
Abstract
Principles of long-term medical management in individuals with urea cycle disorders (UCDs) encompass (1) a low protein diet, (2) supplementation of arginine and/or citrulline along with essential amino acids, nutrients, vitamins and trace elements, and (3) use of nitrogen scavenging agents to reduce recurrent hyperammonemic events (HAEs). These principles aim at providing metabolic stability, elimimation of chronic complications, and achievement of normal development as well as growth. A retrospective comparative analysis was performed by studying 138 individuals with male ornithine transcarbamylase deficiency (mOTC-D), citrullinemia type 1 (CTLN1) and argininosuccinic aciduria (ASA) based on in vitro residual enzymatic activity for severity-adjustment. Results show that individuals with mOTC-D, CTLN1 and ASA are at risk of progressive linear growth impairment, recurrent annual HAEs and an unfavorable neurocognitive outcome despite being under long-term nitrogen scavenging pharmacotherapy. No overall superiority among existing nitrogen scavenging agents with regard to the individual’s metabolic stability, linear growth impairment and poor neurocognitive outcome was observed. Novel therapeutic strategies are urgently needed to ultimately improve health outcomes in individuals with UCDs in order to sufficiently meet guideline-specific goals.
Similar content being viewed by others

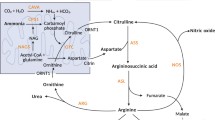
Abbreviations
- ASA:
-
Argininosuccinic aciduria
- BCAAs:
-
Branched chain amino acids
- BZA:
-
Sodium benzoate
- cSDS:
-
Cognitive standard deviation score
- CTLN1:
-
Citrullinemia type 1
- E-IMD:
-
European registry and network for intoxication type metabolic diseases
- IOP(s):
-
Individual observation period(s)
- initial NH4 + max :
-
Initial peak plasma ammonium concentration
- IQR:
-
Interquartile range
- mOTC-D:
-
Male ornithine transcarbamylase deficiency
- PBA:
-
Sodium/glycerol phenylbutyrate
- SD(S):
-
Standard deviation (score)
- UCDC:
-
Urea cycle disorders consortium
- UCD(s):
-
Urea cycle disorder(s)
References
Kolker, S. et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: The initial presentation. J. Inherit. Metab. Dis. 38, 1041–1057. https://doi.org/10.1007/s10545-015-9839-3 (2015).
Kolker, S. et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: The evolving clinical phenotype. J. Inherit. Metab. Dis. 38, 1059–1074. https://doi.org/10.1007/s10545-015-9840-x (2015).
Ruegger, C. M. et al. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J. Inherit. Metab. Dis. 37, 21–30. https://doi.org/10.1007/s10545-013-9624-0 (2014).
Summar, M. L. et al. The incidence of urea cycle disorders. Mol. Genet. Metab. 110, 179–180. https://doi.org/10.1016/j.ymgme.2013.07.008 (2013).
Haberle, J. et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 42, 1192–1230. https://doi.org/10.1002/jimd.12100 (2019).
Posset, R. et al. Long-term effects of medical management on growth and weight in individuals with urea cycle disorders. Sci. Rep. 10, 11948. https://doi.org/10.1038/s41598-020-67496-3 (2020).
Scharre, S. et al. Predicting the disease severity in male individuals with ornithine transcarbamylase deficiency. Ann. Clin. Transl. Neurol. 9, 1715–1726. https://doi.org/10.1002/acn3.51668 (2022).
Zielonka, M. et al. From genotype to phenotype: Early prediction of disease severity in argininosuccinic aciduria. Hum. Mutat. 41, 946–960. https://doi.org/10.1002/humu.23983 (2020).
Zielonka, M. et al. Early prediction of phenotypic severity in Citrullinemia Type 1. Ann. Clin. Transl. Neurol. 6, 1858–1871. https://doi.org/10.1002/acn3.50886 (2019).
Posset, R. et al. Impact of supplementation with L-citrulline/arginine after liver transplantation in individuals with Urea Cycle Disorders. Mol. Genet. Metab. 141, 108112. https://doi.org/10.1016/j.ymgme.2023.108112 (2024).
Posset, R. et al. Severity-adjusted evaluation of liver transplantation on health outcomes in urea cycle disorders. Genet. Med. 26, 101039. https://doi.org/10.1016/j.gim.2023.101039 (2023).
Posset, R. et al. Severity-adjusted evaluation of newborn screening on the metabolic disease course in individuals with cytosolic urea cycle disorders. Mol. Genet. Metab. 131, 390–397. https://doi.org/10.1016/j.ymgme.2020.10.013 (2020).
Zielonka, M. et al. Severity-adjusted evaluation of initial dialysis on short-term health outcomes in urea cycle disorders. Mol. Genet. Metab. 143, 108566. https://doi.org/10.1016/j.ymgme.2024.108566 (2024).
Enns, G. M. Nitrogen sparing therapy revisited 2009. Mol. Genet. Metab. 100(Suppl 1), S65-71. https://doi.org/10.1016/j.ymgme.2010.02.007 (2010).
Enns, G. M. et al. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N. Engl. J. Med. 356, 2282–2292. https://doi.org/10.1056/NEJMoa066596 (2007).
Nagamani, S. C. S. et al. A randomized trial to study the comparative efficacy of phenylbutyrate and benzoate on nitrogen excretion and ureagenesis in healthy volunteers. Genet. Med. 20, 708–716. https://doi.org/10.1038/gim.2017.167 (2018).
Posset, R. et al. Age at disease onset and peak ammonium level rather than interventional variables predict the neurological outcome in urea cycle disorders. J. Inherit. Metab. Dis. 39, 661–672. https://doi.org/10.1007/s10545-016-9938-9 (2016).
Posset, R. et al. Impact of diagnosis and therapy on cognitive function in urea cycle disorders. Ann. Neurol. 86, 116–128. https://doi.org/10.1002/ana.25492 (2019).
Bak, L. K., Schousboe, A. & Waagepetersen, H. S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 98, 641–653. https://doi.org/10.1111/j.1471-4159.2006.03913.x (2006).
Blei, A. T. & Larsen, F. S. Pathophysiology of cerebral edema in fulminant hepatic failure. J. Hepatol. 31, 771–776. https://doi.org/10.1016/s0168-8278(99)80361-4 (1999).
Gropman, A. L., Summar, M. & Leonard, J. V. Neurological implications of urea cycle disorders. J. Inherit. Metab. Dis. 30, 865–879. https://doi.org/10.1007/s10545-007-0709-5 (2007).
Kolker, S. Metabolism of amino acid neurotransmitters: The synaptic disorder underlying inherited metabolic diseases. J. Inherit. Metab. Dis. 41, 1055–1063. https://doi.org/10.1007/s10545-018-0201-4 (2018).
Probst, J. et al. Chronic hyperammonemia causes a hypoglutamatergic and hyperGABAergic metabolic state associated with neurobehavioral abnormalities in zebrafish larvae. Exp. Neurol. 331, 113330. https://doi.org/10.1016/j.expneurol.2020.113330 (2020).
Zielonka, M. et al. Pharmacologic rescue of hyperammonemia-induced toxicity in zebrafish by inhibition of ornithine aminotransferase. PLoS ONE 13, e0203707. https://doi.org/10.1371/journal.pone.0203707 (2018).
Zielonka, M. et al. Bioenergetic dysfunction in a zebrafish model of acute hyperammonemic decompensation. Exp. Neurol. 314, 91–99. https://doi.org/10.1016/j.expneurol.2019.01.008 (2019).
Hediger, N., Landolt, M. A., Diez-Fernandez, C., Huemer, M. & Haberle, J. The impact of ammonia levels and dialysis on outcome in 202 patients with neonatal onset urea cycle disorders. J. Inherit. Metab. Dis. 41, 689–698. https://doi.org/10.1007/s10545-018-0157-4 (2018).
Kido, J. et al. Long-term outcome of urea cycle disorders: Report from a nationwide study in Japan. J. Inherit. Metab. Dis. 44, 826–837. https://doi.org/10.1002/jimd.12384 (2021).
Baruteau, J. et al. Expanding the phenotype in argininosuccinic aciduria: Need for new therapies. J. Inherit. Metab. Dis. 40, 357–368. https://doi.org/10.1007/s10545-017-0022-x (2017).
Castello, A. et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406. https://doi.org/10.1016/j.cell.2012.04.031 (2012).
Erez, A. et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 17, 1619–1626. https://doi.org/10.1038/nm.2544 (2011).
Lerner, S. et al. ASL metabolically regulates tyrosine hydroxylase in the Nucleus Locus Coeruleus. Cell. Rep. 29, e2147. https://doi.org/10.1016/j.celrep.2019.10.043 (2019).
Lerner, S. et al. ASL expression in ALDH1A1(+) neurons in the substantia nigra metabolically contributes to neurodegenerative phenotype. Hum. Genet. 140, 1471–1485. https://doi.org/10.1007/s00439-021-02345-5 (2021).
Seidl, M. J. et al. ASS1 deficiency is associated with impaired neuronal differentiation in zebrafish larvae. Mol. Genet. Metab. 141, 108097. https://doi.org/10.1016/j.ymgme.2023.108097 (2024).
Burrage, L. C. et al. Sodium phenylbutyrate decreases plasma branched-chain amino acids in patients with urea cycle disorders. Mol. Genet. Metab. 113, 131–135. https://doi.org/10.1016/j.ymgme.2014.06.005 (2014).
Kolker, S. et al. Networking across borders for individuals with organic acidurias and urea cycle disorders: The E-IMD Consortium. JIMD. Rep. 22, 29–38. https://doi.org/10.1007/8904_2015_408 (2015).
Posset, R. et al. Transatlantic combined and comparative data analysis of 1095 patients with urea cycle disorders-a successful strategy for clinical research of rare diseases. J. Inherit. Metab. Dis. 42, 93–106. https://doi.org/10.1002/jimd.12031 (2019).
Seminara, J. et al. Establishing a consortium for the study of rare diseases: The urea cycle disorders consortium. Mol. Genet. Metab. 100(Suppl 1), S97-105. https://doi.org/10.1016/j.ymgme.2010.01.014 (2010).
Summar, M. L., Endo, F. & Kolker, S. On the creation, utility and sustaining of rare diseases research networks: Lessons learned from the Urea Cycle Disorders Consortium, the Japanese Urea Cycle Disorders Consortium and the European Registry and Network for Intoxication Type Metabolic Diseases. Mol. Genet. Metab. 113, 105–108. https://doi.org/10.1016/j.ymgme.2014.09.002 (2014).
Cole, T. J., Freeman, J. V. & Preece, M. A. British 1990 growth reference centiles for weight, height, body mass index and head circumference fitted by maximum penalized likelihood. Stat. Med. 17, 407–429 (1998).
Acknowledgements
All UCDC and E-IMD sites contributed to the datasets of the longitudinal studies used in this publication. Principal investigators and personnel with key contributions are listed as UCDC and E-IMD consortia study group members. The Urea Cycle Disorders Consortium (UCDC; U54HD061221) is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The Urea Cycle Disorders Consortium is also supported by the O’Malley Foundation and the Kettering Fund. Members of the UCDC include the following, some of whom have been listed as authors on this manuscript: Nicholas Ah Mew, Matthias R. Baumgartner, Gerard Berry, Susan A. Berry, Margo Breilyn, Lindsay Burrage, Curtis Coughlin, George A. Diaz, Gregory Enns, Can Ficicioglu, Renata C. Gallagher, Andrea Gropman, Cary O. Harding, Georg F. Hoffmann, Laura Konczal, Christina Lam, Shawn E. McCandless, Sandesh C.S. Nagamani, Roland Posset, Andreas Schulze, Jennifer Seminara, Tamar Stricker, Derek Wong, and Greta Wilkening. Furthermore, we gratefully acknowledge the following study coordinators – Saima Ali, Kim Bardillon, Kia Bryan, Liora Caspi, Kiaira Coles, Sara Elsbecker, Debbie Fu, Florian Gleich, Seishu Horikoshi, Elijah Kravets, Genya Kisin, Rhonda Jones, Ursula Kuhn, Jennifer Phillip, Thu Quan, Kara Simpson, Julia Sloan, Tamar Stricker, Hayden Vreugdenhil, Ashley Wilson, and Melissa Zerofsky – and study neuropsychologists – Fabienne Dietrich Alber, Christopher Boys, David Breiger, Benjamin Goodlett, Elizabeth Kerr, Casey Krueger, Eva Mamak, Ami Norris-Brilliant, David Schwartz, Yuri Shishido, Arianna K. Stefanatos, Rachel Tangen, and Magdalena E. Walter. We would also like to acknowledge the contributions of (former) longitudinal study PIs: Mark L. Batshaw, Stephen Cederbaum, Annette Feigenbaum, Douglas S. Kerr, Brendan Lee, Uta Lichter-Konecki, Margretta R. Seashore, and Marshall L. Summar. The authors are grateful for the contributions and leadership of the late Cynthia LeMons. Ms LeMons served as a Co-PI for the UCDC and the Executive Director for the National Urea Cycle Disorders Foundation. Furthermore, we gratefully thank Persephone Augoustides-Savvopoulou for his valuable contribution to the E-IMD. In particular, we are indebted to all our individuals with UCDs and their families for their trust, patience, and participation in both longitudinal registry studies for many years.
Funding
Open Access funding enabled and organized by Projekt DEAL. The Urea Cycle Disorders Consortium (UCDC; U54HD061221) is part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The Urea Cycle Disorders Consortium is also supported by the O’Malley Foundation, the Rotenberg Family Fund, the Dietmar Hopp Foundation, the Kettering Fund, and the National Urea Cycle Disorders Foundation. In addition, support for neuropsychological testing is provided by a NIH grant for Intellectual and Developmental Disability Research Centers (U54HD090257). This work was also supported in part by the Clinical Translational Core at Baylor College of Medicine which is supported by the IDDRC grant number P50 HD103555 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The E-IMD patient registry has received funding by the European Union (E-IMD; EAHC no 2010 12 01; coordinator: Stefan Kölker), in the framework of the Health Programme. After the end of the EU funding period the E-IMD patient registry has been sustained by funding from the Kindness-for-Kids Foundation (Munich, Germany), and the Kettering Fund. This work was supported by a Trainee Research Fellowship Award 2022–2023 provided by the UCDC. S.K., and G.F.H. receive funding from the Dietmar Hopp Foundation (St. Leon-Rot, Germany) for coordinating the study “Newborn Screening 2020/2025” and “Long-term outcome of patients with inherited metabolic diseases identified by expanded newborn screening” (grant numbers 23011220, 23011221, 1DH1911376 and 1DH2011117), which include individuals with UCDs.
Author information
Authors and Affiliations
Contributions
Dr. Posset and Dr. Zielonka contributed to conception and design of the study. Dr. Posset, Mrs. Epp, Dr. Garbade, Mr. Gleich, Dr. Gropman, Dr. Nagamani, Dr. Hoffmann, Dr. Kölker, Dr. Zielonka, as well as all individual contributors from the UCDC and E-IMD (Suppl. Table 7) contributed to the acquisition, analysis, and interpretation of data. Dr. Posset, Dr. Garbade, Mr. Gleich, and Dr. Zielonka contributed to drafting the text and preparing the figures. All authors finally approved the content of the manuscript. Dr. Posset and Dr. Zielonka agree to be accountable for all aspects of the work in ensuring that questions related to the integrity and accuracy of any part of the work are appropriately investigated and resolved.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Posset, R., Epp, F., Garbade, S.F. et al. Impact of long-term nitrogen scavenger therapy on clinical outcome in individuals with urea cycle disorders. Sci Rep (2026). https://doi.org/10.1038/s41598-026-42150-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-026-42150-6
