
Abstract
In recent years, great interest has been committed to the search for alternative clinical treatments for herpetic infections that reduce side effects, overcome drug resistance, and combat the intense inflammatory response triggered by viral infection. Pistachios (Pistacia vera L.) are known to contain polyphenols, pharmacologically active compounds with both immunomodulatory and antiviral activities. The present work investigates the antiviral properties of pistachio extracts against HSV-1 and their potential immunomodulatory effect on human monocytic cells, with a focus on NF-κB signaling. The RT2 Profiler PCR array was used to identify differential expression of chemokines during infection and pretreatment. We discovered that HSV-1 induces potent cytokine and chemokine activation in monocytes, and that this activation is significantly reduced by in vitro treatment with pistachio extracts. Our focus included CXCL10, CXCL11, CCL13, CCL2, CCL4, CCL13 and the receptor CMKLR1, which were particularly expressed after HSV-1 replication and downregulated by pretreatment with pistachio extracts. We further confirmed this inhibitory activity using zeaxanthin, a bioactive carotenoid found in pistachios, which has previously shown to inhibit HSV-1 replication in permissive cells. In addition, by blocking viral replication with phosphonoacetic acid (PAA), we demonstrated that in HSV-1-infected THP-1 cells, activation of CXCL10, CXCL11, CCL13, CCL2, CCL4, CCL13 and CMKLR1 was significantly downregulated, suggesting that chemokine activation is partially dependent on active HSV-1 replication. Lastly, using THP-1-dnIκBα cells, we have demonstrated that chemokine accumulation was correlated with HSV-1-induced NF-κB activation. Importantly, when neuronal (SH-SY5Y) and epithelial (HEp-2) cells were exposed to supernatants derived from pistachio extracts and zeaxanthin-treated infected THP-1 cells, we observed a significant reduction in the production of new HSV-1 viral progeny compared to the untreated infected THP-1 cells. In conclusion, the study highlights the use of pistachio extracts and zeaxanthin as a promising therapeutic approach against HSV-1. Notably, it offers valuable insights into the complex virus-host interaction, demonstrating how HSV-1 modulates the chemokine-mediated cell response, including CXCL10, CXCL11, CCL13, CCL2, and CCL4, and the receptor CMKLR1, to maintain a delicate balance with the host cell, thereby promoting viral persistence.
Similar content being viewed by others
Introduction
Human Herpes Simplex viruses (HSVs) are among the oldest and best-adapted human pathogens1. They can evade the host’s immune surveillance through a well-developed hide-and-seek mechanism that is an evolutionary strategy. A non-specific defense initiates the host’s battle against the invading HSV-1 virion in primary infection. After the initial infection or relapse, virions are transported into the neuronal cell bodies in the trigeminal ganglia by retrograde axonal transport, resulting in a lifelong latent infection2. HSV-1 infection has minimal consequences for healthy adults. However, infection in infants or immunocompromised individuals can lead to fatal encephalitis, highlighting the importance of the corresponding immune response3. Indeed, the causes of such different outcomes following HSV-1 infection are unknown but involve the interplay between the virus and the immune response. Activated macrophages regulate the initial direct antiviral action of the host via the release of cytokines, tumor necrosis factor (TNF) and type I interferon (IFN-α)4. The host cytokines TNF and IFN-1, in turn, induce natural-killer (NK) cells to release IFN-γ. Their positive feedback with synergistic interactions collectively generates nitric oxide (NO) and reactive oxygen species (ROS) against the invading virion5. Simultaneously, the combination of cytokines, macrophages, and other cells activates the immune cells to eliminate the pathogen. Chemotactic cytokines, as well as chemokines, play an important role in the development of immunological resistance to HSV-1 replication and act as a bridge between the innate and adaptive immune recognition of the pathogen and subsequent mobilization of leukocytes6. HSV-1 infection stimulates the production of various cytokines and chemokines, many of which act to suppress viral replication. In some cases, these products produce contradictory outcomes depending on the affected tissue site, while others require further study to determine their role. For instance, HSV-1 was shown to induce the secretion of CCL2, IL-8, IL-6, and TNF in primary genital endometrial epithelial cells7. In vivo, HSV promotes CXCL9 expression in the cervical mucus of HSV-positive women8. Importantly, CXCL9 and CXCL10 play a crucial role in preventing HSV-1 replication in central nervous system infections in a mouse model, likely by recruiting NK cells and cytotoxic T cells to infected tissues9. In addition, CXCL10 is considered a pivotal chemokine in orchestrating the recruitment of various leucocyte subsets, such as CD4 and CD8 T cells, macrophages, dendritic cells, and NK cells to the CNS during HSV-1 infection10. Nevertheless, virus-mediated inhibition of other chemokines could favour the host. Indeed, CXCL2, which is secreted by monocytes in response to HSV, recruit’s neutrophils that elicit damaging inflammatory immune responses against host cells and tissues, specifically neurons11. CXCL10 can also be induced by both IFN-γ and type I IFNs (IFN-α and IFN-β)12. However, the functional relevance of chemokine enhancement in the HSV life cycle and pathogenesis remains unclear. Data from our laboratory have demonstrated the effect of polyphenol-rich extracts from natural shelled (NRRE) pistachio kernels (Pistacia vera L.) on HSV-1 replication13,14. Besides, natural products and their derivatives, such as flavonoids and carotenoids, richly present in vegetables, fruits, cereals, nuts, herbs, seeds, stems, and flowers of numerous plants, possess numerous medicinal properties15,16. Particularly, flavonoids and carotenoids, which are abundant in plant-based foods, possess diverse biological activities, including anti-inflammatory properties17,18. These compounds can modulate inflammatory pathways by inhibiting the production of cytokines, chemokines and inflammatory enzymes19,20. At the same time, some studies have shown that carotenoids such as β-carotene, zeaxanthin and lycopene can regulate inflammatory pathways through MAPK and NF-kB, in endothelial cells21,22,23,24. Their antiviral effects and influence on chemokine regulation during HSV-1 replication remain largely unexplored. Monocytes play a central role in the host response, contributing to antiviral immunity and inflammatory responses. These cells produce various pro-inflammatory cytokines and chemokines, including interleukin-1β, interleukin-6, TNF-α, and monocyte chemotactic protein-1 (MCP-1)25. While the antiviral property of carotenoids in this context, particularly through different inflammatory signal pathways, has not yet been proven, in our study, we will use a monocyte as an in vitro model for HSV-1 replication, and potentially an inducer of the cellular-mediated chemokine response, to test the antiviral properties of natural extracts. Indeed, monocytes and macrophages play a crucial role in the immune response against HSV-1, contributing to reduced viral load in the mouse model6,26. Understanding advances in the immunopathogenesis of HSV-1 infections is essential to these goals. In conclusion, these studies suggest that combination drug therapies for managing HSV-1 infections could be more effective than single-drug therapies27.
Results
Antiviral effects of NRRE and RURE on HSV-1 replication
To assess the direct impact of NRRE and RURE pistachio extracts (detailed in Materials and Methods section) on HSV-1 replication in monocytic THP-1 cells, a cytotoxicity assay was performed at different time points, as shown in Fig. S1a and b. The CC50 value was calculated to determine the range within which the extracts can be used without causing toxic effects (Fig. S1c and d). Based on the cytotoxicity results, THP-1 cells and the virus were pre-treated with concentrations of both NRRE and RURE extracts (0.4 and 0.6 mg/mL) below the toxic threshold (Fig. 1a). The effects at different stages of the viral replication cycle were examined using multiple approaches, including viral titration, quantification of viral DNA, evaluation of representative viral transcripts by quantitative PCR (qPCR), and detection of ICP8 protein accumulation. The results indicated a dose-dependent reduction in viral plaques, suggesting a significant inhibitory effect of pretreatment with both NRRE and RURE extracts (Fig. 1b). Furthermore, qPCR confirmed a decrease in HSV-1 DNA levels (Fig. 1c), reinforcing the extracts’ antiviral activity. Transcriptional analysis of key viral genes ICP0 (alpha), UL42 (beta), and US11 (gamma) showed marked downregulation after pretreatment with NRRE and RURE (Fig. 1d), indicating interference with the viral genetic cascade. Western blot analysis further revealed a significant reduction in ICP8 protein accumulation upon pretreatment with the extracts, as quantified through band intensity measurements normalized to GAPDH levels (Fig. 1e and f). Collectively, these findings demonstrate that NRRE and RURE effectively impair HSV-1 replication by targeting multiple stages of the viral replication program, highlighting their potential as novel antiviral agents.

Antiviral effect of NRRE and RURE pretreatment on HSV-1 replication. (a) Graphical representation of the experimental workflow: THP-1 cells and virus were pre-treated with NRRE and RURE at 0.4 and 0.6 mg/mL for 1 h at 37 °C and then infected at 50 MOI with pretreated HSV-1 at 37 °C. Thus, 1 h later, the viral inoculum was removed and replaced with growth medium containing NRRE and RURE at 0.4 and 0.6 mg/mL. Samples were collected at 24 h p.i. and processed for further analysis. (b) Evaluation of viral title in THP-1 cells following NRRE and RURE pretreatment. Viral titer was determined from the cell-associated virus obtained from infected, treated, and untreated THP-1 cells after three freeze–thaw cycles. Serial dilutions of the resulting viral suspensions (100 µL per dilution) were used to infect VERO cell monolayers. The multiwell plates were incubated for 1 h at 37 °C. Then, the viral inoculum was removed and replaced with a culture medium containing 0.8% methylcellulose. After 72 h, the plaques were visualized and counted at the microscope after staining with a crystal violet solution. (c, d) Infected and uninfected, treated, and untreated THP-1 cells were collected 24 h p.i and processed for viral DNA detection (panel c) and mRNA extraction and quantitative Real-time PCR to analyze ICP0, UL42, and Us11 viral transcripts as representative genes of the sequential genetic cascade of HSV-1 (panel d). Relative quantization of viral DNA and genes was performed using real-time quantitative PCR and analyzed by the comparative Ct method (∆∆Ct). Data are expressed as a mean (± SD) of at least three experiments, and asterisks (**, ***, ****) indicate the significance of p-values less than 0.01, 0.001 and 0.0001, respectively. n.s. means no significant difference. e A representative Western blot of ICP8, a viral protein, was shown. f Densitometric analysis of ICP8 bands was normalized to GAPDH expression and quantified using ImageJ software and represents the mean of two independent experiments.
Modulation of HSV-1-induced inflammatory chemokines by pistachio extracts in human monocytes
Given monocytes’ critical role in orchestrating immune responses, we first investigated whether HSV-1 replication induces chemokines and cytokines, with or without NRRE and RURE extracts, in THP-1 cell lines. We analyzed cytokine and chemokine expression patterns in HSV-1-infected THP-1 cells, both untreated and treated with the extracts, using a Human Cytokines & Chemokines RT2 Profiler PCR Array (Qiagen, Hilden, Germany). Expression profiles were compared (Fig. 2a). The array analysis revealed significant activation in immune-related gene expression following HSV-1 infection. The data were used to generate a heatmap, where red represents high expression and green indicates low expression in each sample. Out of the 84 human cytokines and chemokines analyzed (Table S1), 34 genes were upregulated by HSV-1, many of which are associated with immune cell recruitment and inflammation. Conversely, four genes were downregulated: C5, a key component of the complement system and essential for innate immunity; ACKR2, an immune-regulatory chemokine receptor involved in chemokine clearance and immune homeostasis; and CCL23 and CCL28, chemokines involved in immune cell migration and mucosal immunity, respectively. These data suggest that HSV-1 not only triggers chemokines but may also actively suppress certain immune responses to establish the infection. Among the 34 genes upregulated by HSV-1 replication, 21 were statistically lower than the infected group upon pretreatment with NRRE and RURE (Fig. 2b). These genes were categorized into three groups based on their chemokine motifs: -C-X-C motif chemokines: CXCL1, CXCL8, CXCL9, CXCL10, CXCL11, CXCL12, and CXCL13. -C-C motif chemokines: CCL2, CCL3, CCL4, CCL7, CCL8, CCL13, CCL24, and CCL26.-C-C motif chemokine receptors: CCR1, CCR4, CCR6, CCR7, CCRL1, and CMKLR1. Overall, our findings indicate that HSV-1 promotes a strong response by activating chemokines and cytokines during viral replication. At the same time, pretreatment with NRRE and RURE extracts appears to downregulate key immune-regulatory genes involved in immune cell recruitment and inflammation (Fig. 2).

Expression profile of cytokines and chemokines in THP-1 cells infected with HSV-1 and treated with NRRE and RURE. THP-1 cells and the virus were pretreated with NRRE and RURE at 0.6 mg/mL for 1 h at 37 °C, then infected with pretreated HSV-1 at 50 MOI. 1 h later, the viral inoculum was removed and replaced with growth medium containing NRRE and RURE at 0.6 mg/mL. Total RNA was extracted 24 h p.i. and reverse-transcribed to generate cDNA. (a–b) Expression data of cytokines and chemokines in the THP-1 cells were measured by performing a DNA array (Human Cytokines & Chemokines RT2 Profiler PCR Array; Qiagen) and expressed as relative mRNA normalized to untreated and uninfected THP-1. The chemokines were classified into 3 groups as reported in panel (b). Statistical analysis for panel b. relative to the HSV-1-infected control, was performed using two-way ANOVA followed by Dunnett’s multiple comparisons test (GraphPad Prism v.8.0.1). Significance levels are indicated as follows: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***).
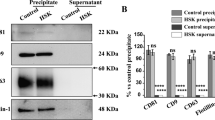
To confirm these findings, the transcriptional levels of 6 the most representative chemokines (CCL2, CCL4, CCL13, CXCL10, CXCL11, CXCL13) and the receptor CMKLR1, were measured by qPCR (Fig. 3a). The findings agreed with the PCR array results (Fig. 2). Importantly, the results obtained by measuring chemokine transcriptional levels were confirmed by Western blot analysis of some of them. In particular, the accumulation of CXCL10 and CXCL13 proteins. The data demonstrated that the accumulation of CXCL10 and CXCL13 proteins, upon HSV-1 infection, was inhibited in treated THP-1-infected cells, indicating that NRRE and RURE influence chemokine expression at both transcriptional (Fig. 3a) and translational levels (Fig. 3b lanes 5–6 vs. lane 2 and Fig. 3c).

Validation of array data by qPCR and evaluation of CXCL10 and CXCL13 proteins following NRRE and RURE treatment in THP-1 cells. THP-1 cells were pretreated, with NRRE and RURE at 0.6 mg/mL for 1 h at 37 °C and then infected with HSV-1 at 50 MOI as described in Material and Methods. One hour later, the viral inoculum was removed and replaced with growth medium containing NRRE and RURE at 0.6 mg/mL. (a) Relative quantization of CCL2, CCL4, CC13, CXCL10, CXCL11, CXCL13 and CMKRL1 genes was performed using real-time quantitative PCR and analyzed by the comparative Ct method (∆∆Ct). (b) Western blot analysis was performed to detect the levels of CXCL10 and CXCL13 proteins as representative cellular proteins. (c) Densitometric analysis of CXCL10 and CXCL13 bands was normalized to β-ACTIN expression and quantified using ImageJ software and graphically represented by GraphPad Prism 8 software. Data are expressed as a mean (± SD) of two independent experiments. ****Indicates the significance of p-values less than 0.0001.
HSV-1-mediated NF-κB activation is involved in regulating chemokines in THP-1-infected cells
One of the key regulators of pro-inflammatory genes, including those encoding chemokines and cytokines, is the NF-κB. It is well-established that HSV-1 recruits NF-κB during viral replication, most likely to ensure a favorable environment for viral survival28,29. Among the chemokines activated by HSV-1, CXCL10 possesses an upstream regulatory sequence that contains several critical regulatory elements for NF-κB and interferon-stimulated response element (ISRE), as well as sites for the binding of proteins such as heat shock (HS) factors30. Therefore, given that NRRE and RURE extracts reduce the expression of chemokines, such as CXCL10, CXCL11, CCL13, CCL2, CCL4, CCL13 and the receptor CMKLR1 activated by HSV-1, we hypothesized that these extracts exert their effects by targeting the HSV-1-mediated NF-κB activation. To test this, we measured NF-κB transcript levels in HSV-1-infected THP-1 cells in the presence or in the absence of NRRE and RURE (Fig. 4a). The results showed that HSV-1 infection caused a significant increase in NF-κB transcript levels (Fig. 4a), which correlates with the accumulation of the phosphorylated form of NF-κB (Phospho-NF-κB p65), as shown in Fig. 4b, lane 2, and graphically in panel c. In contrast, the pretreatment of HSV-1-infected THP-1 cells with NRRE and RURE significantly reduces the NF-κB transcript levels (Fig. 4a) and Phospho-NF-κB p65 (Fig. 4b lane 2 vs. lanes 5 and 7 and c). These data suggest that these natural compounds effectively inhibit NF-κB activation induced by HSV-1 infection.

NF-κB activation in THP-1 infected cells. THP-1 cells were pretreated with NRRE and RURE at 0.6 mg/mL for 1 h at 37 °C, then infected with HSV-1 at 50 MOI as described in Materials and Methods. One hour later, the viral inoculum was removed and replaced with growth medium containing NRRE and RURE at 0.6 mg/mL. (a) Quantitative Real-Time PCR assessed relative mRNA expression of NF-κB at 24 h p.i. (b) Western blot analysis was performed to detect phospho-NF-κB p65 levels at 24 h p.i. (c) Densitometric analysis of phospho-NF-κB p65 bands was normalized to GAPDH expression and quantified using ImageJ software and graphically represented by GraphPad Prism 8 software. Data are expressed as a mean (± SD) of two independent experiments. ** and ***Indicate the significance of p-values less than 0.01 and 0.001, respectively.
Zeaxanthin inhibits chemokine expression in HSV-1-infected THP-1 cells
Previous studies have reported that the pure compound zeaxanthin, a carotenoid present in pistachio extracts, exhibits antiviral activity against HSV-1 in infected cells14. Therefore, we have investigated the specific effects of zeaxanthin on chemokine expression during HSV-1 replication. To this purpose, THP-1 cells and HSV-1 were pretreated with zeaxanthin (10 µM) for 1 h at 37 °C. Following pretreatment, the virus was used to infect THP-1 cells at a multiplicity of infection (MOI) of 50. One hour post-infection, the viral inoculum was removed and replaced with a growth medium containing zeaxanthin (10 µM). The concentration of 10 µM was selected based on THP-1 cell viability assays using increasing zeaxanthin concentrations (1, 2, 5, 10, 25, and 35 µM) for 24 h (Fig. S2). At 24 h post-infection, total RNA was extracted and analysed by quantitative real-time PCR to measure the transcript levels of the chemokines CCL2, CCL4, CC13, CXCL10, CXCL11, CXCL13, and CMKRL1. The results showed that zeaxanthin significantly reduced the levels of chemokines upregulated by HSV-1, as evidenced by decreased transcriptional levels of CCL2, CCL4, CC13, CXCL10, CXCL11, CXCL13, and CMKRL1 (Fig. 5a). To note, CXCL10 protein levels were significantly reduced following HSV-1 zeaxanthin treatment (Fig. 5b, lane 2 vs. 4 panel c), as a consequence a significant reduction (approximately 60%) of US11 viral protein accumulation (Fig. 5b, lane 2 vs. 4 and panel c) and the representative viral transcripts of the HSV-1 gene cascade ICP0 (α gene), UL42 (β gene), US11 ( γ gene) were observed (Fig. 5d).
To confirm the results obtained with NRRE and RURE, we also tested zeaxanthin’s ability to inhibit NF-κB transcript accumulation. Figure 5e shows that zeaxanthin effectively inhibits HSV-1-mediated NF-κB transcriptional activation, correlating with decreased chemokine transcription. The reduction of NF-κB transcripts correlated with reduced activation of phospho-NF-κB p65 (Fig. 5f lane 4 vs. 2 and panel g).

Inhibitory effects of zeaxanthin on HSV-1 replication, chemokines and NF-κB expression. THP-1 cells and virus were pretreated with zeaxanthin (10 µM) for 1 h at 37 °C. Then, the pretreated virus was used to infect THP-1 cells at 50 MOI. 1 h later, the viral inoculum was removed and replaced with growth medium containing zeaxanthin (10 µM). The samples were collected 24 h p.i. and processed. (a) Relative quantization of CCL2, CCL4, CC13, CXCL10, CXCL11, CXCL13 and CMKRL1 genes was performed using real-time quantitative PCR and analyzed by the comparative Ct method (∆∆Ct). Data are expressed as a mean (± SD) of at least three experiments. (b) Western blot analysis was performed to evaluate the expression of CXCL10 and US11. The β-actin was used as a housekeeping gene. (c) Densitometric analysis of CXCL10 and Us11 bands was normalized to β-actin expression and quantified using ImageJ software. (d) Real-time PCR was performed to analyze ICP0, UL42, and Us11 viral transcripts as representative genes of genetic cascade of HSV-1. (e) Quantitative Real-Time PCR assessed relative mRNA levels of NF-κB at 24 h p.i. (f) Western blot analysis was performed to detect the expression levels of phospho-NF-κB p65 and Us11 as a representative viral gene. (g) Densitometric analysis of phospho-NF-κB p65 and Us11 bands was normalized to α-tubulin expression and quantified using ImageJ software. Data are expressed as a mean (± SD) of two independent experiments, and asterisks (*, ***, ****) indicate the significance of p-values less than 0.05, 0.001 and 0.0001, respectively.
Conditioned medium from infected monocytes treated with NRRE, RURE, and zeaxanthin inhibits the production of new HSV-1 viral progeny in neuronal and epithelial cells
To further investigate whether the treatment of HSV-1-infected monocytes with NRRE, RURE, or zeaxanthin can indirectly modulate HSV-1 replication in permissive cells, conditioned supernatants derived from HSV-1-infected THP-1 monocytes treated with NRRE, RURE, or zeaxanthin were used on SH-SY5Y neuroblastoma cells and HEp-2 epithelial cells (Fig. 6a). As part of the experimental design, we also included treatment with PAA, a well-established inhibitor of HSV-1 DNA polymerase. PAA served as a positive control to define the maximal achievable suppression of viral replication in infected monocytes and to provide a reference point for interpreting the antiviral effects of NRRE, RURE, and zeaxanthin.

Treatment of HEp-2 and SH-SY5Y cells with conditioned supernatants from treated and infected monocytes. (a) Schematic representation of the experimental workflow. THP-1 cells and HSV-1 were separately pre-treated for 1 h at 37 °C with NRRE, RURE, or zeaxanthin prior to infection. THP-1 cells were then infected with HSV-1 at a multiplicity of infection (MOI) of 50 using the pre-treated virus. After 1 h of adsorption, the viral inoculum was removed, the cells were washed, and fresh medium containing the corresponding compounds or PAA was added. At 24 h p.i, cell-free supernatants were collected and either used for viral titration in Vero cells (as reported in Table S2) or transferred onto HEp-2 epithelial cells and SH-SY5Y neuroblastoma cells. After 1 h of exposure, supernatants were removed, and recipient cells were incubated for an additional 24 h before viral titer determination. (b) Total HSV-1 titers were measured in HEp-2 cells following exposure to conditioned supernatants from infected THP-1 cells. (c) Cell-free HSV-1 titers released from HEp-2 cells after infection with conditioned supernatants. (d) Total HSV-1 titers measured in SH-SY5Y cells following exposure to conditioned supernatants from infected THP-1 cells. (e) Cell-free HSV-1 titers released from SH-SY5Y cells after infection with conditioned supernatants. Viral titers were determined by standard plaque assay on Vero cells and are expressed as plaque-forming units per milliliter (PFU/mL). Data are presented as mean ± SEM of at least three independent experiments. Statistical significance was assessed using appropriate statistical tests; **p < 0.01, ***p < 0.001, ****p < 0.0001.
Conditioned supernatants containing released free-cell virus were subsequently transferred to SH-SY5Y neuroblastoma cells and HEp-2 epithelial cells separately. After a 1-hour incubation, the supernatants were removed, and the cells were maintained for an additional 24 h in a fresh growth medium. Samples were then collected to determine the production of new viral progeny, and the total virus and cell-free viral titers were measured (Fig. 6).
The results show that the production of new HSV-1-viral progeny was significantly reduced in both HEp-2 (Fig. 6 panels b-c) and SH-SY5Y cells (Fig. 6 panels d-e) exposed to supernatants from NRRE- or RURE-treated THP-1 compared to untreated infected cells, although to a lesser extent than the strong inhibitory effect observed with PAA-treated THP-1 cells. A similar inhibitory effect on the production of new HSV-1 viral progeny was observed when zeaxanthin-treated THP-1 cells were used, with a more pronounced reduction in HEp-2 cells than in SH-SY5Y cells. Specifically, zeaxanthin-conditioned medium reduced the total viral titer of new HSV-1-viral progeny by about 4 logarithmic units in HEp-2 cells (Fig. 6 panel b) and by 1 logarithmic unit in SH-SY5Y cells (Fig. 6 panel d). Overall, the data indicate a consistent antiviral effect of NRRE, RURE, and zeaxanthin, with a markedly stronger impact on HSV-1 replication in permissive HEp-2 epithelial or SH-SY5Y neuronal cells. To verify whether the observed effects were due to reduced viral load or to antiviral factors present in THP-1-derived conditioned media, we quantified the viral titers released by THP-1 cells under all experimental conditions. As shown in Table S2, treatment of THP-1 cells with NRRE, RURE, or zeaxanthin resulted in a significant reduction in cell-free HSV-1 titers in monocyte supernatants compared with untreated infected controls. Consequently, when an equal amount (500 µL) of these supernatants was applied to HEp‑2 and SH‑SY5Y cells, the resulting inoculum corresponded to an estimated MOI of 31 for untreated cells, which is sufficient to establish a productive infection in permissive cells. In contrast, the MOIs calculated for the treated conditions were proportionally lower due to reduced viral titers, resulting in correspondingly lower total and cell-free viral yields in both permissive cell lines. To verify whether the THP‑1 supernatant contained antiviral factors capable of altering viral replication in HEp‑2 or SH‑SY5Y cells, we performed a parallel control infection using wild‑type HSV‑1 viral stock (derived from VERO cells) at an MOI of 31. After 24 h, we quantified both cell‑free and total viruses under both conditions (Figure S3). The viral titers produced by HEp‑2 and SH‑SY5Y cells infected at MOI 31 were essentially identical to those produced by cells exposed to the THP‑1-derived supernatants.
Chemokine accumulation is partially dependent on HSV-1 replication and virus-induced NF-κB activation
The data reported to this point demonstrate that HSV-1 induces a strong activation of the chemokine response in host immune cells by recruiting NF-κB. However, it remains unclear whether the expression of these chemokines depends solely on viral entry or also requires active viral replication. Additionally, it remains unclear whether HSV-1-induced chemokine expression is a direct consequence of NF-κB activation or if it occurs independently.
Therefore, to determine whether chemokine induction depends on viral replication, THP-1 cells were infected in the presence of PAA (300 µg/mL), an inhibitor of HSV-1 DNA polymerase. PAA treatment significantly downregulates the accumulation of all measured chemokine transcripts (CXCL10, CXCL11, CCL13, CCL2, CCL4, CCL13 and CMKLR1) compared to untreated infected controls (Fig. 7a), indicating that active viral replication is required for the full induction of chemokines. To confirm the inhibitory effect of PAA on viral replication, we assessed the mRNA levels of Us11, a late viral gene whose expression depends on newly synthesized viral DNA and is therefore blocked when DNA replication is inhibited31. PAA treatment resulted in a substantial decrease in Us11 transcripts (Fig. 7b), validating that the observed reduction in chemokine expression was associated with impaired HSV-1 replication. In addition, CXCL10 protein levels and NF-κB activation were monitored by Western blot analysis (Fig. 7c). Consistent with the transcriptional data, protein levels of CXCL10 were markedly reduced upon PAA treatment, indicating that their induction is dependent on HSV-1 replication (Fig. 7c lane 2 vs. lane 4). Moreover, the expression of phosphorylated NF-κB p65, which was strongly increased in infected THP-1 cells, as also shown in Fig. 4b lane 2, was drastically reduced upon PAA treatment (Fig. 7c, lane 2 vs. 4 and graphically reported in panel d). These findings further support the conclusion that NF-κB activation is closely linked to viral replication and is significantly impaired when HSV-1 DNA synthesis is inhibited.

Comparative evaluation of chemokine transcript levels in THP-1 and THP-1 dnIκBα cell lines upon HSV-1 infection. (a) THP-1 cells were infected with HSV-1 (50 MOI) in the presence or absence of PAA (300 µg/mL), an inhibitor of viral DNA replication. After 24 h post-infection (p.i.), total RNA was extracted and analyzed by quantitative Real-Time PCR to assess the mRNA levels of CCL2, CCL4, CCL3, CXCL10, CXCL11, CXCL13, and CMKRL1. (b) The efficacy of viral replication inhibition by PAA was confirmed by measuring the transcriptional levels of the viral gene Us11. (c) Western blot analysis was performed to analyze the expression of phospho-NF-Kb p65, CXCL10 and US11 in THP-1 cells untreated and treated with PAA. The α-tubulin was used as a housekeeping gene. The grouping blots were cropped from different gels, as indicated by the horizontal line in the figure, and were captured using a ChemiDoc Touch Imaging System (Bio-Rad). (d) Densitometric analysis of phospho-NF-Kb p65, CXCL10 and Us11 bands was normalized to α- tubulin expression and quantified using ImageJ software. (e) Comparison of chemokine transcript levels in parental THP-1 and THP-1 dnIκBα cells infected with HSV-1 (50 MOI) for 24 h. (f) Transcriptional levels of viral genes (ICP0, UL42, and Us11) were measured in THP-1 and THP-1 dnIκBα cells to assess the impact of NF-κB inhibition on viral replication. Data are presented as mean ± SD from at least three independent experiments and analyzed using the ΔΔCt method. (g) THP-1 and THP-1 dnIκBα cells were infected with HSV-1 (50 MOI) in the presence or absence of PAA. The samples were collected at 24 h p.i., and the virus yield was evaluated by titrating total viral particles. *,**, *** and *** indicate the significance of p-values less than 0.05, 0.01, 0.001 and 0.0001. b.d. = below detection limit.
To assess the specific contribution of NF-κB to the pro-inflammatory signaling triggered by HSV-1, we compared chemokine expression in parental THP-1 cells with chemokine expression in THP-dnIKBa cells, which are engineered to inhibit NF-κB activation29.
HSV-1 infection in THP-dnIκBα cells resulted in a significant reduction in chemokine mRNA levels compared to wild-type infected THP-1 cells (Fig. 7e), indicating that NF-κB is a critical driver of HSV-1-induced chemokine transcription. Notably, NF-κB inhibition-mediated suppression of chemokines rescued HSV-1 replication. Indeed, in THP-dnIκBα cells, a significant increase in mRNA levels of key viral genes, including ICP0, UL42, and Us11, was observed when compared to infected wild-type THP-1 cells (Fig. 7f). This suggests that inhibiting NF-κB recruitment supports HSV-1 replication, presumably by promoting a pro-viral cellular environment. Consistently, the titer levels of HSV-1 were significantly higher in THP-dnIκBα cells compared to wild-type THP-1 cells (Fig. 7g).
Conclusion
This study demonstrates that Pistacia vera L. extracts, specifically NRRE and RURE, exert potent antiviral effects against HSV-1 infection in monocytic THP-1 cells. Treatment with non-cytotoxic concentrations of the extracts significantly reduced HSV-1 replication, as evidenced by decreased viral titers, reduced levels of viral DNA, massive reduction in the accumulation of key viral gene transcripts (ICP0, UL42, US11), and diminished accumulation of the viral protein ICP8 (Fig. 1). Concomitantly, HSV-1-induced upregulation of multiple chemokines, including CCL2, CCL4, CCL13, CXCL10, CXCL11, CXCL13 and the receptor CMKLR1, was markedly attenuated by pistachio extract pretreatment (Figs. 2 and 3). Mechanistically, these effects correlated with a significant reduction in HSV-1-mediate NF-κB activation (Fig. 4). Importantly, the carotenoid zeaxanthin reproduced the extracts’ antiviral effects, leading to decreased viral gene expression, chemokine production, and NF-κB activation (Fig. 5). As demonstrated in our previous study14, using the Vero cell model, NRRE, RURE, and zeaxanthin exhibit a partial neutralizing effect on HSV‑1 infectivity when pre‑incubated with viral particles. Although this activity does not result in complete virion inactivation, it may nonetheless contribute to the antiviral effects observed in the present work, where extracts and virus were added simultaneously to THP‑1 cells. Under these conditions, the reduction in viral replication and the consequent downstream decrease in NF‑κB activation and chemokine production may therefore reflect a combination of partial virion neutralization and intracellular interference.
Beyond their direct antiviral effects, conditioned supernatants from pistachio extracts–treated HSV-1-infected monocytes were able to impact the production of new HSV-1-viral progeny in fully permissive neuronal and epithelial cell lines (Fig. 6). Moreover, our findings indicate that chemokine induction by HSV-1 is primarily dependent on the active viral replication and NF-κB. Indeed, inhibition of HSV-1 replication by PAA or by blocking NF-κB activation with THP-1-dnIκBα reduces the accumulation of chemokine transcripts (Fig. 7). Consistent with our observations, a similar response in CCL4 expression has been documented in U937-dnIκBα cells 24 h post-infection, further supporting the critical role of NF-κB in regulating this chemokine during HSV-1 replication32. Thus, while NF-κB activation robustly induces multiple chemokines, suppressing NF-κB reduces chemokine levels. However, this event does not suppress viral replication and may even enhance it. On the other hand, elevated NF-κB and chemokine levels coexist with active viral replication. This paradox suggests that the activation of chemokines during HSV-1 infection may serve dual and context-dependent roles, both antiviral and proviral, and that the virus may exploit the host’s response to its advantage6. Indeed, while chemokine upregulation is typically associated with antiviral immune responses, our findings, along with those of others, suggest that specific chemokines may play a proviral role. For example, CXCL10 and CCL2, though known for recruiting immune effector cells, can paradoxically promote viral spread by attracting permissive or inflammatory cells to the site of infection33. CXCL10 is upregulated in infected corneal and neuronal tissues. It supports immune cell recruitment, but excessive or dysregulated chemokine production may exacerbate tissue damage and facilitate viral dissemination through a mechanism dependent on cellular PKR and viral ICP034. These findings reinforce the idea that the virus may tolerate, or even benefit from, a certain level of chemokines, or gain access to immune cells to facilitate spread or modulate the immune environment. During HSV-1 infection, TLR2 engagement mediates CCL2 production in infected neurons, thereby coordinating macrophage recruitment26. HSV-1 and HSV-2 induced expression of the CC chemokine RANTES/CCL5 in murine macrophage cell lines and peritoneal cells35. A non-directed link between HSV-1 and CMKLR1 has been identified so far. However, its expression and activity are modulated in HIV, where CMKLR1 signaling is involved in viral control through the recruitment of plasmacytoid dendritic cells36. It has been proposed to function as a minor co-receptor, promoting infection by selecting HIV and SIV isolates37. In other viral models, chemokine-mediated proviral effects have also been described. For instance, HIV-1 uses CCR5 and CXCR4 as coreceptors, and chemokines that bind these receptors can paradoxically enhance infection under certain conditions38. In influenza A virus infection, CCL2 has been shown to exacerbate lung pathology and enhance viral spread by recruiting inflammatory monocytes39. These findings emphasize that chemokine production, although part of the host defense, can be subverted by viruses to support their own replication and persistence. In the case of HSV-1, the potential benefit of chemokine potentiation is currently unknown. Existing hypotheses include the recruitment of more susceptible cells to the site of infection and the induction of an activated proviral state in neighboring cells40. Our data show that the chemokine storm is not only a consequence of infection but an NF-κB-dependent, virus-facilitated process that may be proviral. In summary, the induction of chemokines and the recruitment of NF-κB during HSV-1 replication reflect a complex interplay between these factors. However, they may also be coopted by HSV-1 to facilitate replication and spread. The paradoxical effect in the cellular model in which NF-κB is blocked (THP-1-dnIκBα), resulting in reduced chemokines and subsequent increases in viral replication, may indicate that the chemokine-induced immune response exerts partial control over the virus and that HSV-1 has developed mechanisms to balance this. Further studies are necessary to elucidate the specific role of individual chemokines in HSV-1 infection, as their diverse functions may differentially influence viral replication, immune cell recruitment, and tissue inflammation. In conclusion, our study highlights the potential of Pistacia vera L.-derived compounds as promising agents that target HSV-1 replication. Therefore, based on our findings and literature data, we propose a mechanism to describe the impact of viral replication on monocytes characterized by upregulation of several chemokines involved in viral pathogenesis and immune activation (Fig. 8a). Natural compounds, such as Pistacia vera L. extracts (NRRE and RURE) and zeaxanthin, which interfere with viral replication, consequently attenuate chemokine-mediated immune activation (Fig. 8b). This effect suggests a potential therapeutic relevance for managing herpetic-related complications. In particular, they may represent a promising experimental approach to modulate HSV-1 replication and associated inflammatory responses in immune-privileged or highly sensitive tissues, such as the brain, eyes, and genital tract, where inflammation is a major contributor to overall pathology. The identification of zeaxanthin as an active constituent that not only reproduces but also enhances, in some aspects, the effects of the crude extracts underscores the relevance and impact of our findings, highlighting the value of bioactive compounds as a more effective and pharmacologically tractable strategy for future therapeutic development.

HSV-1-induced monocyte signaling and its modulation by pistachio extracts. (a) Proposed mechanism describing the impact of HSV-1 replication on monocytes signaling, (b) and its modulation by natural products. The red arrows illustrate the mechanism of HSV replication, which culminates in the accumulation of chemokines while the green arrows represent the “decrease mechanism” mediated by pistachio extracts. (Created in https://BioRender.com)
Materials and methods
Cells and virus
THP-1 (human acute monocytic leukemia) and VERO (African green monkey kidney), HEp-2 (human epithelial laryngeal carcinoma), and SH-SY5Y (human neuroblastoma) cells were originally obtained from ATCC (https://www.atcc.org/). THP-1 cells were cultured in RPMI-1640 medium supplemented with 10% FBS (Euroclone), 1mM Sodium Pyruvate (Sigma-Aldrich), 10 mM Hepes buffer (Sigma-Aldrich). DN IκBα THP-1 cells, stably transfected with a dominant negative mutant IκBα29 were cultured in RPMI-1640 medium and maintained under selection with 400 µg/ml of Geneticin (Gibco). VERO and HEp-2 cells were cultured in Dulbecco’s Modified Eagle’s High Glucose Medium (DMEM, Euroclone, Pero, MI, Italy) supplemented with 6% and 10% FBS fetal bovine serum (FBS), respectively. SH-SY5Y cells were grown in RPMI-1640 medium supplemented with 10% FBS, 1% of Non-Essential Amino Acid Solution (NEAA) 1X and 2 mM of L-Glutamine.
All mediums are supplemented with 100 U/mL penicillin, and 100 mg/mL streptomycin (Lonza, Belgium). All cell lines were grown at 37 °C in a 5% CO2 incubator.
The prototype HSV-1 (F) strain, used for the in vitro experiments, was kindly provided by Dr. Bernard Roizman (University of Chicago, Chicago, IL, USA), and the virus stock was produced and titered in Vero cells.
Materials
Californian natural raw polyphenols-rich extract (NRRE, Pistacchi Sgusciati California Pissgsu01/BSV1, L67316221262) and roasted unsalted polyphenols-rich extract (RURE, Pistacchi Sgusciati Tostati California Pissgstu01/BSV1) pistachio polyphenols-rich extracts were kindly supplied by the American Pistachio Growers (Fresno, CA, USA). NRRE or RURE (10 g) were extracted with n-hexane as previously described14 and the quantification of phenolic compounds was performed by RP-HPLC-DAD.
Antibodies and reagents
Primary antibodies used included GAPDH (sc-32233), α Tubulin (DM1A) (sc-32293), CXCL10 (sc-101500), and CXCL13 (sc-73740) from Santa Cruz Biotechnology; ICP8 and US11, kindly provided by Professor Bernard Roizman; β-actin (ab8226) from Abcam; and phospho-NF-κB p65 (Ser536) (#3033) from Cell Signaling Technology. HRP-conjugated goat anti-mouse and anti-rabbit IgG secondary antibodies were obtained from Merck Millipore.
Phosphonoacetic acid (PAA) from Sigma-Aldrich, was used as an inhibitor of DNA synthesis in HSV-infected cells and was dissolved in the medium and used at 300 µg/mL during and after HSV-1 adsorption41.
Viability assay
To evaluate cell viability after exposure to pistachio extracts, the ViaLight™ Plus Cell Proliferation and Cytotoxicity Bioassay (Lonza Group Ltd., Basel, Switzerland) was utilized. THP-1 cells were plated in 96-well microplates and treated with different concentrations of NRRE and RURE (0.8, 0,6, 0.4, 0,3 and 0.2 mg/mL). Following the incubation period, cell viability was determined at 24 h, 48 h, and 72 h using the GloMax® Multi Microplate Luminometer (Promega Corporation, Madison, WI, USA) with the ViaLight™ Plus assay, which detects luminescence generated by ATP breakdown.
The luminescent signals were subsequently translated into a cell viability percentage using the following formula:
where:
A represents the mean luminescence of treated samples,
B denotes the background luminescence, and.
C is the mean luminescence of untreated control samples.
The CC50 values of pistachio extracts and zeaxanthin were estimated at 24 h post treatment exposing THP-1 cells to 0.4,0.8,1,1.5,3,6,9 mg/mL of NRRE and RURE and to 1,2,5,10,25 and 35 µM of zeaxanthin. The absorbance recorded in each well was normalized to the average absorbance of nontreated control wells to calculate the percentage of cell viability. The CC50 values were calculated via non-linear regression with GraphPad Prism 8.0.1 using a bottom constraint of 0 and the formula Y = Top/(1 + 10^((LogCC50-X) * HillSlope)) as reported in Fig. S1.
Pre-treatment and infection protocol in THP-1 cells
THP-1 cells were seeded in appropriate culture flasks and maintained under standard conditions. Prior to infection, both THP-1 cells and the HSV-1 viral suspension were separately pre-treated with NRRE, RURE, and zeaxanthin at various concentrations, depending on the specific experimental setup. The pre-treatment was carried out for 1 h at 37 °C. Following this step, THP-1 cells were infected with HSV-1 virus at a multiplicity of infection (MOI) of 50, using the virus previously exposed to NRRE, RURE, and zeaxanthin. The infection was allowed to proceed for 1 h at 37 °C. After the infection period, the viral inoculum was carefully removed, and the cells were washed with growth medium to eliminate residual unbound viruses. Fresh growth medium containing NRRE, RURE, and zeaxanthin at the same concentrations used during the pre-treatment phase was then added to the cells. The samples were incubated for an additional 24 h post-infection (p.i.) and then collected and processed for subsequent analyses, depending on the objectives of each experimental condition (e.g., reduction plaque assay, gene expression analysis, or protein quantification).
Standard plaque assay on VERO cells
The plaque assay was performed on VERO cell monolayers. Depending on the experimental setting, cell‑free virus titers were quantified from clarified supernatants, whereas total virus titers were obtained by subjecting infected cells and their corresponding supernatants to three freeze-thaw cycles to release intracellular virions from infected cells. Serial ten‑fold dilutions of each sample were inoculated to VERO cell monolayers and allowed to absorb for 1 h at 37 °C under gentle agitation. After absorption, the viral inoculum was removed and replaced with culture medium containing 0.8% methylcellulose. After 72 h, viral plaques were visualized and counted under a microscope following staining with a crystal violet solution.
Western blot analysis
An Immunoblot analysis was performed to assess the accumulation of viral proteins, as previously described41. Briefly, total cell lysates were prepared by resuspending cells in 1X SDS sample buffer (62.5 mM Tris-HCl, pH 6.8; 50 mM dithiothreitol (DTT); 10% glycerol; 2% sodium dodecyl sulfate (SDS); 0.01% Bromophenol Blue; 1X EDTA-free Protease Inhibitor Cocktail (Roche). The samples were then boiled for 5 min. Equal amounts of protein were separated on gels containing different percentages of SDS-polyacrylamide, 15% to resolve CXCL10 and CXCL13, and 10% to resolve phospho-NF-κB p65 and transferred to nitrocellulose membrane. After incubation in blocking buffer (5% non-fat dry milk in TBS) at 37 °C, membranes were probed overnight with specific primary antibody, and then incubated with secondary antibodies. Immunoreactive bands were visualized using the LiteUP WB Chemiluminescent Substrate (Euroclone) and captured using a ChemiDoc Touch Imaging System (Bio-Rad) where indicated. Quantification of band intensities was conducted using ImageJ software and Bio-Rad Image Lab 6 software. Target protein levels were normalized to housekeeping, and results were graphically represented using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA).
Viral DNA extraction and real-time PCR
Viral DNA was extracted using TRIzol® (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
The procedure, based on the phenol/chloroform extraction method, allows for the precipitation of viral DNA from the organic phase42. The DNA pellet was washed twice in a solution containing 0.1 M trisodium citrate in 10% ethanol and then dissolved in 8 mM NaOH. DNA concentration was determined using a fluorometer with the Qubit double-stranded DNA (dsDNA) HS (High Sensitivity) Assay Kit, according to the manufacturer’s instructions. The amplification of viral DNA was carried out by TaqMan™Universal Master Mix II (Applied Biosystems™, Foster City, CA, USA) in a 50 µL reaction mixture containing the following: TaqMan Universal Master Mix II, DNA (100 ng); HSV-1 forward (10 µM) and HSV-1 reverse (10 µM) primers (Table S3); and the TaqMan probe (5 µM) (Table S3). The amplification was carried out on Applied Biosystems 7300 Real-Time PCR System under the following conditions: 10 min at 95 °C, 60 s at 95 °C for 40 cycles, 30 s at 60 °C, and 30 s at 72 °C. Each amplification run contained one negative control. The primers were designed on a large catalytic subunit of HSV-1 DNA polymerase holoenzyme (UL30 gene). The relative quantitation of HSV-1 DNA was performed by the comparative Ct method using GAPDH as a housekeeping gene (Table S3). One-way analysis of variance (ANOVA) and the GraphPad Prism 6 software (GraphPad Software, San Diego, CA, USA) was used to perform statistical analysis and graphical representations, respectively.
RNA extraction, reverse transcription, and real-time PCR
Total RNA was extracted using TRIzol® (Life Technologies, Carlsbad, USA), according to the manufacturer’s instructions, and DNase-treated before cDNA transcription as follows: 1 µg of RNA was incubated at 37 °C for 2 h with 5 µL 10X DNase I Buffer, 2 µL Recombinant RNase-free DNase I (10U) (2270 A TaKaRa, Dalian, China) and RNase inhibitor (20U) (N251A Promega). The procedures were published previously43. The concentration of extracted RNA was determined using Qubit™ RNA HS Assay (Invitrogen). Total RNA (0.5 µg) was reverse transcribed using avian myeloblastosis virus reverse transcriptase (Promega, Madison, WI) under the following conditions: denaturation at 70 °C for 10 min, followed by 42 °C for 45 min, 52 °C for 45 min and 95 °C for 5 min. The cDNAs were used for quantitative Real-Time PCR by using QuantiNova SYBR Green PCR Kit (Qiagen) carried out on Qiagen QIAquant 96 2plex PCR Thermal Cycler. The thermal profile consists of a 2 min incubation at 95 °C followed by 40 cycles of 5 s denaturation at 95 °C, 10 s annealing/extension at 60 °C. The cDNA copy numbers were normalized to GAPDH. The analytic primers for RT-PCR are listed in Table S3. Each quantitative Real-time PCR experiment includes a minus-reverse transcriptase control.
RT2 profiler PCR arrays
A total of 750 ng of cDNA was used for the RT² Profiler PCR Array (QIAGEN, Cat. no. PAHS-033Z), in combination with the RT² SYBR® Green qPCR Mastermix (Cat. no. 330502) in RNase/DNase-free water. Equal volumes (25 µL) of the reaction mixture were dispensed into each well of the array plate, which contained pre-aliquoted, gene-specific primer sets. PCR amplification and detection were carried out using the Applied Biosystems 7300 Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) with the following thermal cycling conditions: initial denaturation at 95 °C for 10 min, followed by 40 cycles of 15 s at 95 °C (denaturation) and 60 s at 60 °C (annealing/extension).
Each 96-well plate included controls for genomic DNA contamination, reverse transcription efficiency, and PCR performance. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the reference (housekeeping) gene. Data analysis was performed using the Qiagen GeneGlobe online platform, comparing all experimental conditions: infected vs. uninfected and treated vs. untreated samples. A cycle threshold (Ct) value of 35 was used as the cut-off for detectable gene expression. Supplementary Table S1 presents the gene expression profiles of HSV-1-infected THP-1 cells, either untreated or treated with NRRE and RURE, in comparison to uninfected, untreated controls.
Conditioned medium transfer assay
THP-1 cells were seeded at 1 × 10^6 cells per t25 flasks. Cells were infected with HSV-1 at a multiplicity of infection (MOI) of 50. Before infection, both cells and virus were pretreated with NRRE or RURE extracts (0.6 mg/mL) or zeaxanthin (10 µM), as described in paragraph 4.5. Infection was performed by adding the pretreated virus to the cells for 1 h to allow viral adsorption. After adsorption, cells were washed three times with fresh medium to remove unbound virus and residual treatments. A parallel infected sample was treated with PAA to block viral replication and served as a control for replication-dependent effects.
After 24 h post-infection, cell-free conditioned supernatants were collected by centrifugation to remove cellular debris. An aliquot of 100 µL of the clarified supernatant was used to determine the cell-free viral titer in Vero cells. These were transferred onto SH-SY5Y neuroblastoma cells and HEp-2 epithelial cells, seeded at 4 × 105 cells per well in 6-well plates. The cells were exposed to the cell-free conditioned supernatants for 1 h under gentle rocking. Following incubation, supernatants were removed. Cells were washed twice with fresh medium and then cultured in growth medium for an additional 24 h. Cell-free virus, measured directly from the supernatant, and total virus titer, obtained by freeze–thawing the cell monolayer to release intracellular virions, were quantified in Vero cells using the standard plaque assay procedure described in paragraph 4.6.
Statistical analysis
Data are presented as mean ± standard deviation (SD) from at least three independent experiments. Statistical analysis was performed using GraphPad Prism version 8.0.1 (GraphPad Software Inc., San Diego, CA, USA). For array datasets, statistical significance was assessed by two-way ANOVA followed by Dunnett’s multiple comparisons test. For all other analyses, one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc correction was used. Array data were calculated using values for three technical and one biological replicates. For qPCR analysis, means ± standard deviations were calculated from two biological replicates and three technical replicates. For all assays, the detection limits were determined based on the minimum reliably measurable values (Ct > 40). Data points falling below these limits were labeled as “b.d.” (below detection) in the figures, and this is indicated in the corresponding figure legends.
Data availability
All data supporting the findings of this study are included in the article and its supplementary materials.
References
Arvin, A. C.-F. G. M. E. M. P. R. B. W. R. Y. K. editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. (2007).
Whitley, R. J. & Roizman, B. Herpes simplex virus infections. Lancet 357, 1513–1518 (2001).
Saleh, D. Y. S. S. S. Herpes simplex type 1. (2025). https://www.ncbi.nlm.nih.gov/books/NBK482197/
Toews, G. B. Macrophages. in Asthma and COPD 133–143Elsevier, (2009). https://doi.org/10.1016/B978-0-12-374001-4.00011-0
Ellermann-Eriksen, S. Macrophages and cytokines in the early defence against herpes simplex virus. Virol. J. 2, 59 (2005).
Smith, J. B., Herbert, J. J., Truong, N. R. & Cunningham, A. L. Cytokines and chemokines: the vital role they play in herpes simplex virus mucosal immunology. Front. Immunol. 13, 936235 (2022).
Wang, L., Wang, R., Xu, C. & Zhou, H. Pathogenesis of herpes stromal keratitis: immune inflammatory response mediated by inflammatory regulators. Front Immunol 11, 766 (2020).
Thapa, M., Welner, R. S., Pelayo, R. & Carr, D. J. J. CXCL9 and CXCL10 expression are critical for control of genital herpes simplex virus type 2 infection through mobilization of HSV-specific CTL and NK cells to the nervous system. J. Immunol. 180, 1098–1106 (2008).
Shin, H. & Iwasaki, A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491, 463–467 (2012).
Hussain, M. S. et al. Immunopathology of herpes simplex virus-associated neuroinflammation: Unveiling the mysteries. Rev. Med. Virol. 34(1), e2491 (2024).
Suazo, P. A. et al. Evasion of early antiviral responses by herpes simplex viruses. Mediators Inflamm (2015). (2015).
McKimmie, C. & Michlmayr, D. Role of CXCL10 in central nervous system inflammation. Int. J. Interferon Cytokine Mediat Res. 1 https://doi.org/10.2147/IJICMR.S35953 (2014).
Musarra-pizzo, M. et al. In vitro anti-HSV-1 activity of polyphenol-rich extracts and pure polyphenol compounds derived from pistachios kernels (Pistacia vera l). Plants 9(2), 267 (2020).
Pennisi, R. et al. Mechanistic understanding of the antiviral properties of Pistachios and Zeaxanthin against HSV-1. Viruses 15, 1651 (2023).
Zheng, X., Zhang, X. & Zeng, F. Biological functions and health benefits of flavonoids in fruits and vegetables: a contemporary review. Foods 14, 155 (2025).
Yi, Y. S. Regulatory roles of flavonoids in caspase-11 non-canonical inflammasome-mediated inflammatory responses and diseases. Int. J. Mol. Sci. 24, 10402 (2023).
Al-Khayri, J. M. et al. Flavonoids as potential anti-inflammatory molecules: a review. Molecules 27, 2901 (2022).
Saini, R. K., Nile, S. H. & Park, S. W. Carotenoids from fruits and vegetables: chemistry, analysis, occurrence, bioavailability and biological activities. Food Res. Int. 76, 735–750 (2015).
Leyva-López, N., Gutierrez-Grijalva, E., Ambriz-Perez, D. & Heredia, J. Flavonoids as cytokine modulators: a possible therapy for inflammation-related diseases. Int. J. Mol. Sci. 17, 921 (2016).
Ribeiro, D., Freitas, M., Lima, J. L. C. & Fernandes, E. Flavonoids inhibit the production of cytokines/chemokines and induce apoptosis in human neutrophils. Free Radic Biol. Med. 75, S46 (2014).
Bai, S. K. et al. β-Carotene inhibits inflammatory gene expression in lipopolysaccharide-stimulated macrophages by suppressing redox-based NF-κB activation. Exp. Mol. Med. 37, 323–334 (2005).
Di Tomo, P. et al. β-Carotene and lycopene affect endothelial response to TNF‐α reducing nitro‐oxidative stress and interaction with monocytes. Mol. Nutr. Food Res. 56, 217–227 (2012).
Li, R., Hong, P. & Zheng, X. β-carotene attenuates lipopolysaccharide‐induced inflammation via inhibition of the NF‐κB, JAK2/STAT3 and JNK/p38 MAPK signaling pathways in macrophages. Anim. Sci. J. 90, 140–148 (2019).
Lin, H. W. et al. Regulation of virus-induced inflammatory response by β-carotene in RAW264.7 cells. Food Chem. 134, 2169–2175 (2012).
Dash, S. P., Gupta, S. & Sarangi, P. P. Monocytes and macrophages: origin, homing, differentiation, and functionality during inflammation. Heliyon 10, e29686 (2024).
Brun, P. et al. Herpes simplex virus type 1 engages toll like receptor 2 to recruit macrophages during infection of enteric neurons. Front Microbiol. 9, 2148 (2018).
Garber, A., Barnard, L. & Pickrell, C. Review of whole plant extracts with activity against herpes simplex viruses in vitro and in vivo. J Evid. Based Integr. Med 26, 2515690X20978394 (2021).
Teresa Sciortino, M. et al. Signaling Pathway Used by HSV-1 to Induce NF‐κB Activation. Ann. N Y Acad. Sci. 1096, 89–96 (2007).
Venuti, A. et al. HSV-1\EGFP stimulates miR-146a expression in a NF-κB-dependent manner in monocytic THP-1 cells. Sci. Rep. 9, 5157 (2019).
Elemam, N., Talaat, I. & Maghazachi, A. CXCL10 chemokine: a critical player in RNA and DNA viral infections. Viruses 14, 2445 (2022).
Honess, R. W. & Watson, D. H. Herpes simplex virus resistance and sensitivity to phosphonoacetic acid. J. Virol. 21, 584–600 (1977).
Marino-Merlo, F. et al. HSV-1-induced activation of NF-κB protects U937 monocytic cells against both virus replication and apoptosis. Cell. Death Dis. 7, e2354–e2354 (2016).
Carr, D. J. J., Chodosh, J., Ash, J. & Lane, T. E. Effect of anti-CXCL10 monoclonal antibody on herpes simplex virus type 1 keratitis and retinal infection. J. Virol. 77, 10037–10046 (2003).
Carr, D. J. J. & Tomanek, L. Herpes simplex virus and the chemokines that mediate the inflammation. in Chemokines and Viral Infection 47–65 (Springer Berlin Heidelberg, Berlin, Heidelberg). https://doi.org/10.1007/978-3-540-33397-5_3
Melchjorsen, J., Pedersen, F. S., Mogensen, S. C. & Paludan, S. R. Herpes simplex virus selectively induces expression of the CC chemokine RANTES/CCL5 in macrophages through a mechanism dependent on PKR and ICP0. J. Virol. 76, 2780–2788 (2002).
Yoshimura, T. & Oppenheim, J. J. Chemokine-like receptor 1 (CMKLR1) and chemokine (C–C motif) receptor-like 2 (CCRL2); Two multifunctional receptors with unusual properties. Exp. Cell. Res. 317, 674–684 (2011).
Samson, M. et al. ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur. J. Immunol. 28, 1689–1700 (1998).
Faivre, N., Verollet, C. & Dumas, F. The chemokine receptor CCR5: multi-faceted hook for HIV-1. Retrovirology 21, 2 (2024).
Lai, C. et al. C-C Motif Chemokine Ligand 2 (CCL2) mediates acute lung injury induced by lethal influenza H7N9 virus. Front. Microbiol. 8, 587 (2017).
Pontejo, S. M., Murphy, P. M. & Pease, J. E. Chemokine subversion by human herpesviruses. J. Innate Immun. 10, 465–478 (2018).
Colao, I. et al. The ERK-1 function is required for HSV-1-mediated G1/S progression in HEP-2 cells and contributes to virus growth. Sci. Rep. 7, 9176 (2017).
Pennisi, R. et al. Analysis of antioxidant and antiviral effects of olive (Olea europaea L.) leaf extracts and pure compound using cancer cell model. Biomolecules 13, 238 (2023).
Pennisi, R. & Sciortino, M. HSV-1 triggers an antiviral transcriptional response during viral replication that is completely abrogated in PKR–/– cells. Pathogens 12, 1126 (2023).
Acknowledgements
We are deeply grateful to Professor Bernard Roizman for his invaluable scientific support and for inspiring us to explore the hidden world behind HSV infection, shaping our research with his insight and passion.
Funding
This work was funded by American Pistachio Growers (APG), Fresno, CA (USA).
Author information
Authors and Affiliations
Contributions
Conceptualization: M.T.S., G.M. and R.P.; software: R.P.; investigation: R.P., P.T., M.P.T and M.C; data curation: R.P.; and M.T.S., supervision: M.T.S., G.M. and R.P.; project administration: M.T.S. and G.M.; funding acquisition: M.T.S. and G.M. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pennisi, R., Costa, M., Tamburello, M.P. et al. Restriction of HSV-1 replication by Pistacia vera L. extracts reveals a promising strategy for regulating virus-mediated chemokine response in monocytic cells. Sci Rep 16, 10800 (2026). https://doi.org/10.1038/s41598-026-43975-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-43975-x
