad: Supervision, Writing – original draft, Validation, Software, Resources, Project administration, Methodology, Investigation, Formal analysis, Data curation, and Conceptualization. Mohamed A.M. Ali: Writing – original draft, Supervision, Funding acquisition, Conceptualization. Anis Ahmad Chaudhary: Writing – review & editing, Writing – original draft, Visualization, Data curation. Reem H. Obaydo: Writing – original draft, Project administration, Data curation, Sustainability evaluation. Safaa F. Saleh and Angum M.M. Ibrahim: Writing – original draft, Visualization, Resources, Project administration, Formal analysis, Data curation. Wedad Mawkili and Nujud Maslamani: Writing – review & editing, Writing – original draft, Software, Project administration, Data curation, Conceptualization.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All experimental procedures, including analyses performed on spiked human plasma and urine samples, were reviewed and approved by the Institutional Ethics Committee for Scientific Research, Faculty of Pharmacy, Al-Azhar University, Assiut, Egypt. All methods were performed in accordance with the relevant guidelines and regulations. The investigators affirm that the work complied with the ethical principles outlined in the Declaration of Helsinki (1975) and its 2008 amendment.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hamad, A.A., Ali, M.A.M., Chaudhary, A.A. et al. Coupling MgSO4-assisted SALLE with a fluorimetric turn-off strategy for the determination of cinacalcet HCl in pharmaceutical and human matrices. Sci Rep 16, 13756 (2026). https://doi.org/10.1038/s41598-026-41887-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-41887-4
