
Abstract
The comprehensive effects of cross-industry Bioaugmentation with Aspergillus oryzae CICC 2339, derived from bean-based fermented foods, on traditional medium-high temperature Daqu and its fermented crude Baijiu were evaluated, with the aim of providing a scientific basis for optimizing the quality of Texiangxin Baijiu. Application of Aspergillus oryzae CICC 2339 in Daqu fortification effectively regulated the growth of thermotolerant dominant bacteria and significantly increased the contents of flavor compounds (particularly those with sauce and floral aromas) in fortified Daqu and its fermented crude Baijiu. This study provides an innovative approach for producing high-quality Texiangxin Baijiu using medium-high temperature Daqu, and the findings hold both scientific and practical value for advancing Baijiu quality.
.
Similar content being viewed by others


Introduction
Chinese Baijiu, renowned globally for its distinct aromatic profiles, relies on Daqu—a unique sacchariferous and fermentative agent—as the cornerstone of its production1,2. Daqu serves multifunctional roles in Baijiu brewing, acting as a reservoir of microorganisms, enzymes, and precursors of flavor compounds3, thereby integrating saccharification, fermentation, and aroma generation processes4. The stylistic diversity of Baijiu is predominantly shaped by the synergistic interactions within Daqu’s complex microbial consortia5, which dictate the metabolic pathways responsible for flavor compound biosynthesis. Traditional Daqu production employs raw materials, spontaneous microbial inoculation, and manual cultivation techniques. While this method yields Daqu with unique regional characteristics, its quality consistency remains suboptimal due to uncontrolled microbial succession and environmental fluctuations6. To address this limitation, fortified Daqu (FD) has emerged as a promising solution. FD is a specialized strain of Daqu (Chinese traditional fermentation starter, GB/T 15109 − 2021) produced through the incorporation of high-performance ripe starter for inoculation or selected microbial inocula into the raw material matrix during the Daqu-making process, followed by controlled microbial cultivation and fermentation to achieve superior functional properties. For instance, FD fortified with Bacillus halotolerans exhibits elevated acidity, saccharifying power, fermentation efficiency, and enhanced concentrations of volatile organic compounds (VOCs) such as alcohols and esters compared to conventional high-temperature Daqu (HTD)7. The functional microorganisms used in FD production primarily belong to three taxonomic groups: molds, yeasts, and bacteria8. These strains are typically introduced as seed suspensions or bran-based inocula, either through direct incorporation into raw materials or surface spraying onto Daqu bricks, followed by natural fermentation and storage. However, current FD development relies heavily on isolating functional strains from Baijiu-related matrices (e.g., Daqu, fermented grains, pit mud), limiting microbial diversity and innovation potential. In contrast, Foods and beverages relying on microbial fermentation are highly popular among consumers, traditional Asian bean-based fermented foods (BBFFs), such as soy sauce, doubanjiang, and miso, harbor a rich repertoire of beneficial microorganisms9, Modern technologies are employed to refine the production processes of traditional fermented foods, elevate product quality, and strengthen their sustainability10. Among these, Aspergillus oryzae stands out as a key functional species, equipped with multiple enzyme-encoding genes related to flavor biosynthesis pathways, including lipases and proteases, which facilitate the production of aromatic compounds11. While BBFFs emphasize the critical role of microbial inoculation in enhancing food quality and safety—a practice systematically adopted only in the 20th century—A. oryzae CICC 2339 (Huniang 3.042) remains the most widely used starter culture, particularly in doubanjiang production, due to its non-aflatoxigenic nature and high safety profile12. However, despite its well-established efficacy in food fermentation, the application of A. oryzae CICC 2339 in Daqu enhancement has been largely overlooked, with no prior studies reporting its use for cross-industry Daqu fortification. This study, therefore, investigates the impact of A. oryzae CICC 2339 on the microbial community structure and Volatile Organic Compounds profile in medium-high temperature Daqu and Crude Baijiu, while elucidating the underlying microbial interaction mechanisms. The findings aim to validate the feasibility of A. oryzae in flavor-oriented Baijiu production and provide a theoretical foundation for its cross-disciplinary application in the Baijiu brewing industry.
Materials and methods
Strains and sample collection
Fortified strain
Aspergillus oryzae CICC 2339, also known as Aspergillus oryzae 3.042, Hunaing 3.042 was preserved in our laboratory. The strain was first activated on potato dextrose agar (PDA) at 30 °C for 72 h, then cultivated on sterilized wheat bran under solid-state fermentation at 30 °C for 48 h to produce the bran-based inoculum (FuQu, a bran-based starter culture prepared by solid-state fermentation, which is commonly used in traditional Chinese fermentation processes). The inoculum was then incorporated into the Daqu raw materials at a ratio of 0.5% (w/w) based on total wheat and bran weight.
Technical route
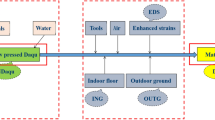
The Critical procedures in the technical route during the research were illustrated in Fig. 1, which was divided into two main processes: Daqu-making and Baijiu brewing.

Critical procedures of medium-high temperature Daqu fortified by Aspergillus oryzae CICC 2339 and its crude Baijiu brewing within the technical pathway.
Daqu sample collection
Medium-high temperature Daqu was produced at a distillery in Jiangxi Province using conventional methods and served as the control group (CG). Simultaneously, in the experimental group (AOG), An additional 0.5% (w/w, calculated based on the total weight of wheat and bran) of Aspergillus oryzae CICC 2339 FuQu seed was added, which was incorporated into the traditional DaQu-making raw materials. After 6 months of storage, matured Daqu samples were collected from each group. For each group, samples were taken from the upper, middle, and lower layers of the Daqu pile, with 10 pieces collected from each layer. This stratified sampling approach (upper, middle, and lower layers) was adopted to account for potential vertical gradients in microbial and physicochemical properties within the Daqu pile, while the pooling of 10 bricks per layer and three independent batches ensured that the composite sample accurately represented the entire batch. The collected pieces were then ground and thoroughly mixed. The Daqu powder was stored at -20 °C for subsequent analysis of physicochemical properties, enzyme activity, and volatile compounds. A portion of the powdered Daqu was stored at -80 °C for DNA extraction.
Crude Baijiu sample collection
24 fermentation pits continuously operated by the same brewing team were selected as experimental units and divided into a control group and an experimental group using a random number table method, with 12 pits in each group. During grouping, it was ensured that the two groups were comparable in terms of potential influencing factors such as fermentation cycle, historical crude baijiu quality, and microbial environment (p > 0.05). Both groups adopted refermentation grains and pit-entry process. Specifically, Contents of one distilling pot of refermentation grains was taken, cooled, and then 10.0 ± 0.1 kg of Daqu was added. After thorough mixing of the fermented grains and Daqu, the mixture materials was laid at the bottom of the fermentation pits, with strict consistency maintained in process parameters. Solid-state fermentation was carried out for 30 days according to the traditional special-flavor Baijiu brewing process. After fermentation, the pits were opened, and the bottom layer of alcoholic fermentative materials from the refermentation grains was collected. Those materials from each pit were distilled separately in a distilling pot, with cutting-out both end of the distillate, and the middle fraction was used as the crude Baijiu. The crude Baijiu from each pit was stored separately. The experiments were carried out consecutively in three independent batches. After 6 months of storage at room temperature, the crude Baijiu samples were randomly selected from three pits in each group. For each sample, 660 mL of the crude Baijiu was taken and divided into 250 mL glass bottles. The samples were stored at 4 °C for the detection of volatile compounds.
Physicochemical indicators analysis
Moisture content was determined using the constant temperature drying method. Daqu samples were dried at 101–105 °C, and the moisture content was calculated based on the mass loss after drying (Chai et al., 2021). For each sample, 5 g of Daqu powder was suspended in 50 mL of sterile distilled water, and the suspension was centrifuged at 6,000×g for 15 min. The supernatant was collected for further analysis. Titratable acidity was determined by titration with a standard NaOH solution (0.1 mol/L). Protease activity was measured using the formaldehyde method as per the SB/T 10,317 − 1999 standard, which involves the catalytic hydrolysis of proteins into amino acids, followed by formaldehyde fixation of the amino groups. The amount of amino acids produced was determined by titration with NaOH, and the enzyme activity was expressed as “mg amino nitrogen/100 g dry basis” Fermentation power was determined by the CO2 loss method, following the method used in the Xiangjiao test. The fermentation power was defined as the grams of CO2 produced by fermentable sugars per gram of Daqu in 24 h at 30 °C, expressed as “g/g·24 h.” Liquefaction ability was measured using the iodine decolorization method, based on QB/T 4257 − 2011, and was defined as the grams of starch liquefied per gram of Daqu per hour at 35 °C and pH 4.6, expressed as “g/g·h.” Saccharification ability was determined by the Fehling’s method, also following QB/T 4257 − 2011. It was defined as the milligrams of glucose produced from soluble starch per gram of Daqu in one hour at 35 °C and pH 4.6, expressed as “mg/g·h.” Esterification ability was measured using the saponification method, following QB/T 4257 − 2011, and was defined as the milligrams of ethyl ester synthesized from acetic acid and ethanol per 50 g of Daqu over 7 days at 35 °C, expressed as “mg/50 g·7d.”
Volatile organic compounds analysis
One gram of Daqu powder was combined with 4 mL of saturated sodium chloride solution and 10 µL of internal standard (2-octanol, 20 mg/L) in a 25 mL vial. The sample was sealed with a silicone stopper. Volatile compounds were extracted using DVB/CAR/PDMS fibers (50/30 µm, Supelco, USA) at 50 °C for 50 min and then desorbed at 230 °C for 5 min for separation. Flavor chemicals were analyzed using HS-SPME-GC-MS (7890a-5975c, Agilent, USA) with a DB-WAX column (30 m × 0.25 mm × 0.5 μm, Agilent, USA) and a flame ionization detector. The GC oven temperature was initially held at 40 °C for 5 min, then increased at a rate of 5 °C/min to 160 °C, held for 5 min, and further increased at a rate of 20 °C/min to 210 °C without holding, followed by a final increase at 5 °C/min to 230 °C, held for 5 min. The helium carrier gas flow rate was 1 mL/min. Mass spectrometry (MS) was performed with 70 eV electron ionization, and data were collected in the range of 33–450 m/z with a scan rate of 1 scan/second (Yang et al., 2024). Volatile compounds were identified by comparing their mass spectra with the NIST17 spectral database (Agilent).
DNA extraction and sequencing
Genomic DNA from microbial communities in Daqu samples was extracted using the E.Z.N.A.® Soil DNA Kit (Omega Bio-tek, USA) following the manufacturer’s instructions. The DNA’s purity and concentration were assessed with a Nanophotometer N50 Touch spectrophotometer, and its integrity was verified by 1% agarose gel electrophoresis at 6 V/cm for 30 min. To analyze the microbial community structure of Daqu, primers 338 F (5’-ACTCCTACGGGAGGCAGC-3’)/806R (5’-GGACTACHVGGGTWTCTAAT-3’) were used to amplify the V3-V4 region of 16 S rDNA, and primers ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′)/ITS2R (5′-GCTGCGTTCTTCATCGATGC-3′) were used to amplify the ITS1-ITS2 region of fungal internal transcribed spacers (ITS). The resulting amplicon libraries were sequenced on the Illumina HiSeq platform (2 × 250 bp) for paired-end high-throughput sequencing. Sequencing data were analyzed using QIIME2. Raw sequences underwent sample demultiplexing and quality filtering as previously described. High-quality sequences were denoised with DADA2 and converted into amplicon sequence variants (ASVs). Taxonomic annotation of fungal ASVs was conducted by comparing representative sequences against the UNITE database (v10.0) at an 80% confidence level. Bacterial ASVs were classified by comparing representative sequences to the SILVA database (v13.8) at an 80% confidence level.
Data analysis
The relative abundance of ASVs at multiple taxonomic levels was calculated using the microeco package (v1.14.0) in R. After rarefying the ASV table to a uniform sequencing depth with the vegan package (v2.6-10) in R (v4.2.1), community richness (Chao1), diversity (Shannon), and Bray-Curtis distances were calculated (Jing et al., 2024). Statistical analyses were performed using the rstatix package (v0.7.2) in R. VIP values for PLS-DA analysis were calculated with the ropls package (v1.40.0). Visualization of microbial community data, physicochemical properties, and volatile compounds was accomplished using the ggplot2 package (v3.5.2) in R. Correlations between microbiota and physicochemical properties or volatile compounds were visualized using network tools available at https://www.omicstudio.cn/tool. Metabolic pathway analysis of metabolites was conducted by querying the KEGG database. Microbial core genome analysis was performed using the comparative genomics tool Panacota (v1.4.0), and image layout was done using Adobe Illustrator.
Results and discussion
Effect of Aspergillus oryzae bioaugmentation on microbial community structure and diversity of Daqu
A comparative analysis was conducted to examine the diversity and structural changes in bacterial (Fig. 2A) and fungal (Fig. 2B) communities within Daqu samples from the experimental group (AOG) inoculated with Aspergillus oryzae and the control group (CG).

Diversity and taxonomic composition of microbial communities in Daqu. (A-B) Stacked bar charts showing the relative abundance of bacterial (A) and fungal (B) communities at the genus level in the Control Group (CG) and the Aspergillus oryzae Added Group (AOG). Genera with relative abundance < 1% are grouped as “Others”. (C-D) Boxplots comparing the relative abundance of dominant bacteria (C) and fungi (D) between CG and AOG. Only genera with an average relative abundance > 1% are shown, Detection rate > 60%. Key bacterial genera: Acetobacter (醋酸杆菌属), Bacillus (芽孢杆菌属), Companilactobacillus (伴生乳杆菌属), Enterobacter (肠杆菌属), Kroppenstedtia (克罗彭斯特菌属), Lactobacillus (乳杆菌属), Limosilactobacillus (拉莫斯乳杆菌属), Oceanobacillus (大洋芽孢杆菌属), and Weissella (魏斯氏菌属). Key fungal genera: Aspergillus (曲霉属) and Thermoascus (嗜热子囊菌属). Statistical significance was determined by Wilcoxon rank-sum test (ns: not significant). (E-H) Venn diagrams and Pie charts displaying the number and proportion of shared and unique OTUs for bacteria (E, F) and fungi (G, H) across the two groups.
Effect of Aspergillus oryzae bioaugmentation on microbial community structure a of Daqu
Changes in bacterial community structure
The results revealed that, compared to the control group (CG), the Aspergillus oryzae-enhanced Daqu (AOG) exhibited a relative abundance exceeding 1% for nine bacterial genera, including Bacillus Oceanobacillus and Acetobacter, among which Bacillus was significantly different (P < 0.05) Oceanobacillus significantly increased (P > 0.05). Notably, the relative abundance of the genus Bacillus some increased, while that of several other genera, such as Lactobacillus, Weissella and Enterobacter, some decreased. Consequently, bacterial diversity declined in the AOG group, as illustrated in Fig. 2A and C. Venn diagram analysis (Fig. 2E) indicated that the AOG and CG groups shared 92 bacterial operational taxonomic units (ASVs), accounting for 18% of the total ASVs. The AOG group possessed 211 unique ASVs, representing 42% of its bacterial community, but the CG group possessed 40%, which suggested that A. oryzae inoculation significantly influenced bacterial species richness and community structural diversity. Furthermore, the bacterial community in the AOG group underwent succession, with Bacillus emerging as the core dominant genus (Fig. 2F).
Changes in fungal community structure
Regarding the fungal community, the AOG group demonstrated a substantial increase in the relative abundance of Aspergillus, which became the dominant genus. The fungal community exhibited a centralized diversity pattern, with Aspergillus playing a leading role. Two fungal genera, Aspergillus and Thermoascus, each had a relative abundance exceeding 1% (Figs. 2D). Notably, the relative abundance of Thermoascus decreased (P > 0.05), however, there was an increase in the relative abundance of Aspergillus (P > 0.05). The Venn diagram (Fig. 2E) also revealed that the AOG and CG groups shared 15% of their fungal ASVs, with the AOG group having 38% unique ASVs and CG group having 47% unique ASVs. This indicates an alteration in fungal species composition diversity due to A. oryzae inoculation (P > 0.05). The community composition pie chart (Fig. 2H) demonstrated that the proportion of dominant fungal genera (e.g., Thermoascus) in the AOG group increased, while the proportions of Pichia and Kodamaea decreased. Additionally, the co-occurrence of Aspergillus-related taxa within the community suggests that, following A. oryzae inoculation, the fungal community structure in Daqu underwent succession towards specific dominant genera.
Effect of Aspergillus oryzae bioaugmentation on microbial community diversity in Daqu
As shown in Fig. 3A and B, the alpha diversity of bacterial and fungal communities was evaluated. Specifically, the Chao1 index was utilized to assess the richness, and the Shannon index was employed to evaluate the diversity. After Aspergillus oryzae (AOG) was added, a comparison was made between the AOG group and the control group (CG). For both bacteria and fungi, no significant differences in the Chao1 and Shannon indices were detected between these two groups (P > 0.05). When augmented fermentation with A. oryzae was carried out, no significant influence was exerted on the richness (as measured by Chao1) and diversity (as measured by Shannon) of the bacterial and fungal communities in Daqu.

Alpha diversity, Beta diversity and co-occurrence networks of microbial communities. (A-B) Alpha diversity indices (Chao1 and Shannon) for bacterial (A) and fungal (B) communities in AOG and CG. Boxplots show the median and interquartile range (ns: not significant). (C-D) Principal Coordinate Analysis (PCoA) based on Bray-Curtis distances revealing the structural differences in bacterial (C) and fungal (D) communities. The percentage of variation explained by PCo1 and PCo2 is indicated on the axes. (E-F) Spearman correlation network analysis of bacterial (E) and fungal (F) genera. Nodes represent genera, and edges represent strong correlations (Thresholds: Spearman |r| > 0.4, p < 0.05, Relative abundance > 0.01%). The size of the node is proportional to its degree of connectivity. Red lines indicate positive correlations. Key hub genera identified include: Thermoactinomyces (高温放线菌属), Pediococcus (片球菌属), Staphylococcus (葡萄球菌属) for bacteria; and Rhizopus (根霉属), Aspergillus (曲霉属) for fungi.
Principal coordinate analysis (PCoA)
The PCoA, based on the Bray-Curtis distance, was employed to visualize the dissimilarities in microbial community structures. For the bacterial community (Fig. 3C), PC1 and PC2 collectively accounted for 73.1% of the total variation. Although certain differences were observed in the bacterial community structures between the AOG and CG groups, these differences did not reach statistical significance (P > 0.05). In contrast, a relatively more pronounced, yet still non-significant, difference was noted in the fungal community structures between the AOG and CG groups (P > 0.05). These results collectively suggest that the addition of A. oryzae induced directional shifts in both the bacterial and fungal community structures in Daqu, but the inter-group differences were not statistically significant (P > 0.05).
Microbial correlation network analysis
The Spearman correlation network analysis provided valuable insights into the intricate interaction patterns within the bacterial and fungal communities present in Daqu. In the bacterial community (Fig. 3E), core genera like Bacillus and Lactiplantibacillus were found to have high degrees of connectivity. This high connectivity indicates their central roles in the microbial network, potentially acting as keystone species that influence the overall community structure and function. Moreover, these core genera exhibited significant positive correlations with acid - producing genera, including Lactiplantibacillus and Weissella. Positive correlations between these genera suggest that they may co-exist in a mutually beneficial manner, perhaps through the exchange of metabolic products. For example, acid - producing genera can create an acidic microenvironment that may inhibit the growth of some competing microorganisms, while the core genera might provide other essential nutrients or signaling molecules.
Within the fungal community (Fig. 3F), strong correlations were observed among genera such as Aspergillus, Rhizopus, and Thermoascus. These robust correlations imply synergistic interactions among fungal species in terms of enzyme secretion and environmental adaptation. Aspergillus species are well - known for their ability to secrete a wide range of enzymes, including amylases, proteases, and cellulases, which are crucial for the breakdown of complex organic compounds in Daqu13. Rhizopus can also produce various enzymes, and its growth characteristics may complement those of Aspergillus. Thermoascus, being a thermophilic fungus, can adapt to the relatively high - temperature conditions during Daqu fermentation. The synergistic interactions among these fungi, in terms of enzyme secretion, can enhance the degradation of raw materials and the formation of flavor - related compounds. Additionally, their coordinated environmental adaptation strategies, such as tolerance to different pH and temperature ranges, contribute to the overall stability of the fungal community in Daqu. These synergistic interactions among fungi are likely to contribute to the enhancement of biotransformation processes in Daqu. Biotransformation is a key step in Daqu fermentation, where microorganisms convert raw materials into various flavor and aroma compounds. The efficient enzyme - mediated reactions facilitated by the fungal community can accelerate this biotransformation process.
Both the bacterial and fungal communities demonstrated positive correlation network characteristics centered around core functional genera. This network structure provides a fundamental interaction basis for the stable metabolism and functional expression of the microbial community in Daqu. A stable microbial community with well - defined interaction patterns is essential for maintaining the consistent quality of Daqu. It ensures that the various metabolic processes occur in a coordinated manner, which in turn supports the overall quality and efficiency of the Daqu fermentation process.
Aspergillus oryzae plays a pivotal role in the koji-making and fermentation processes of soy sauce production. The koji prepared using Aspergillus oryzae 3.042 exhibits high activities of protease, glucoamylase, and cellulase14. Bacillus species constitute one of the most abundant microbial groups in the brewing of strong-aroma Baijiu and are major contributors to its characteristic sauce-like flavor. In previous studies on Daqu, core microbial communities in high-temperature Daqu have been identified as comprising Bacillus, Oceanobacillus, Kroppenstedtia, Aspergillus, Thermoascus, and Thermomyces. In medium-high-temperature Daqu fortified with Aspergillus oryzae, not only does Aspergillus oryzae emerge as the dominant strain (P > 0.05), but it also promotes the targeted enrichment of thermotolerant Bacillus (P < 0.05) and Oceanobacillus (P > 0.05), among other dominant bacteria. Additionally, it moderately alters the abundance ratios of thermotolerant fungi such as Thermoascus and Thermomyces, while overall maintaining a relatively stable microbial structure in the fortified Daqu.
The findings of this study align with those reported by Yang et al.15., which indicated that during the storage of high-temperature Daqu, the relative abundances of Bacillus, Oceanobacillus, and Aspergillus significantly increase, whereas those of Thermoascus, Thermomyces, and Kroppenstedtia markedly decrease. Notably, a particular pattern emerged in the microbial abundances. An increase in the abundances of bacilli and aspergilli was detected. At the same time, a decrease in the abundances of lactic acid bacteria and Kroppenstedtia species was observed. There was an inverse relationship between these two sets of changes. This observation precisely explains the observed decline in lactic acid bacteria and Weissella levels in this study. Daqu is primarily composed of three systems: microbial flora, enzyme systems, and substrate substances. Among them, the microbial flora occupies a central position. Its growth and metabolic activities directly determine the composition and activity of enzyme systems, thereby exerting a crucial influence on the transformation and utilization of substrate substances.
Analysis of changes in physicochemical properties and enzyme activities of Daqu enhanced with Aspergillus oryzae, and correlation analysis with dominant microorganisms
Changes in physicochemical properties and enzyme activities of Daqu enhanced with Aspergillus oryzae
A systematic comparative analysis was conducted to evaluate the disparities in physicochemical parameters and enzymatic profiles between AOG and CG, with experimental data visualized in Fig. 4 (A-G) .Statistical evaluation revealed that AOG exhibited a significant reduction in acidity (Δ = 20.0%, P < 0.05) compared to CG, while moisture content (Δ = 2.2%, (P > 0.05)) and α-amylase activity (Δ = 9.9%, (P > 0.05) showed non-significant increases(P > 0.05). Notably, AOG demonstrated marked improvements in glucoamylase activity (Δ = 5.5%, P < 0.01), neutral protease activity (Δ = 33.1%, P < 0.05), esterification capacity (Δ = 47.2%, P < 0.001), and fermentation power (Δ = 15.8%, P < 0.05).

The physicochemical and enzyme activity characteristics of the control group (CG) and Aspergillus oryzae (AOG) enhanced fermentation Daqu, as well as the correlation between physicochemical and enzyme activity and dominant microorganisms. (A-G): The box plot showed the distribution of total acid content (A), water content (B), amylase activity (C), glucoamylase activity (D), neutral protease activity (E), esterification ability (F) and fermentation ability (G) among treatments. Mantel test was used to test the correlation between microbial genera and physicochemical/enzyme variables, and the H Heatmap of Pearson correlation coefficient between purple and green physicochemical parameters and enzyme activity. The color gradient represents the intensity of correlation (red: positive; blue: negative). Mantel test was used to test the correlation between microbial genera and physicochemical / enzymatic variables. The purple and green lines (r. sign) represented positive (r > 0) and negative (r < 0), the solid line (P) represented significant (P < 0.05 ), and the dotted line represented non-significant (P > 0.05). The line thickness (r. abs) corresponds to the absolute value of the correlation coefficient (| r |).
The findings of this study exhibit consistency with those of Xu et al., as both demonstrate a common trend of enhanced enzymatic activities in microbially - fortified Daqu. Bacillus licheniformis and Bacillus velezensis were inoculated into medium - high temperature Daqu, resulting in increases of 59.7%, 124.1%, 396.9%, and 32.0% in the saccharifying power, liquefying power, fermenting power, and esterifying power of the fortified Daqu, respectively16. Therefore, selecting appropriate microbial strains for Daqu fortification can enhance its quality.
The correlation between dominant microorganisms and physicochemical properties and enzyme activity in Daqu
The results of the correlation analysis (Fig. 4F) indicated that fermentation ability exhibited a significant positive correlation with amylase activity, glucoamylase activity, and proteinase activity. Conversely, the content of total acids showed a significant negative correlation with the activities of these three enzymes. Additionally, a close positive correlation was observed among amylase activity, glucoamylase activity, and proteinase activity. To further elucidate the connections between microbial communities and environmental factors, this study employed Mantel tests to analyze the correlations between dominant microbial genera and physicochemical properties. The results revealed that the genus Bacillus exhibited significant positive correlations with total acids, amylase activity, and proteinase activity. The genus Limosilactobacillus showed a significant positive correlation with total acids. The genus Lederbergia demonstrated significant positive correlations with glucoamylase activity and proteinase activity. Both Thermoascus and Aspergillus exhibited significant positive correlations with moisture and proteinase activity.
This study unveiled the complex interplay among physicochemical factors and between microorganisms and physicochemical factors during the fermentation process. Firstly, the correlation analysis among physicochemical factors aligns with the fundamental biochemical logic of fermentation. Fermentation ability, as a comprehensive indicator of fermentation progress and efficiency, shows a positive correlation with amylase, glucoamylase, and proteinase activities. This clearly indicates that substrate degradation is a key step driving fermentation. Amylase and glucoamylase synergistically hydrolyze starches and other polysaccharides into fermentable sugars, while proteinase degrades proteins to release free amino acids, providing essential carbon and nitrogen sources for microbial growth and metabolism. The coordinated activities of these three enzymes (which also exhibit positive correlations with each other) collectively promote fermentation. Conversely, as a fermentation product, the accumulation of total acids typically exerts feedback inhibition on microbial growth and related enzyme activities, which explains the negative correlation between total acids and the activities of these three hydrolytic enzymes.
Secondly, the results of the Mantel tests provide evidence for the functional positioning of specific microbial genera within the fermentation ecosystem. The strong positive correlation between the genus Bacillus and amylase and proteinase activities corroborates its characteristic as a dominant enzyme-producing strain. Bacillus spp. is a “workhorse” in industrial production for various hydrolytic enzymes (such as amylase and proteinase), efficiently degrading macromolecules in the early stages of fermentation to provide nutrients for subsequent microbial communities. Its positive correlation with total acids may suggest that certain Bacillus strains possess acid tolerance or remain active in acidic environments17. The significant positive correlation between Limosilactobacillus, a member of the Lactobacillaceae family, and total acids is expected. Bacteria of this genus are typical lactic acid bacteria that effectively utilize sugars for fermentation and produce substantial amounts of acidic substances, making them a major contributor to acidity formation in fermentation systems18. Lederbergia, Thermoascus, and Aspergillus: These three genera (the latter two being fungi) all show significant positive correlations with proteinase activity, highlighting the universal importance of protein degradation in the fermentation process. Lederbergia, Thermoascus, and Aspergillus are renowned for their robust proteinase production capabilities. The positive correlations of Thermoascus and Aspergillus with moisture content align with the physiological characteristics of fungi, which require suitable moisture conditions for growth and enzyme production.
In summary, these correlations reveal that key enzymatic reactions—particularly saccharification and proteolysis—serve as core drivers of fermentation, while also clarifying the ecological niches of microorganisms such as Bacillus and Limosilactobacillus in substrate conversion and flavor formation.
Effect of bioaugmented Aspergillus oryzae on volatile compounds in Daqu and crude Baijiu samples
Changes in volatile compounds after enhancement of Daqu with A. oryzae
Analysis of species and contents of volatile compounds
As illustrated in Fig. 4A, a total of 42 volatile flavor components were detected in both the augmented Daqu sample (AOG) and the control group Daqu sample (CG), including 7 alcohols, 5 aldehydes, 15 esters, 4 phenols, 8 pyrazines, and 3 pyrroles. The average total content of volatile flavor components in the augmented Daqu sample AOG was 7392.65 mg/1000 g, whereas it was 2111.19 mg/1000 g in the control group Daqu sample CG (Table 1). Analysis of variance revealed that, compared to the control group CG, the total content of volatile compounds in the augmented group AOG was significantly increased (P < 0.01). Specifically, the content of phenols was extremely significantly increased (P < 0.01), while the contents of esters and pyrazines were significantly increased (P < 0.05). These findings indicate that Aspergillus oryzae augmented fermentation can influence the formation of volatile compounds in Daqu, enhancing its aroma and laying a rich material foundation for the subsequent flavor formation in Baijiu brewing.
Differential analysis of volatile flavor substances
To analyze the differences between samples across groups, PCA analysis of volatile compounds was conducted, with results shown in Fig. 4C. According to Fig. 4C, the two principal components (PC1 and PC2) explained 99.80% of the total variance contribution, with the differences primarily manifested in PC1. There were notable differences between the sample points of AOG and CG, indicating substantial variations in aroma. To identify the characteristic compounds in Daqu after augmented fermentation, PLSDA analysis was performed on the volatile compounds, and VIP scores were calculated. Compounds with VIP > 1 were considered potential characteristic compounds for the discriminant model. Among the 42 compounds in Daqu, 13 substances were selected as potential differential markers between the augmented group sample AOG and the control group Daqu sample CG, the aroma and flavor characteristics of VOCs are presented in Table 2. Among these, 12 substances had higher contents in AOG than in CG, while 1 substance had a lower content in AOG. However, the differences between the two groups were not statistically significant (P > 0.05), suggesting that Aspergillus oryzae augmentation can directionally regulate the production of volatile compounds in Daqu. These compounds possess distinct aroma and flavor characteristics, laying a material foundation for the subsequent flavor formation in Baijiu brewing.
Changes of volatile compounds in fermented crude Baijiu samples after Aspergillus oryzae strengthening Daqu
Based on the analysis of volatile compounds in the crude Baijiu samples from the control group (CG) and the 0.5% Aspergillus oryzae-enhanced group (AOG) (Fig. 5B), a total of 117 volatile components were detected. These were categorized into 56 esters, 25 alcohols, 12 aldehydes, 5 pyrazines, 11 ketones, 2 acids, 2 phenols, and 4 others, with esters and alcohols constituting the predominant fractions of the volatile compounds. The results revealed increases in the contents of various compounds after Aspergillus oryzae reinforcement: Esters (from 6.91 g/L in CG to 8.22 g/L in AOG), Alcohols (from 0.90 g/L in CG to 1.21 g/L in AOG), Acids (from < 0.01 g/L in CG to 0.55 g/L in AOG), Aldehydes (from 0.09 g/L in CG to 0.13 g/L in AOG), Pyrazines (from 0.06 g/L in CG to 0.07 g/L in AOG), and Ketones (from 0.03 g/L in CG to 0.06 g/L in AOG). Principal coordinate analysis (PCoA, Fig. 5D) revealed significant differences in the volatile compound profiles between the CG and AOG groups (Permanova test, P = 0.05). T-test analysis demonstrated that after Aspergillus oryzae treatment, the contents of 2,3,5,6-tetramethylpyrazine, 2,5-dimethylpyrazine, and 2,6-dimethylpyrazine in the crude Baijiu significantly increased (P < 0.05). The most notably changed components include 2,3,5,6-tetramethylpyrazine (with a 2.7-fold increase in concentration after treatment), 2,5-dimethylpyrazine (with a 4.3-fold increase), and 2,6-dimethylpyrazine (with a 2.4-fold increase), among others.

The volatile compounds in the control group (CG) and Aspergillus oryzae (AOG) enhanced fermentation Daqu (A) and the crude Baijiu (B). A principal coordinate analysis (PCA) map was established to visualize the inter-group differences between Daqu (C) and the crude Baijiu (D) communities. PLS-DA analysis of volatile compounds in fermented Daqu (F) and the crude Baijiu samples (G) showed compounds with VIP values greater than 1.
According to the principal coordinate analysis (PCoA) plot derived from β-diversity analysis in Fig. 5D, the samples from the CG group and the AOG group are located above and below the PC2 axis, respectively. This indicates that Aspergillus oryzae reinforcement has exerted a significant impact on the overall flavor profile of the Baijiu body.
Partial least squares discriminant analysis (PLS-DA, Fig. 5F) identified 34 key differential compounds with VIP values greater than 1. Among these, components such as hexanoic acid, phenylethyl alcohol, isoamyl acetate, and ethyl laurate exhibited significant enrichment in the AOG group (P < 0.05). The results reveal significant findings. The A. oryzae enhancement strategy was applied.This strategy expanded the diversity of volatile compounds in the crude Baijiu samples. Moreover, it prominently promoted the accumulation of several flavor substances. These include soy sauce-like aroma substances (pyrazines), floral flavor substances (phenylethyl alcohol), and fruity flavor substances (esters).
In this study, among the 117 compounds produced during the fermentation of the fortified Daqu for crude Baijiu production, 56 were esters, showing consistency with Xu et al.‘s research in terms of increasing the total ester content. However, there were differences in specific components. Xu et al. utilized a mixed strain consisting of Bacillus subtilis, Staphylococcus epidermidis, and Millerozyma farinosa to produce medium- high temperature fortified Daqu19. They found that among the 113 compounds in the crude Baijiu of the experimental group, 50 were esters. This study revealed that fortification with Aspergillus oryzae CICC 2339 could enhance the ethyl acetate content, while Xu et al.‘s study observed a decrease in ethyl acetate content. Such differences in ester composition were mainly determined by the microbial and enzymatic compositions of Daqu as well as the pit microecology.
Comparison of volatile compounds between fortified Daqu (AOG) and its brewed crude Baijiu (AOG)
By comparing the volatile compound profiles of AOG Daqu in Fig. 5A and AOG-brewed crude Baijiu in Fig. 5B, a total of 124 volatile compounds were detected in both. Specifically, 39 compounds were found in AOG Daqu and 95 in AOG crude Baijiu. Among them, 29 compounds were unique to AOG Daqu, 85 were unique to AOG crude Baijiu, and 10 were common to both. According to the analysis, a total of 36 compounds exhibited significant changes in both Daqu and the resulting Baijiu in this study. However, only Tetramethyl pyrazine showed a significant increase in both Daqu and crude Baijiu(P < 0.05). Among the compounds unique to AOG Daqu, components such as vinyl guaiacol, 2,3-dimethyl-5-ethylpyrazine, and 2,3-dimethylpyrazine exhibited soy sauce-like aroma characteristics. The analysis focused on common and unique compounds. Among them, pyrazine compounds were of particular interest. These included 2,5-dimethylpyrazine, 2,6-dimethylpyrazine, tetramethylpyrazine, and trimethylpyrazine. These pyrazine compounds exhibited soy sauce-like aroma characteristics. Furthermore, significant differences in these compounds were observed between the two groups (p < 0.05). The results indicate that some volatile compounds with soy sauce-like aroma characteristics from the Aspergillus oryzae-fortified Daqu exhibit continuity in the brewed crude Baijiu. Notably, the number of volatile compound types in AOG Daqu is significantly lower than that in the brewed crude Baijiu, suggesting that the synergistic action of Daqu microorganisms and enzyme systems in the pit fermentation environment promotes the generation of more flavor compounds in the fermented grains. These compounds enter the crude Baijiu through the distillation process, thereby significantly enriching the flavor profile of the crude Baijiu.
The results of this study provide clear evidence. It was found that this reinforcement with Aspergillus oryzae has a more profound and extensive impact. Specifically, the impact is on the flavor profile of the crude Baijiu, rather than on Daqu itself. This amplification effect is in line with the logic of fermentation engineering. Aspergillus oryzae serves as a saccharifying and fermenting agent. Changes occur in the physicochemical properties of Daqu, including enzyme systems and flavor precursors. These changes are inherited during the subsequent solid-state fermentation process. Moreover, they are further amplified during the distillation process.
A significant increase in 10 ester compounds was observed in the crude Baijiu (but not in Daqu), which represents a core finding of this study. Esters are key components contributing to the main aroma of the crude Baijiu, especially fruity and cellar-like aromas. The reinforcement with A. oryzae likely promotes ester formation through two pathways:
The content of 3-Methylbutanol (isoamyl alcohol) increased in Daqu reinforced with A. oryzae, and its corresponding ester, Isoamyl acetate (isoamyl acetate, with a banana-like odor), showed a significant increase in Baijiu. This reveals a clear conversion chain: the reinforced Daqu provides a richer pool of alcohol precursors, which are further esterified during the fermentation process.
Aspergillus oryzae is renowned for its powerful hydrolytic enzyme system, particularly amylases, proteases, lipases, and esterifying enzymes. The high-activity esterifying enzymes and lipases introduced by A. oryzae into the fermentation system greatly catalyze the esterification reactions between alcohols and acids (such as the elevated Caproic acid) during fermentation, leading to the accumulation of a series of key esters, including ethyl acetate and ethyl caproate, in the Crude Baijiu.
Pyrazine compounds typically impart “baked” or “nutty” aromas to food and are important aroma components in sauce-flavored and strong-flavored Baijiu. Tetramethylpyrazine showed a significant increase in both the Daqu and the crude Baijiu(P < 0.05), indicating that the introduction of A. oryzae continuously promotes its formation, which may be related to the strain’s involvement in Maillard reactions or specific amino acid metabolic pathways. Interestingly, the decrease of 2-Ethenyl-6-methyl pyrazine in Daqu coexisted with the increase of other pyrazines (such as 2,3-Dimethyl pyrazine), suggesting that (A) oryzae not only increases the total amount of pyrazines but also reshapes their internal composition and proportions. In the biosynthesis of trimethylpyrazine, pyruvate is converted into acetoin through the action of EC:2.2.1.6 and EC:4.1.1.5. Previous studies have shown that during Daqu production and Baijiu fermentation, Bacillus licheniformis and Bacillus velezensis promote the accumulation of pyrazine compounds20. Considering that microbial functional characteristics often vary at the strain level, we conducted a comparative genomics analysis on the core genomes (detection rate > 80%) of (B) licheniformis and B. velezensis. The results revealed that both strains possess core genes encoding EC:1.1.1.103, EC:2.2.1.6, and EC:4.1.1.5.
Only 12 substances increased in Daqu, while 23 substances increased in the crude Baijiu, with most of the increased substances being esters that play a decisive role in the flavor of Baijiu. This indicates that the reinforcement effect of Aspergillus oryzae exhibits a “cascade amplification” characteristic: it first optimizes the enzyme system and flavor precursor pool (such as 3-Methylbutanol) during the Daqu-making stage, then catalyzes more efficient bioconversions (such as esterification reactions) during the brewing fermentation stage, ultimately achieving a significant improvement in the flavor quality of the finished Baijiu. Therefore, the conclusion that “the reinforcement treatment with Aspergillus oryzae has a greater impact on the crude Baijiu” is strongly supported by the data.
Conclusion
An interdisciplinary fortification strategy was developed by incorporating Aspergillus oryzae CICC 2339 into medium- high temperature Daqu, achieving significant enhancements. Microbial community & enzyme activity: AOG exhibited a stable microbial structure dominated by thermotolerant genera (Aspergillus, Bacillus et al.), with significantly elevated glucoamylase (P < 0.01), protease (P < 0.05), fermentation power (P < 0.001), and esterification capacity (P < 0.05), while acidity decreased (P < 0.05). A positive correlation between A. oryzae and key enzyme activities was confirmed. Flavor compound enrichment: AOG and its fermented crude Baijiu contained richer volatile profiles, with notably higher concentrations of pyrazines, esters, and other flavor-active compounds. The crude Baijiu displayed greater compound diversity than Daqu, highlighting fermentation-driven flavor enhancement. Mechanistic insights: A. oryzae modulated carbon-nitrogen metabolism and microbial niches, synergizing with dominant microbial consortia and enzyme systems to boost the accumulation of sauce-aroma, floral-aroma, and fruity-aroma compounds in Daqu and the crude Baijiu.
Overall, this study demonstrates the potential of using A. oryzae-fortified Daqu as an innovative approach to modulate the microbial-enzyme system and flavor profile of Te-flavor Baijiu, providing a scientific basis for process optimization.
However, this work primarily focused on microbial community and flavor chemistry outcomes. The sensory attributes of the final product and the comprehensive evaluation system for the crude Baijiu require further establishment and validation. Additionally, the applicability of A. oryzae CICC 2339 to other Daqu types remains to be investigated. Future research should delve into the underlying molecular mechanisms, such as the specific regulatory pathways through which A. oryzae influences microbial interactions and metabolite networks, to enable more precise and robust application of this bioaugmentation strategy.
Data availability
The datasets generated and/or analyzed during this study are available in the [SRA] repository, [with data persistent accession numbers: PRJNA1417074/PRJNA1417070].
References
Xu, M. L., Yu, Y., Ramaswamy, H. S. & Zhu, S. M. Characterization of Chinese liquor aroma components during aging process and liquor age discrimination using gas chromatography combined with multivariable statistics. Sci. Rep. 7 (1), 39671 (2017).
Deng, J. et al. Characterization of physicochemical properties, volatile compounds and microbial community structure in four types of Daqu. LWT 184, 115064 (2023).
Gong, L. et al. Traceability between microbial community and environmental microbial Community in Maotai-flavor Daqu. Food Chemistry: X. 27, 102321 (2025).
Yan, S., Tong, Q. & Guang, J. Yeast dynamics and changes in volatile compounds during the fermentation of the traditional Chinese strong-flavor Daqu. LWT 106, 57–63 (2019).
Lu, Y. et al. Effect of Environmental Microorganisms on Fermentation Microbial Community of Sauce-Flavor baijiu, Foods 12(1), 10 (2023).
Shi, X. et al. Tetramethylpyrazine in Chinese baijiu: Presence, analysis, formation, and regulation. Front. Nutr. 9, 1004435 (2022).
Cheng, W. et al. Effects of the improved application of Bacillus halotolerans on the microbial community and volatile components of high-temperature daqu. Front. Microbiol. 16, 1626160 (2025).
Zhang, J. et al. Fortified Jiuqu of the Chinese Baijiu: A review on its functional microorganisms, strengthening effects, current challenges, and perspectives. Food Bioscience. 55, 103045 (2023).
Nurmilah, S., Frediansyah, A., Cahyana, Y. & Utama, G. L. Biotransformation and health potential of isoflavones by microorganisms in Indonesian traditional fermented soy products: A review. J. Agric. Food Res. 18, 101365 (2024).
Auchtung, J. M., Hallen-Adams, H. E. & Hutkins, R. Microbial interactions and ecology in fermented food ecosystems. Nat. Rev. Microbiol. 23 (10), 622–634 (2025).
Yang, Y. et al. Metagenomics unveils microbial roles involved in metabolic network of flavor development in medium-temperature daqu starter. Food Res. Int. 140, 110037 (2021).
Zhu, Y. et al. A comparative secretome analysis of industrial Aspergillus oryzae and its spontaneous mutant ZJGS-LZ-21. Int. J. Food Microbiol. 248, 1–9 (2017).
Zhou, Q. et al. Exploring the diversity of the fungal community in Chinese traditional Baijiu daqu starters made at low-, medium- and high-temperatures. LWT 162, 113408 (2022).
Mu, Y. et al. The key proteolytic enzyme analysis of industrial Aspergillus oryzae at solid-state koji fermentation with a local database extension. Food Bioscience. 58, 103738 (2024).
Yang, L., Fan, W. & Xu, Y. Effects of storage period and season on the microecological characteristics of Jiangxiangxing high-temperature Daqu. Food Res. Int. 196, 115034 (2024).
Xu, B. Y. et al. Analysis of the microbial community and the metabolic profile in medium-temperature Daqu after inoculation with Bacillus licheniformis and Bacillus velezensis. LWT-Food Sci. Technol. 160, 113214 (2022).
Falkenberg, F., Voß, L., Bott, M., Bongaerts, J. & Siegert, P. New robust subtilisins from halotolerant and halophilic Bacillaceae. Appl. Microbiol. Biotechnol. 107 (12), 3939–3954 (2023).
Namshir, B. et al. Fermentation and Functional Properties of Plant-Derived Limosilactobacillus fermentum for Dairy Applications. Fermentation 11 (5), 286 (2025).
Xu, P. et al. Application of fortified Daqu to optimize the core fermentation microbial flora and improve the ester profile of Nongxiangxingbaijiu, LWT 117711 (2025).
Shi, W. et al. Spatial heterogeneity of the microbiome and metabolome profiles of high-temperature Daqu in the same workshop. Food Res. Int. 156, 111298 (2022).
Funding
This research was supported by the Special Fund Project for the Construction of an Innovation-Driven Province in Hunan Province (Grant No. 2025RC3283), the Central Government-Guided Local Development Funding Program (Grant No. 2024ZYC029), the Science and Technology Innovation Team Support Program for General Higher Education Institutions in Hunan Province (Xiang Jiao Tong [2023] No. 233), and the Shaoyang University-Enterprise Cooperation Project (Grant No. 2024HX23, 2026HX01, 2025hx291 and 2025hx292).
Author information
Authors and Affiliations
Contributions
You-Gui Yu: Data curation, Formal analysis, Investigation, Methodology, Writing-original draft. Tao Yang: Formal analysis, Visualization. Dong-Cai Zhu: Investigation, Methodology. Jie Li: Conceptualization, Supervision. Li-Pin Wu: Investigation, Methodology. Xin-She Li: Conceptualization, Investigation, Supervision. Xiuqiong Huang: Formal analysis, Methodology. Zheng-Pei Chen: Conceptualization, Funding acquisition, Supervision, Writing-review & editing. Xiangyang Tang: Conceptualization, Supervision, Writing-review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Y., Yang, T., Zhu, D. et al. Fortification of Daqu with the soy sauce-functional fungus Aspergillus oryzae CICC 2339: investigations into Daqu properties and Baijiu brewing outcomes. Sci Rep 16, 14906 (2026). https://doi.org/10.1038/s41598-026-44158-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-44158-4
