
Abstract
This study isolated, characterized, and identified rhizosphere-associated soil bacteria from sorghum-cultivated soils in Bhubaneswar, India, using serial dilution and spread plate techniques on Nutrient Agar. Morphological and biochemical analyses (Gram staining, catalase, oxidase, MR, and indole tests) identified 13 bacterial isolates, predominantly Gram-positive rods (Bacillus, Paenibacillus) and cocci (Staphylococcus, Micrococcus), with one Gram-negative isolate. Molecular characterization involved 16S rRNA gene amplification (~ 1500 bp), sanger sequencing, and phylogenetic analysis. BLASTN confirmed AG3 as Bacillus licheniformis (99.37% identity) (GenBank: PV590072), and AG11 as Bacillus cereus (100% identity) (GenBank: PV590099). The phylogenetic tress analysis of bacterial isolates AG3 and AG11 revealed distinct evolutionary rate variations. For AG3, site-specific rates ranged from 0.07 to 3.64 substitutions/site (nucleotide frequencies: A = 24.50%, T/U = 19.99%, C = 24.70%, G = 30.81%), while AG11 exhibited more uniform rates ranged from 0.90 to 1.10 substitutions/site (nucleotide frequencies: A = 25.79%, T/U = 20.86%, C = 22.71%, G = 30.64%). Maximum likelihood trees (log-likelihood: − 4077.853 for AG3, − 1928.562 for AG11) with 1,531 (AG3) and 1,402 (AG11) aligned positions resolved their phylogenetic relationships. This study provides valuable insights into the diversity and evolutionary dynamics of rhizosphere-associated Bacillus spp. in sorghum-cultivated soils, contributing to a better understanding of their ecological significance and potential application in sustainable agriculture and plant growth promotion.
Similar content being viewed by others


Introduction
Sorghum (Sorghum bicolor L.) ranks as the world’s fifth-most important cereal crop, with global production exceeding 60 million tons annually across 45 million hectares, thriving especially in arid and semi-arid regions due to its exceptional drought tolerance, yielding up to 40% more than maize under water-limited conditions1. Its rhizosphere supports a diverse microbiome, where plant growth-promoting rhizobacteria (PGPR) like Bacillus species boost nutrient uptake by 20–50%, enhance drought and salinity stress tolerance, and suppress pathogens via antagonism, reducing disease incidence by up to 70%2. Notably, Bacillus licheniformis and Bacillus cereus stand out for their varied roles, i.e., promoting root elongation and yield increases of 15–30%3. Recent studies have reported that B. licheniformis can improve phosphate solubilization (increasing available P by up to 35%) and produce auxins (up to 12 µg/ml)4, while B. cereus exhibits biocontrol potential against fungal pathogens, reducing disease incidence by 40–60%5. However, despite these benefits, precise identification and characterization of these isolates remain challenging due to overlapping phenotypic traits and genomic similarities among Bacillus spp., necessitating advanced morpho-biochemical and molecular techniques for accurate discrimination6.
Traditional identification methods, such as Gram staining, catalase tests, and carbohydrate fermentation assays, often yield ambiguous results due to high intra-species variability, with misidentification rates exceeding 15% in some studies7. Modern molecular techniques, including 16S rRNA sequencing and species-specific PCR (e.g., gyrB and rpoB gene markers), have improved accuracy, with phylogenetic analyses showing > 98% bootstrap support for correct classification8. Despite these advancements, inconsistencies persist in correlating phenotypic traits with genotypic data, particularly in environmental isolates, where horizontal gene transfer and niche adaptation may blur taxonomic boundaries9. While numerous studies have explored Bacillus spp. in agricultural systems, a critical gap exists in the comprehensive, multi-method identification of B. licheniformis and B. cereus specifically from the sorghum rhizosphere10. Most research has focused on either biochemical or molecular methods in isolation, leading to incomplete strain characterization11,12. Furthermore, comparative statistical analyses of morpho-biochemical and genomic datasets are lacking, particularly in distinguishing beneficial versus pathogenic strains within these species13. Therefore, this research addresses the critical gap of integrating morpho-biochemical assays with molecular diagnostics for the precise identification and comparative analysis of Bacillus sp. specifically from the sorghum rhizosphere, where such a consolidated approach is lacking.
Materials and methods
Isolation and purification of soil bacteria
Soil samples were collected from three distinct locations in Bhubaneswar, India, representing diverse land uses: SS1 at Smrutivan Park (N 20°14′ 34.98″ E 85° 45′ 43.74″), an urban green space; SS2 at ICAR—Indian Institute of Pulses Research Regional Station (N 20° 11′ 18.26″ E 85° 37′ 22.58″), an agricultural research site; and SS3 at C.V. Raman Global University Campus (20.220493° N 85.736457° E), an academic campus with research facilities. Sorghum seeds (Specialty, Sweet, and Fodder CSV 33MF varieties) were procured from a local market (Satya Seeds and Nursery; 20.26470° N 85.8400° E) and sown in polybags containing a soil-coco peat mixture. Each sorghum variety was cultivated in its corresponding soil sample (SS1, SS2, and SS3, respectively) to assess microbial interactions and soil-specific growth effects. After that, soil were gathered from the rhizosphere of each sorghum plant without destroying the secondary and tertiary root regions about 0–15 cm with a sterile spatula14. Samples were placed in sterile, airtight containers, stored at 4 °C, and processed within 24–48 h to minimize changes in microbial composition15. Isolation of bacteria was carried out using the serial dilution and spread plate technique16. About 1 g of each soil sample was diluted in 9 ml of sterile distilled water and serially diluted up to 10⁻⁶. From the appropriate dilutions (10−4 to 10−6), 0.1 ml was plated on sterile Nutrient Agar (NA) plates and incubated at 28 ± 2 °C for 24–48 h17.
Morphological and biochemical characterization
The isolation and purification of bacterial strains was performed through selective sub-culturing of morphologically distinct colonies that showed variation in phenotypic characteristics including colony color, size, shape, and margin formation. Selected colonies were repeatedly streaked onto fresh NA plates to obtain axenic cultures, followed by preservation on agar slants at 4 °C for subsequent morphological, biochemical and molecular characterization18. The Gram staining protocol was performed on purified bacterial isolates to characterize their cell wall properties and morphology according to the standardized method. Briefly, heat-fixed bacterial smears were sequentially stained with crystal violet (primary stain), Gram’s iodine (mordant), ethanol (decolorizer), and safranin (counterstain). Following each staining step, slides were gently rinsed with distilled water19.
Biochemical characterization included the Methyl Red (MR), Indole Test, Catalase, and Oxidase tests as per standard protocols20. For the Catalase test, a fresh colony was exposed to 3% hydrogen peroxide, and the rapid evolution of oxygen bubbles indicated catalase activity. The Oxidase test was performed using oxidase reagent on filter paper. A color change to deep purple within 30 s confirmed a positive result. The MR tests were performed in MR broth. After 48 h of incubation at 37 °C, methyl red reagent was added to detect mixed acid fermentation for MR test. A red ring in either case was considered a positive result21. Moreover, the indole test was performed to detect the ability of bacterial isolates to produce indole from tryptophan using Kovac’s reagent. After incubating the cultures in tryptone broth for 24–48 h, Kovac’s reagent was added, forming a red ring at the surface for indole-positive strains.
Molecular characterization
Genomic DNA was extracted from bacterial isolates using the HiPurA Bacterial Genomic DNA Purification Kit (HiMedia, India). The optimized protocol involved enzymatic and mechanical cell lysis, followed by DNA binding to a silica membrane and final elution in nuclease-free water. The concentration and purity of the extracted DNA were assessed via NanoDrop spectrophotometry (A260/280 ratios of 1.8–2.0), and its integrity was confirmed by 0.8% agarose gel electrophoresis to visualize high molecular weight genomic DNA. The 16S rRNA gene was amplified using universal primers, including the forward primer SD13F_16srRNA_FP (5'-TGGAGAGTTTGATCATGGCTC-3') and reverse primer SD13F_16srRNA_RP (5'-ACGGCTACCTTGTTACGACTT-3'). The 25 µL PCR reaction mixture contained 1X PCR buffer, 1.5 mM MgCl₂, 200 µM of each dNTP, 0.5 µM of each primer, 1 unit of Taq DNA polymerase, and approximately 50 ng of template DNA. Amplification was performed under the following conditions: initial denaturation at 95 °C for 5 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 45 s, and extension at 72 °C for 1 min 30 s; followed by a final extension at 72 °C for 10 min22. Amplicons were analyzed by 1.5% agarose gel electrophoresis using a 1 kb DNA ladder as a size standard and visualized with ethidium bromide under UV light. Only samples exhibiting a single, intense band at the expected size (~ 1500 bp) were selected for downstream processing23. Target bands were excised and purified using a QIAquick Gel Extraction Kit (Qiagen) to remove primer dimers, non-specific products, and enzyme contaminants, thereby ensuring high-quality template for sequencing24.
Sanger sequencing, BLAST identification, and phylogenetic analysis
Bidirectional Sanger sequencing of the purified PCR amplicons was performed by Eurofins Genomics (India) employing BigDye Terminator v3.1 chemistry and capillary electrophoresis, yielding high-quality reads exceeding 800 bp in length. Prior to consensus sequence assembly, these reads were subjected to a quality trimming process, typically involving the removal of terminal bases with Phred quality scores (Q) below 30, to minimize sequencing errors25. For bioinformatic processing, the raw sequence chromatograms underwent rigorous quality control, including the trimming of low-quality regions. Forward and reverse reads were then assembled into contigs to generate high-fidelity consensus sequences. Taxonomic assignment was achieved via BLASTN queries against the NCBI nucleotide database, applying stringent statistical thresholds (E-value < 1e-5, percent identity ≥ 97%) to ensure reliable identification26. Phylogenetic reconstruction was conducted using MEGA 12 (https://www.megasoftware.net/) software. Multiple sequence alignment was performed with the CLUSTAL W algorithm. A neighbor-joining phylogenetic tree was constructed based on the Kimura 2-parameter nucleotide substitution model using 1000 bootstrap replicates27.
Maximum likelihood estimation of gamma-distributed site rates
The among-site rate heterogeneity within the alignment was modeled using a discrete gamma distribution. The shape parameter (α) of the gamma distribution was optimized via maximum likelihood estimation (MLE) within MEGA 12 software, with the mean substitution rate constrained to 1.028. The likelihood function was numerically maximized using MEGA’s iterative optimization algorithms. Confidence intervals for the estimated α were derived from the inverse of the observed Fisher information matrix or, alternatively, through bootstrap resampling of the alignment. A low α value (< 1) indicated pronounced rate variation across sites, whereas α > 1 suggests a more uniform rate distribution29.
Estimating the mean relative evolutionary rates for each site
Mean relative evolutionary rates per site were estimated, with the rate scale normalized such that an average rate of 1.0 across all sites represents the phylogenetic mean. Consequently, site-specific rates below 1 indicate evolution slower than the mean, while rates exceeding 1 denote accelerated evolution. This analysis was implemented under the General Time Reversible (GTR) nucleotide substitution model, incorporating a discrete Gamma distribution (+ G) to model among-site rate heterogeneity. Rate variation was approximated using five discrete categories. The mean relative rate for a given site was then derived as the weighted average across these categories, based on their respective probabilities and the mean rate of each category30.
Results
Morphological parameters analysis
The morphological characterization of 13 bacterial isolates identified from sorghum rhizosphere revealed distinct gram reactions and cellular morphologies, enabling preliminary taxonomic classification (Table 1). Gram-positive rod-shaped bacteria (AG1, AG3, AG4, AG7, AG8, AG10, AG11, AG12, and AG13) were predominant and tentatively identified as Bacillus, Paenibacillus, Lactobacillus, Clostridium, or Enterococcus species (Fig. 1), while AG4 exhibited pleomorphic rod/cocci morphology, suggesting Arthrobacter (Fig. 2). Gram-positive cocci (AG2, AG5, and AG6) were likely Staphylococcus, Micrococcus, or related genera, with AG6 displaying oval-shaped cocci typical of Micrococcus. The sole Gram-negative isolate (AG9) exhibited cocci morphology, potentially belonging to Neisseria or Acinetobacter. These findings aligned with standard Gram staining protocols and phenotypic observations, providing a basis for further biochemical and molecular analysis.

Taxonomic groups; (A) Staphylococcus; (B) Lactobacillus/Clostridium; (C) Neisseria/Acinetobacter; and (D) Enterococcus.

Gram staining and colony morphology; (A) Gram + ve Bacteria, (B) Gram -ve Bacteria, (C) Rod shape and (D) Spherical Shape.
Biochemical parameters assessment
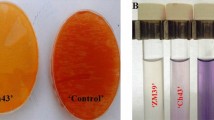
The biochemical characterization of the isolates revealed distinct metabolic profiles that aligned with their morphological classification. The study presented morphological characterization for all 13 bacterial isolates from the sorghum rhizosphere, enabling preliminary taxonomic grouping, while biochemical tests were reported only for 7 selected isolates (AG3, AG6, AG7, AG9, AG11, AG12, and AG13). This selective biochemical analysis was performed based on predefined criteria prioritizing diverse morphological representatives, such as both Gram-positive (rods, cocci, and oval) and the sole Gram-negative isolate (AG9), to efficiently validate likely taxonomic affiliations without redundant testing on morphologically similar Gram-positive rods (i.e., Bacillus-like AG1, AG8, AG10 excluded as they clustered with tested AG3/AG11). The AG3, AG6, AG9, AG11, AG12, and AG13 were catalase-positive, while AG7 was catalase-negative. Only AG12 and AG13 were tryptophanase-positive (indole test), while the rest were negative. In the MR test, AG3, AG11, AG12, and AG13 showed positive (red) results, indicating mixed acid fermentation, whereas AG6, AG7, and AG9 were negative (yellow), suggesting non-fermentative or neutral end products (Table 2).
AG3 and AG11, both Gram-positive, rod-shaped isolates likely belonging to Bacillus/Paenibacillus, exhibited catalase-positive reactions, indicating the presence of catalase enzyme for hydrogen peroxide detoxification, a common trait in aerobic/facultative anaerobic bacteria. Both were tryptophanase-negative, suggesting an inability to degrade tryptophan into indole, and methyl red (MR) test-positive, confirming mixed acid fermentation, a feature consistent with Bacillus spp. In contrast, AG6, AG7, and AG9 were MR-negative, indicating butanediol fermentation, while AG12 and AG13, though MR-positive, were tryptophanase-positive, aligning with Enterococcus spp (Fig. 3). AG3 and AG11 were prioritized for molecular characterization because they share identical biochemical profiles, both catalase-positive, tryptophanase-negative, and MR test positive (red), distinguishing them from the diverse profiles of other isolates like AG6, AG7, and AG9.

Biochemical activity of bacterial isolates (A) Catalysts + ve; (B) Catalysts – ve; (C) Indole + ve; (D) Indole –ve; (E) MR + ve; and (F) MR − ve.
Molecular parameters assessment
PCR amplification targeting the 16S rRNA gene was performed on genomic DNA extracted from pure cultures of isolates AG3 and AG11. Electrophoretic separation of the amplicons yielded a single, intense band at approximately 1500 base pairs for each isolate (Fig. 4C), corresponding to the expected length of the 16S rRNA gene fragment. The 1 kb DNA ladder provided a size standard, confirming the specific amplification of the target region, a prerequisite for reliable bacterial identification.

Isolation and molecular characterization of bacterial isolates; (A and B) pure culture isolation by streak plate method; (C) Agarose gel electrophoresis of 16S rRNA gene PCR products from bacterial isolates AG3 and AG11, with a 1 kb DNA ladder as size reference. Bands at ~ 1500 bp confirmed successful amplification of target sequences. An uncropped agarose gel electrophoresis image of 16S rRNA gene PCR products from bacterial isolates AG3 and AG11 is also indexed in Figure S1.
Sanger sequencing and BLAST analysis
The bidirectional Sanger sequencing raw chromatograms for bacterial isolates AG3 (Figure S2A) and AG11 (Figure S2B) were generated using fluorescent dye-terminator chemistry, with forward and reverse strands sequenced to ensure accuracy. The chromatograms displayed peaks corresponding to A (green), C (blue), G (black), and T (red) nucleotides, with a read length of 1045 bases and a scaling factor of 1.0x. Key sequence regions, such as “ACACGTGGGTAACCTGCC” in AG3 and “TTGAAAGGGCGGCTTCGGCT” in AG11, exhibited high signal-to-noise ratios, indicating robust sequencing. The bidirectional coverage allowed for cross-validation, resolving ambiguities in homopolymeric regions. The processed data confirmed high-confidence consensus sequences for both isolates, suitable for downstream phylogenetic or functional analyses. The BLAST analysis revealed that, for AG3 (Figure S3A), the top matches were with Bacillus licheniformis strains, with the highest Max Score of 2601 and 99.37% identity for strains DSM 13 and BCRC 11702. Other strains, such as ATCC 14580 and NBRC 12200, showed slightly lower Max Scores (2590) and identities (99.23% and 99.16%, respectively). All matches had 100% query coverage and an E value of 0.0, confirming high similarity. For AG11 (Figure S3B), the results indicated a 100% identity match with multiple Bacillus cereus strains, including CCM 2010, NBRC 15305, and ATCC 14579, all with a Max Score of 2590, 100% query coverage, and an E value of 0.0. Additionally, Bacillus proteolyticus and Bacillus albus showed near-perfect matches (99.93% identity) with Max Scores of 2584. These results confirmed AG3 as Bacillus licheniformis and AG11 as Bacillus cereus.
Phylogenetic tree construction
A phylogenetic tree was constructed to identify the evolutionary relationship of the isolate AG3 (GenBank accession no. PV590072) (Fig. 5). The isolate was identified as a member of the Bacillus licheniformis group based solely on 16S rRNA gene sequence similarity, which has limited resolution for distinguishing strains or closely related species within this group. The analysis grouped 115 Bacillus licheniformis strains into two major clusters: Cluster I and Cluster II. Cluster I comprised the majority of the strains, with PV590072 clearly falling within this cluster, suggesting its close evolutionary relationship to other strains in the same group. In contrast, Cluster II included a smaller set of divergent strains (e.g., CP016842.1:9–1447 to CP058453.1:3–1451), indicating greater genetic variation from those in Cluster I. The bootstrap values, indicated at the branch points, provided statistical support for the branching patterns; values above 70% were observed for several clades, indicating high confidence in those nodes. The scale bar represented a genetic distance of 0.10 substitutions per nucleotide position, reflecting evolutionary divergence.

Phylogenetic tree of isolate AG3: Bacillus licheniformis (Genbank accession no. PV590072).
The phylogenetic tree presented in Fig. 6 was constructed to determine the evolutionary relationship of the isolate AG11 (GenBank accession no. PV590099). The isolate was identified as a member of the Bacillus cereus group based solely on 16S rRNA gene sequence similarity, which has limited resolution for distinguishing strains or closely related species within this group. The tree was generated using multiple sequence alignment of 16S rRNA gene sequences from various Bacillus cereus group strains and was categorized into two major clusters: Cluster I and Cluster II. Cluster I was further divided into five sub-clusters (IA, IB, IC, ID, IE), while Cluster II was subdivided into IIA and IIB. Accession no. PV590099 was positioned within Cluster IIB, forming a closely related group with other strains such as OK060371.1, CP092361.1, and OQ858998.1. This sub-cluster (IIB) was distinct from others in both Cluster I and II, indicating a moderate level of genetic divergence.

Phylogenetic tree of isolate AG11: Bacillus cereus (Genbank accession no. PV590099).
Maximum likelihood estimate of gamma parameter, and the mean relative evolutionary rates for each site
The shape parameter for the gamma distribution of the phylogenetic tree in reference to sample AG3 was estimated at 0.34. Substitution rates were calculated using the General Time Reversible (GTR) model, with among-site rate variation modeled via a discrete Gamma distribution (5 categories, [+ G]). The mean evolutionary rates across these categories were 0.00, 0.07, 0.31, 0.97, and 3.64 substitutions per site (Table S1). Nucleotide frequencies were determined as 24.50% (A), 19.99% (T/U), 24.70% (C), and 30.81% (G) (Table 3). A maximum likelihood (ML) tree topology was automatically generated, yielding a maximum log-likelihood score of -4,077.853. The analysis included 101 nucleotide sequences comprising 1,531 aligned positions (Table S1). The shape parameter of for the gamma distribution of the phylogenetic tree in reference to sample AG11 was estimated at 200.00. Substitution rates were derived under the GTR model, with among-site rate variation modeled using a discrete gamma distribution (5 categories, [+ G]). The mean evolutionary rates for these categories were 0.90, 0.96, 1.00, 1.04, and 1.10 substitutions per site (Table S2). Nucleotide frequencies were 25.79% (A), 20.86% (T/U), 22.71% (C), and 30.64% (G) (Table 3). A maximum likelihood (ML) tree topology was automatically generated, resulting in a maximum log-likelihood of -1,928.562. The analysis included 101 nucleotide sequences with 1,402 aligned positions (Table S2).
Discussion
The morphological analysis of 13 bacterial isolates from the sorghum rhizosphere revealed distinct Gram reactions and cellular morphologies, providing preliminary taxonomic insights. The predominance of Gram-positive rods (AG1, AG3, AG4, AG7, AG8, AG10, AG11, AG12, and AG13) aligns with previous studies highlighting Bacillus and Paenibacillus as common rhizosphere colonizers due to their spore-forming ability and plant growth-promoting traits31. The pleomorphic rod/cocci morphology of AG4 suggests Arthrobacter, a genus known for its metabolic versatility and stress tolerance in soil environments32. The Gram-positive cocci (AG2, AG5, AG6) likely belong to Staphylococcus or Micrococcus, genera frequently isolated from rhizospheres due to their adaptability to plant exudates33. The oval-shaped cocci of AG6 further support its classification as Micrococcus, consistent with recent findings on its prevalence in agricultural soils34. The sole Gram-negative cocci isolate (AG9) may represent Acinetobacter, a genus often associated with nutrient cycling and plant interactions, though Neisseria is less common in soil microbiomes5. These morphological findings, while preliminary, are consistent with contemporary studies on rhizosphere microbial diversity and set the stage for molecular confirmation35.
The biochemical characterization of the isolates revealed distinct metabolic profiles consistent with their morphological classification, providing insights into their potential ecological roles. The catalase-positive reactions in AG3, AG6, AG9, AG11, AG12, and AG13 suggest their ability to tolerate oxidative stress, a trait commonly associated with aerobic and facultative anaerobic bacteria such as Bacillus and Enterococcus spp36. The indole-positive results for AG12 and AG13, along with their MR-positive reactions, align with Enterococcus spp., which are known for mixed acid fermentation and indole production in certain strains37. In contrast, AG3 and AG11, being catalase-positive, MR-positive, and indole-negative, exhibit metabolic traits typical of Bacillus/Paenibacillus38. The MR-negative results for AG6, AG7, and AG9 suggest butanediol fermentation or non-fermentative metabolism, possibly indicating Pseudomonas or other Gram-negative bacteria, while AG7’s catalase-negative profile may hint at Lactobacillus-related taxa39. Given their metabolic consistency with beneficial rhizobacteria, AG3 and AG11 were prioritized for molecular characterization, as their biochemical profiles and rod-shaped morphology suggest strong rhizosphere colonization potential, unlike the other isolates with divergent metabolic traits. This selection aligns with recent studies emphasizing Bacillus spp. as key candidates for sustainable agriculture due to their plant-growth-promoting and stress-alleviating properties40.
The molecular characterization of isolates AG3 and AG11 provided robust taxonomic and evolutionary insights. PCR amplification of the 16S rRNA gene yielded distinct ~ 1500 bp bands, confirming successful target amplification, a critical step for bacterial identification41. Sanger sequencing generated high-quality chromatograms with strong signal-to-noise ratios, enabling accurate consensus sequences42. BLAST analysis identified AG3 as Bacillus licheniformis (99.37% identity with DSM 13; E-value: 0.0) and AG11 as Bacillus cereus (100% identity with CCM 2010; E-value: 0.0), aligning with recent studies highlighting these species’ prevalence in agricultural microbiomes43. Phylogenetic analysis further resolved AG3 within a conserved B. licheniformis clade (Cluster I, bootstrap > 70%), while AG11 clustered with B. cereus strains in sub-cluster IIB, corroborating genomic stability in these lineages44. Maximum likelihood estimates revealed evolutionary rates (AG3: gamma shape = 0.34,AG11: gamma shape = 200.00 and nucleotide biases, consistent with Bacillus spp. Heterogeneity45.
Evolutionary rate analysis and gamma parameter interpretations were employed in this study to rigorously characterize the phylogenetic diversity and evolutionary dynamics of 16S rRNA gene sequences from rhizosphere bacteria isolated across diverse Bhubaneswar, India soil types interacting with sorghum varieties46. By estimating the gamma shape parameter (α) via maximum likelihood in MEGA 12 under the GTR model, among-site rate heterogeneity were identified, where a low α (< 1) reveals pronounced variation in substitution rates across alignment positions, critical for validating the robustness of neighbor-joining trees against uneven evolutionary pressures in microbial communities shaped by urban, agricultural, and academic campus with research facilities soils, used in the present study47. Complementing this, mean relative evolutionary rates per site justified the conserved versus hypervariable regions in the 16S rRNA, enabling precise taxonomic resolution via BLAST (≥ 97% identity) and enhancing phylogenetic accuracy for identifying soil-specific bacterial consortia48. This approach, grounded in established protocols29,30, directly supports the study’s scope by discerning adaptive microbial evolution in sorghum rhizospheres.
This study has several notable limitations. Firstly, the identification of Bacillus isolates relied heavily on 16S rRNA gene sequencing, which offers limited resolution for discriminating between closely related species within the B. cereus group or distinguishing B. licheniformis from its nearest relatives49. This single-locus approach cannot fully capture genomic diversity, potential for horizontal gene transfer, or accurately differentiate between beneficial plant-growth-promoting strains and those with pathogenic lineages, particularly for B. cereus50. Secondly, the functional characterization of the identified isolates is absent,while B. licheniformis and B. cereus were identified, their actual plant-growth-promoting rhizobacteria (PGPR) potential, such as phosphate solubilization, auxin production, or antifungal activity, remains unassessed51. Furthermore, the sampling was confined to a specific geographic region with a small sample size, limiting the generalizability of the findings regarding the true diversity and prevalence of these species in the sorghum rhizosphere52.
To address these limitations, future research should employ multi-locus sequence typing (MLST) or whole-genome sequencing (WGS) for precise species- and strain-level identification, enabling accurate discrimination within closely related Bacillus groups and the detection of genes associated with both beneficial and pathogenic traits53. Subsequent work must include comprehensive in vitro and in planta assays to functionally characterize the PGPR potential of these isolates, quantifying specific traits like phosphate solubilization and auxin production54. To improve representativeness, expanded sampling across diverse geographic regions and agricultural practices is essential55. Methodologically, the use of selective and differential media alongside culture-independent techniques would provide a more complete view of the rhizosphere microbiome, reducing cultural bias56. Moreover, robust phylogenetic and comparative genomic analyses with larger sequence datasets and replication are needed to statistically validate the evolutionary patterns and population structure observed, linking genetic diversity to functional ecological roles57.
However, while robust species-level identification is provided by 16S rRNA gene sequencing, which has been widely validated for initial microbial profiling in plant pathology studies, its limitations in predicting strain-specific pathogenic potential or beneficial traits are recognized58. This is complemented by integrated morpho-biochemical assays, by which functional insights into traits like enzyme activity, biofilm formation, and antagonism are offered; these traits have been shown to correlate well with biocontrol efficacy in prior studies on similar rhizobacterial consortia59. Future study should focus on advanced analyses such as virulence gene diagnostics to validate these distinctions and enhance predictive accuracy60.
Conclusion
The study isolated, characterized, and identified rhizosphere-associated bacteria from sorghum-cultivated soils in Bhubaneswar, India, using morphological, biochemical, and 16S rRNA gene sequencing approaches. These analyses revealed a predominance of Bacillus species, including B. licheniformis (GenBank accession no. PV590072) and B. cereus (GenBank accession no. PV590099), with phylogenetic analysis providing insights into their taxonomic placement and evolutionary relationships. Future studies could explore the functional traits of these isolates, such as phosphate solubilization, nitrogen fixation, or antifungal activity, through targeted assays. Metagenomic approaches might further reveal associated microbial diversity, while field evaluations could assess their performance in sorghum cultivation.
Data availability
The 16S rRNA gene sequences generated and analysed during the current study are available in the NCBI GenBank repository under accession numbers PV590072 (https://www.ncbi.nlm.nih.gov/nuccore/PV590072) and PV590099 (https://www.ncbi.nlm.nih.gov/nuccore/PV590099).
References
Hossain, M. S., Islam, M. N., Rahman, M. M., Mostofa, M. G. & Khan, M. A. R. Sorghum: A prospective crop for climatic vulnerability, food and nutritional security. J. Agric. Food Res. 8, 100300 (2022).
Rizvi, A., Ahmed, B., Umar, S. & Khan, M. S. Comprehensive insights into sorghum (Sorghum bicolor) defense mechanisms unveiled: Plant growth-promoting rhizobacteria in combating Burkholderia-induced bacterial leaf stripe disease. Plant Stress 11, 100397 (2024).
Kulkova, I., Dobrzyński, J., Kowalczyk, P., Bełżecki, G. & Kramkowski, K. Plant growth promotion using *Bacillus cereus*. Int. J. Mol. Sci. 24(11), 9759 (2023).
Mahdi, I., Fahsi, N., Hafidi, M., Allaoui, A. & Biskri, L. Plant growth enhancement using rhizospheric halotolerant phosphate solubilizing bacterium Bacillus licheniformis QA1 and Enterobacter asburiae QF11 isolated from Chenopodium quinoa Willd. Microorganisms 8(6), 948 (2020).
Ehling-Schulz, M., Lereclus, D. & Koehler, T. M. The *Bacillus cereus* group: *Bacillus* species with pathogenic potential. Microbiol. Spectr. 7(3), 10–1128 (2019).
Kumar, C. et al. Sorghum rhizosphere bacteriome studies and generation of multistrain beneficial bacterial consortia. Microbiol. Res. 292, 128036 (2025).
Dabban, I. A. et al. Isolation techniques used for molecular characterization of beneficial microorganisms: Cultural, biochemical and molecular characterization. Handb. Agric. Biotechnol. 6, 491–545 (2024).
Almasian-Tehrani, N. et al. Overview of typing techniques as molecular epidemiology tools for bacterial characterization. Cell. Mol. Biomed. Rep. 1(2), 69–77 (2021).
Flörl, L., Meyer, A. & Bokulich, N. A. Exploring sub-species variation in food microbiomes: A roadmap to reveal hidden diversity and functional potential. Appl. Environ. Microbiol. https://doi.org/10.1128/aem.00524-25 (2025).
Prasad, B., Sharma, D., Kumar, P. & Dubey, R. C. Biocontrol potential of Bacillus spp. for resilient and sustainable agricultural systems. Physiol. Mol. Plant Pathol. 128, 102173 (2023).
Khan, A. R. et al. Bacillus spp. as bioagents: Uses and application for sustainable agriculture. Biology 11(12), 1763 (2022).
Santos-Medellín, C., Edwards, J., Liechty, Z., Nguyen, B. & Sundaresan, V. Drought stress results in a compartment-specific restructuring of the rice root-associated microbiomes. MBio 8, e00764-17 (2017).
Etesami, H., Jeong, B. R. & Glick, B. R. Potential use of Bacillus spp. as an effective biostimulant against abiotic stresses in crops—A review. Curr. Res. Biotechnol. 5, 100128 (2023).
Huang, Z. et al. Isolation and characterization of *Bacillus cereus* bacteriophage DZ1 and its application in foods. Food Chem. 431, 137128 (2024).
She, R. C. & Petti, C. A. Procedures for the storage of microorganisms. Manual Clin. Microbiol. 8, 161–168 (2015).
Davis, K. E., Joseph, S. J. & Janssen, P. H. Effects of growth medium, inoculum size, and incubation time on culturability and isolation of soil bacteria. Appl. Environ. Microbiol. 71(2), 826–834 (2005).
Thomas, P., Sekhar, A. C., Upreti, R., Mujawar, M. M. & Pasha, S. S. Optimization of single plate-serial dilution spotting (SP-SDS) with sample anchoring as an assured method for bacterial and yeast cfu enumeration and single colony isolation from diverse samples. Biotechnol. Rep. 8, 45–55 (2015).
Sousa, A. M., Machado, I., Nicolau, A. & Pereira, M. O. Improvements on colony morphology identification towards bacterial profiling. J. Microbiol. Methods 95(3), 327–335 (2013).
Aslim, B., Sağlam, N. & Beyatli, Y. Determination of some properties of Bacillus isolated from soil. Turk. J. Biol. 26(1), 41–48 (2002).
Behera, S. Physiochemical and Microbial Analysis of Soil and Water of Dhobiajharan Village (Proposed Coal Mine Site-Tubed). Doctoral dissertation (2013).
Kones, C., Mwajita, M., Kariuki, L., Kiirika, L. & Kavoo, A. Isolation and characterization of rhizospheric microorganisms from bacterial wilt endemic areas in Kenya. Afr. J. Microbiol. Res. 14(7), 349–360 (2020).
Wang, X., Heazlewood, S. P., Krause, D. O. & Florin, T. H. J. Molecular characterization of the microbial species that colonize human ileal and colonic mucosa by using 16S rDNA sequence analysis. J. Appl. Microbiol. 95(3), 508–520 (2003).
Nikunjkumar, B. D. Molecular Identification of Bacteria Using 16s rDNA Sequencing (Gujarat University, 2012).
Janda, J. M. & Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 45(9), 2761–2764 (2007).
Sting, R., Eisenberg, T. & Hrubenja, M. Rapid and reasonable molecular identification of bacteria and fungi in microbiological diagnostics using rapid real-time PCR and Sanger sequencing. J. Microbiol. Methods 159, 148–156 (2019).
Chen, L. et al. Rapid Sanger sequencing of the 16S rRNA gene for identification of some common pathogens. PLoS ONE 9(2), e88886 (2014).
Crossley, B. M. et al. Guidelines for Sanger sequencing and molecular assay monitoring. J. Vet. Diagn. Invest. 32(6), 767–775 (2020).
Buckley, T. R., Simon, C. & Chambers, G. K. Exploring among-site rate variation models in a maximum likelihood framework using empirical data: Effects of model assumptions on estimates of topology, branch lengths, and bootstrap support. Syst. Biol. 50(1), 67–86 (2001).
Mello, B. Estimating timetrees with MEGA and the TimeTree resource. Mol. Biol. Evol. 35(9), 2334–2342 (2018).
Tamura, K. et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28(10), 2731–2739 (2011).
Soni, R. & Keharia, H. Phytostimulation and biocontrol potential of Gram-positive endospore-forming bacilli. Planta 254(3), 49 (2021).
Hernández-Fernández, G., Galán, B., Carmona, M., Castro, L. & García, J. L. Transcriptional response of the xerotolerant Arthrobacter sp. Helios strain to PEG-induced drought stress. Front. Microbiol. 13, 1009068 (2022).
Chen, Y. et al. Current studies of the effects of drought stress on root exudates and rhizosphere microbiomes of crop plant species. Int. J. Mol. Sci. 23(4), 2374 (2022).
Wang, Y. et al. Evaluation of Rpf protein of Micrococcus luteus for cultivation of soil actinobacteria. Syst. Appl. Microbiol. 44(5), 126234 (2021).
Ansari, F. A., Ahmad, I. & Khan, A. S. Recent molecular tools for analyzing microbial diversity in rhizosphere ecosystem. In Microbial Diversity in the Genomic Era 233–246. (Academic Press, 2024).
Soares, M. B. et al. Sporeforming probiotic bacteria: Characteristics, health benefits, and technological aspects for their applications in foods and beverages. Trends Food Sci. Technol. 138, 453–469 (2023).
Ma, B. et al. Evaluation of in vitro production capabilities of indole derivatives by lactic acid bacteria. Microorganisms 13(1), 150 (2025).
Bal, S. Amelioration of plant biotic stress by mycorrhiza helper bacteria. In Bio-control Agents for Sustainable Agriculture: Diversity, Mechanisms and Applications 359–384 (Springer, 2025).
Lies, A., Delteil, A., Prin, Y., and Duponnois, R. (2018). Using mycorrhiza helper microorganisms (MHM) to improve the mycorrhizal efficiency on plant growth. In Role of Rhizospheric Microbes in Soil: Volume 1: Stress Management and Agricultural Sustainability 277–298.
Hussain, A. & Ibrahim, M. Molecular characterization of *Bacillus subtilis* OKR isolated from wheat rhizosphere reveals key outer membrane proteins associated with plant growth promotion: A step towards climate-smart agriculture. Int. J. Appl. Exp. Biol. 4(1), 95–106 (2025).
Patil, B. L., Gopalkrishna, A. M. & Gm, S. K. Molecular characterization of an endophytic strain of Bacillus subtilis with plant growth-promoting properties from a wild relative of papaya. J. Appl. Microbiol. 136(1), lxaf010 (2025).
Ibrahim, R., Aranjani, J. M., Prasanna, N., Biswas, A. & Gayam, P. K. R. Production, isolation, optimization, and characterization of microbial PHA from *Bacillus australimaris*. Sci. Rep. 15(1), 8395 (2025).
Liu, Y. et al. Unraveling the ecological interactions between dairy strains Bacillus licheniformis and Bacillus cereus during the dual-species biofilm formation. Food Microbiol. 128, 104716 (2025).
Sousa, E. G. et al. The research on the identification, taxonomy, and comparative genomics analysis of nine Bacillus velezensis strains significantly contributes to microbiology, genetics, bioinformatics, and biotechnology. Front. Microbiol. 16, 1544934 (2025).
da Silva, P. T. L. et al. Biological modulatorns with Bacillus licheniformis and Bacillus subtilis in compost barn. Cad. Pedagog. 22(5), e14872–e14872 (2025).
Das, S. et al. Bacillus ayatagriensis sp. nov., a novel plant growth-promoting rhizobacteria strain isolated from mulberry rhizosphere. Sci. Rep. 15, 26693 (2025).
Murag, S. et al. Molecular analysis of Bacillus anthracis isolates from Karnataka’s ruminant Anthrax outbreaks reveals genetic relationships and environmental factors Influencing spore persistence. Res. Microbiol. 8, 104343 (2025).
Nikolaidis, M. et al. A comparative analysis of the core proteomes within and among the Bacillus subtilis and Bacillus cereus evolutionary groups reveals the patterns of lineage-and species-specific adaptations. Microorganisms 10(9), 1720 (2022).
Gonçalves, L. B. et al. Microbial inoculants and fertilizer reduction in Sorghum cultivation: Implications for sustainable agriculture. Microbiol. Res. 16, 115 (2025).
Petkar, G. V., Giri, G. K. & Charpe, A. M. Antagonistic potential of phylloplane Bacillus subtilis PBs4 isolate against grain mold fungi of sorghum in India. Int. J. Environ. Agric. Biotechnol. 10(3), 619305 (2025).
Uchegbu, N., Adepeju, A. B. & Fasuan, T. O. Bio-nutritional and microbiological quality assessment of tiger-nut-enriched instant sorghum kunu-zaki beverage as potential milk-analogue. Nutr. Food Sci. 55(7), 1202–1211 (2025).
Muhammad, S. A. et al. Effect of phosphate solubilizing Bacillus on the growth of Sorghum (Sorghum bicolor [L.] Moench). Niger. J. Basic Appl. Sci. 32(2), 58–66 (2024).
Adeleke, B. S., Ayangbenro, A. S. & Babalola, O. O. Genomic analysis of endophytic Bacillus cereus T4S and its plant growth-promoting traits. Plants 10(9), 1776 (2021).
Imtiyaz, T. Bioprospecting of Bacterial Endophytes Isolated from Himalayan Cold Desert for Abiotic Stress Tolerance in Plants. Doctoral dissertation, University of Agricultural Sciences, GKVK, Bangalore (2022). https://krishikosh.egranth.ac.in/server/api/core/bitstreams/3bcba48a-b721-4845-a0a8-f0c62d47047e/content
Aehsas, S. et al. Morpho-biochemical and molecular characterization of Bacillus subtilis and Priestia megaterium isolates from eastern Indian farmlands. Mol. Biol. Rep. 52(1), 706 (2025).
Satapathy, S. S., Priyadarshini, B., Subhadarshini, S., Nayak, J., Kuldip, R. & Sahoo, J. P. Pangenomics for studying plant-microbe interactions. In Plant Pangenomes and Pangenomics 347–360. (Academic Press, 2025).
Mishra, A. P., Sahoo, J. P., Padhi, P. P., Pattnaik, S. & Jena, L. Applications of horizontal gene transfer in soil bioremediation. In Bioremediation and Phytoremediation Technologies in Sustainable Soil Management 171–203. (Apple Academic Press, 2022).
Raimi, A. & Adeleke, R. 16S rRNA gene-based identification and plant growth-promoting potential of cultivable endophytic bacteria. Agron. J. 115(3), 1447–1462 (2023).
Okechukwu, E. C. et al. Pea-saving partners: Bacillus and pseudomonas combat downy mildew in pea crops. Plant Pathol. 75(1), e70095 (2026).
Patra, D., Mishra, S., Mohanty, J., Subhadarsani, S. & Sahoo, J. P. Harnessing the plant microbiome: a genomic and metagenomic perspective for sustainable agriculture. Acad. Mol. Biol. Genom. https://doi.org/10.20935/AcadMolBioGen7892 (2025).
Acknowledgements
The authors are indebted to the Department of Biotechnology, Faculty of Agriculture and Allied Sciences, C.V. Raman Global Universiity, Bhubaneswar, India, and Regional Institute of Biotechnology, Bhubaneswar, India, for providing laboratory support for undertaking the experiments.
Author information
Authors and Affiliations
Contributions
Conceptualization, data curation, formal analysis, investigation, methodology, resources, software, visualization, writing-original draft: Jyoti Prakash Sahoo, and Angeline G. Jurry; writing-review and editing: Jyoti Prakash Sahoo, Siddhartha Shankar Sharma, Jannila Praveena, Shaikh Nausad Hossain, Nandini Sahu, Subhadarsini Pradhan, Priyanka Nayak, Swarnalata Tripathy, Dhaarani Vijayakumar, Sashanka Sekhar Dash, Biswajit Jena, Pranaya Pradhan, Samikshya Sankalini Pradhan; project administration, supervision, validation: Jyoti Prakash Sahoo. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jurry, A.G., Sahoo, J.P., Sharma, S.S. et al. Morpho-biochemical and molecular identification of Bacillus licheniformis and Bacillus cereus isolates from sorghum (Sorghum bicolor L.) rhizosphere. Sci Rep 16, 8983 (2026). https://doi.org/10.1038/s41598-026-42932-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-42932-y
