
Abstract
A cost-effective surface plasmon resonance (SPR)-based sensing platform was developed to evaluate alterations in albumin binding capacity under clinically relevant conditions. This ex vivo approach enables real-time assessment of albumin-ligand interactions using albumin directly isolated from plasma, thus overcoming key limitations of conventional in vitro approaches. The sensing surface was prepared by covalently immobilizing a polyclonal anti-albumin antibody onto a CM5 chip, followed by a single-step immunocapture of albumin from patient plasma samples. Mass spectrometry confirmed the selective retrieval of both native and structurally modified albumin forms, preserving their relative abundance and disease-associated microheterogeneity. The sensing surface demonstrated high reusability and analytical reproducibility over ~ 500 capture-release cycles, significantly lowering per-sample costs. Functional validation was performed using ligands targeting the three main albumin binding sites. As proof of application, the system was used to investigate albumin binding properties in plasma from (i) type 2 diabetic patients with (n = 10) and without (n = 10) moderate kidney impairment, and (ii) patients with cirrhosis and acute-on-chronic liver failure (n = 6), a condition associated with extensive albumin damage. The proposed approach provides a robust analytical framework for the functional characterization of circulating albumin in healthy and diseased conditions.
Similar content being viewed by others


Introduction
Human serum albumin (HA) is the most abundant plasma protein and plays a central role in maintaining oncotic pressure, exerting antioxidant effects, and transporting a wide variety of endogenous and exogenous ligands, including fatty acids, hormones, metal ions, and pharmaceutical compounds1,2,3. Its ligand-binding versatility arises from a flexible, multidomain structure that features several binding pockets, most notably Sudlow sites I and II, and site III4,5,6. HA-ligand interactions contribute to drug solubility and distribution, influencing pharmacokinetics and therapeutic efficacy7. Because these functional properties depend on HA’s structural integrity, structural alterations can impact its folding, binding affinity, and clearance8.
HA exists in plasma as a heterogeneous mixture of native and modified forms. This microheterogeneity is dynamic and reflects physiological and pathological conditions. Structural modifications, including oxidation, glycation, cysteinylation, and terminal truncations, are known to increase in settings of systemic inflammation and oxidative stress, such as liver or kidney disease and diabetes9,10,11.
Structural changes have been reported to affect the binding capacity of HA toward clinically relevant ligands and drugs, including warfarin12,13,14, nonsteroidal anti-inflammatory drugs15, sulfonylureas16, and others17, which may compromise drug efficacy and safety.
Most studies of HA binding have relied on in vitro models using chemically modified, commercially available HA. These models typically assess the impact of single alterations, often without detailed structural characterization of the modified forms18. As a result, they fail to replicate the complex mixture of HA forms present in clinical samples, where multiple modifications coexist and evolve in response to disease severity and patient condition. Additionally, in vitro assays generally use defatted HA, whereas HA circulates bound to fatty acids, which are known to influence ligand binding19,20. These limitations have led to divergent findings across studies21. While some ex vivo approaches have been proposed, including direct plasma studies or HA isolation before binding assays, these are either confounded by the presence of other binding proteins or involve time-consuming purification steps. There remains a need for analytical strategies capable of assessing HA binding in clinically relevant samples, preserving structural microheterogeneity, and enabling efficient, reproducible measurements21,22,23,24.
In this context, the present study aimed to develop a cost-effective and reusable sensing surface for the real-time assessment of HA binding properties under clinically relevant conditions. A sensor chip was functionalized with a covalently immobilized anti-HA antibody, enabling selective, single-step immunocapture of HA from patient plasma without any extensive pretreatment. To ensure broad applicability, the setup was designed to support high-throughput use, minimal sample preparation, and low per-analysis costs.
Surface plasmon resonance (SPR) was employed for binding measurements due to its sensitivity and real-time monitoring capabilities. Although SPR has been extensively applied to the study of biomolecular interactions, its use with complex biofluids remains limited25. Prior SPR studies involving HA have largely relied on commercial preparations or covalent immobilization methods that do not capture circulating microheterogeneity26,27,28,29,30,31. Here, as proof-of-concept of application, we applied the newly developed sensing surface to assess HA binding in two patient cohorts characterized by distinct levels of HA structural damage: (i) patients with type 2 diabetes mellitus (T2DM), with or without moderate kidney impairment (n = 10 each), and (ii) patients with cirrhosis and acute-on-chronic liver failure (n = 6), a condition associated with extensive HA modification11,32,33,34.
Results and discussion
Design of the sensing surface
The strategy for designing an analytical device capable of capturing albumin in a single step from patient plasma was defined, considering its final application: performing in vitro binding studies on a sensing surface that reflects patient-specific HA heterogeneity.
Hence, SPR technology was selected because it is one of the most suitable methods for studying biorecognition phenomena in real-time, with a mid-throughput wizard-supported workflow that is well-suited to the aims of this work. The ability to detect both weak and strong interactions, along with easy access to affinity and kinetic data on complex formation/disruption, makes this technique highly informative.
To generate the SPR sensing surface, we opted for immunocapture-based immobilization of HA, as this approach enables the selective immobilization of the plasma target protein without pre-purification while minimizing the risk of contamination from other plasma proteins. Indeed, direct immobilization of circulating HA through covalent coupling would not provide the necessary selectivity, as competing, abundant plasma proteins would also be bound to the sensor chip surface, leading to a multi-protein sensing surface.
Additionally, the immunocapture approach also ensures the device is cost-effective. Indeed, immunocapture, as a reversible immobilization process, allows for the removal of the target molecule from the sensing surface through a suitable surface regeneration procedure without damaging the capturing antibody. This allows for the repeated use of the same “activated” sensing surface over multiple analysis cycles.
To this end, we first prepared an anti-HA sensor chip by covalently immobilizing a polyclonal anti-HA antibody using a well-established covalent immobilization procedure35.
Then, the anti-HA sensor chip was used to capture HA from plasma samples without any pre-purification step. The amount of plasma needed to assess HA affinity toward an analyte is limited to a few dozen microliters. After the analysis cycle is completed, the capture surface is regenerated. Surface regeneration prepares the sensing surface for reuse in another capture cycle, enabling multiple analysis cycles. The general design and workflow are summarized in Fig. 1.

Sensing surface design. Schematic representation of the experimental design, along with the illustration of the instrumental setup for SPR binding measurement with the Biacore X100 system.
Validation of polyclonal anti-HA antibody for quantitative capture of HA microheterogeneity
To effectively sample the various forms of HA under both physiological and pathological conditions, it is essential that the immunocapture method allows for the comprehensive collection of all circulating HA forms while preserving their relative abundances. To evaluate this, we assessed the capturing performance of the selected commercial polyclonal anti-HA antibody against circulating HA using liquid chromatography–mass spectrometry (LC–MS). This technique is crucial for distinguishing among the structurally altered forms of albumin, which differ in mass.
Since SPR cannot be directly interfaced with mass spectrometry, the polyclonal anti-HA antibody was covalently bound to a short monolithic column (CIMac-αHSA, 5.0 mm × 5.2 mm I.D.), and the antibody’s capturing performance was evaluated using plasma samples characterized by varying albumin integrity and microheterogeneity profile (namely samples from patients with T2DM, with and without renal impairment). The relative abundances of the immunocaptured HA forms were profiled by LC-MS/MS and compared to those in untreated samples.
As depicted in Fig. 2, no significant differences (P > 0.5) were observed between the relative levels of HA forms before and after extraction for both sets of samples, although their HA microheterogeneity profiles were markedly different. In addition, mass spectrometric analysis confirmed that no other plasma proteins were co-captured under the applied conditions.
These results demonstrate that the selected anti-HA antibody effectively captures native and modified HA forms, preserving their relative abundance and, hence, reproducing disease-associated HA microheterogeneity profiles.

Ability of anti-HA antibody to maintain HA microheterogeneity. Relative amount of HA forms before (white bars) and after (grey bars) extraction from (a) T2DM patients without renal impairment (n = 5) and (b) T2DM patients with renal impairment (n = 5) by using an affinity column with the selected anti-HA antibody. The different forms of HA were identified, and relative abundances were assessed using a previously developed LC–MS method36. HA-DA: truncation at the N-terminal portion; HA-L: truncation at the C-terminal portion; HA+Cys-DA: N-terminal truncated form cysteinylated at Cys34; HA: native albumin; HA-SO2H: sulfonylated form at Cys34; HA+Cys: cysteinylation at the level of Cys34; HA+Glyc: monoglycation; HA+Cys+Glyc: cysteinylated form carrying one glycation; HA+2Glyc: glyc; HA+Cys+2Glyc: cysteinylated form carrying two glycations.
Development and validation of the sensing surface
As mentioned above, the sensing surface was developed in two steps: first, the anti-HA antibody was covalently bound to the surface to provide a stable immunocapturing sensor chip; second, this anti-HA sensor chip was used to reversibly immunocapture HA. In the first step, an anti-HA sensor chip was obtained by covalently immobilizing the selected anti-HA antibody onto the carboxymethyl dextran layer of a CM5 sensor chip using an established amine coupling reaction37. Optimal preconcentration conditions were achieved at pH 5.0. The final immobilization level was 18,000 RU.
The immunocapture procedure for obtaining the anti-HA sensing surface was initially optimized using the commercial HA. To maximize capture level, the concentration of the HA solution, flow rate, and contact time were optimized (Supplementary Fig. S1). The maximum capture level was achieved when a 50 µM HA solution was infused over the anti-HA sensor chip at 5 µL/min for 840 s. Based on these data, for ex vivo studies, the total HA concentration was normalized to 50 µM prior to immunocapture to achieve similar immobilization levels across all samples (Fig. 3).

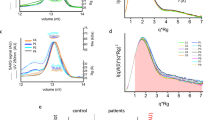
Immunocapture profiles of HA from different sources. Overlaid representative sensorgrams depicting the immunocapture profiles of commercial HA (black), HA from a plasma control (orange), and from a patient with decompensated cirrhosis and acute-on-chronic liver failure (ACLF, red) under optimized experimental conditions. All samples were diluted to a final concentration of 50 µM and infused at 5.0 µL/min for 840 s. Under the given conditions, the same final capture level was achieved with both the plasma samples and commercial HA solutions.
Surface regeneration was achieved by injecting glycine buffer at pH 2.0, followed by 0.1 N NaOH. This step enabled the removal of HA from the chip surface without damaging the antibody, thereby restoring the chip surface for subsequent capture and analysis cycles. After regeneration, blank injections were performed to monitor the baseline stability and account for systematic and random variations. Regeneration conditions did not significantly affect antibody binding capacity, as evidenced by the lack of a significant difference in the capture efficacy across two subsequent capture cycles (Supplementary Fig. S2).
The HA sensing surface was validated prior to its application to plasma samples from the study participants. False-positive interactions were ruled out by analyzing galantamine, a non-binding drug, confirming no interaction with HA27. On the other hand, to assess whether HA maintains its unaltered binding capacity at its three high-affinity binding sites, three reference binders were analyzed, namely, (i) phenylbutazone (PBZ), a marker for Sudlow site I37; (ii) dansyl-L-phenylalanine (DPA), a marker for Sudlow site II38; and (iii) biliverdin (BVD), a marker for site III39. For all markers, a good concentration-response relationship was observed (Supplementary Fig. S3). Notably, for BVD, significant interactions with the anti-HA antibody were observed, as confirmed by injecting BVD on the bare anti-HA sensor chip without any immobilized HA. Hence, to properly account for those nonaspecific interactions, investigations involving BVD were performed using a sensor chip with the antibody immobilized in both sample and reference flow cells. The KD values obtained for PBZ, DPA, and BVD were in good agreement with those reported in the literature (PBZ: 14.0 ± 0.5 µM vs. 0.12–3.3 µM40,41; DPA: 1.7 ± 0.3 µM vs. 6.0 µM5; BVD: 13.0 ± 3.0 µM vs. 10−6–10−8 M42).
Application of the HA-sensing surface for the evaluation of HA-binding properties in cirrhotic patients and diabetic patients with renal impairment
Ex vivo assessment of HA binding functions in patients with decompensated cirrhosis
The validated sensing surface was used in a pilot study examining HA binding in patients with decompensated cirrhosis and ACLF, a syndrome characterized by elevated systemic inflammation and oxidative stress, as well as reduced levels of circulating HA. Indeed, several reports have highlighted that severe HA damage occurs in advanced stages of liver cirrhosis and is mainly associated with high levels of oxidation11,32,43. Due to the accumulation of these altered molecular forms, the amount of native, fully functional HA significantly decreases44. Furthermore, clinical evidence of altered HA binding properties in ACLF patients was observed together with alterations in its conformation20,22.
Based on these observations and as proof of concept of the application of the proposed approach to real samples, plasma samples from 6 hospitalized patients with ACLF (2 males and 4 females, aged 68–72) and 6 age-matched healthy volunteers (CTRL, 5 males and 1 female, aged 38–73) were selected.
HA microheterogeneity in the samples under study was initially profiled by LC-MS analysis (Supplementary Table S1)36,45. Analysis showed that, compared with healthy individuals, ACLF patients presented a drastically lower relative amount of the native form of HA (nHA) (8.5% vs. 43.1%) and a concomitant significant increase in the oxidized forms of HNA1 (from 37.9 to 87.9%) and glycated forms (from 11.6 to 23.0%). For each subject, the HA binding capacity at the three high-affinity binding sites was evaluated by deriving KD values for PBZ, DPA, and BVD (Supplementary Table S2). Furthermore, the affinity of teicoplanin (TEICO), a non-site-specific HA binder46 commonly administered to cirrhotic patients to counteract infections, was also evaluated (Supplementary Fig. S4 and Supplementary Table S2).
To the sake of completeness, it must be noted that albumin captured directly from plasma may retain endogenous ligands, such as fatty acids. Hence, the KD values obtained should be interpreted as apparent KD values, which aligns with common practice when albumin is studied under physiologically relevant conditions.
HA from ACLF patients showed a slightly greater affinity for PBZ at Sudlow site I than did HA from CTRL subjects (P = 0.0280) (Fig. 4a). On the other hand, no significant differences in affinities for DPA or BVD were detected at sites II and III, respectively (Fig. 4b and c). Indeed, despite the extensive structural changes HA undergoes in ACLF patients, binding properties at high-affinity sites are quite preserved, although caution is warranted regarding drug binding at the Sudlow site I.
Importantly, these findings should be considered preliminary because the study was undertaken as a proof-of-application for the developed device, and thus involved a limited number of patients.
Interestingly, a significant alteration in TEICO binding capacity was observed in ACLF patients, in which structural changes in HA reduced its binding affinity (Fig. 4d). To our knowledge, this is the first study to investigate the binding of TEICO in ACLF patients. Since teicoplanin is one of the antibiotics of choice for treating infections in cirrhotic patients, these results are potentially relevant and require further evaluation, especially considering that ACLF patients also exhibit reduced plasma levels of HA (hypoalbuminemia)44,47,48.

Impact of structural damages associated with ACLF on albumin binding capacity. Individual values plots showing average KD values with standard error of the mean (SEM) for the three site-specific binding markers and teicoplanin (TEICO) for controls (CTRL, black) and for patients with decompensated cirrhosis with ACLF (red). Column graphs for (a) PBZ as a marker for Sudlow site I, (b) DPA as a marker for Sudlow site II, (c) BVD as a marker for site III, and (d) TEICO as a non-site-specific HA binder.
Ex vivo assessment of HA binding functions in patients with T2DM and renal impairment
A second pilot study involved a small cohort of type-2 diabetic patients with diabetic kidney disease (T2DM + DKD). Indeed, exacerbated inflammation and oxidative stress are also typical pathological conditions in patients suffering from DKD and are major causes of morbidity and mortality in patients with diabetes mellitus. In agreement with this observation, more oxidized forms have been observed in albumin from T2DM patients13,14. Additionally, patients may also experience hypoalbuminemia due to alterations in glomerular filtration, a condition that may exacerbate HA dysfunction49.
As a pilot screening, 20 diabetic patients were selected: 10 subjects without renal damage (T2DM-DKD) (4 females and 6 males; aged 46–87 years), and 10 subjects classified according to KDIGO classification parameters50 as subjects with very high risk renal impairment (T2DM + DKD) (1 female and 9 males; aged 28–71 years)50. Previous investigations have shown that HA from T2DM + DKD patients also undergoes structural damage, although to a more limited extent when the glycemic level is under drug control51. In agreement with these findings, MS analysis revealed a significant, although not extensive, decrease in the relative abundance of native HA, from 58.5 to 51.7%, in T2DM + DKD patients with a concomitant increase in (mainly) oxidized forms (details in Supplementary Table S3). Notably, after treatment with glucose-lowering drugs, the levels of the glycated forms of HA were not significantly greater than those in the controls (P = 0.089; Supplementary Table S3).
Since in ACLF patients the affinity of TEICO for HA resulted impaired, this drug was chosen as a pilot marker to assess whether the albumin binding capacity in T2DM + DKD patients was also affected (single KD values in Supplementary Table S4). Results show that the binding capacity of TEICO did not significantly affect this cohort of patients (Fig. 5). This finding suggests that only substantial alterations to the HA structure can lead to significant changes in the protein’s binding properties.

Impact of structural damages associated with T2DM + DKD on albumin binding capacity towards TEICO. Individual values plot showing average KD values with SEM for teicoplanin binding to HA in control T2DM-DKD patients (black) and T2DM + DKD patients (orange).
To conclude, these preliminary investigations demonstrate that the developed and optimized chip-based tool enables patient-specific binding studies.
Sensing surface stability
The stability of the sensing surface was monitored by measuring HA capture levels at each analysis cycle throughout the sensor chip lifetime (Fig. 6). Adding a protease inhibitor cocktail during plasma dilution was essential to prevent HA and/or antibody degradation over repeated loading-regeneration cycles, thereby extending chip longevity. Under optimized conditions, a single sensor chip could be reused for more than 500 analyses.
Because HA capture gradually decreases with repeated use, we assessed whether this might affect affinity measurements. KD determination in SPR is based on equilibrium (isothermal) binding curves and, according to classical binding theory and SPR methodology, the KD obtained is independent of the absolute amount of immobilized analyte, provided that equilibrium is reached, the system is not mass-transport-limited, and the injected ligand concentrations are in excess over the captured target52,53,54. Under these conditions, a reduction in capture capacity lowers the response amplitude and signal-to-noise ratio but should not bias KD.
To ensure that these requirements were met throughout the chip’s lifetime, each analytical session began with a quality-control run in which the KD of a reference ligand (PBZ) toward commercial HA, captured under identical conditions, was measured. Only when the reference KD matched the expected value within normal analytical variability were plasma-derived samples analyzed; when this criterion was no longer met (i.e., a too-high standard deviation after an ~ 80% loss of capture capacity), the chip was discarded. To put it in numbers, PBZ binding to commercial HA yielded comparable KD values at early, mid-, and late-life phases (15.6 ± 0.1 µM, 15.1 ± 0.2 µM, and 16.1 ± 0.2 µM, respectively), confirming that reduced capture capacity did not bias affinity determination but mainly affected signal-to-noise.

Stability study. Percentage variation in the amount of commercial HA captured by the anti-HA antibody across sequential HA-capture cycles, considering both commercial and plasma sample runs.
Materials and methods
Chemicals
Anti-human albumin antibody produced in rabbits (whole antiplasma, product code A3293; antigen 66.437–66.600 kDa), standard HA (essentially fatty acid-free, ≥ 96%, product code A1887; MW: 66.4 kDa), sodium dihydrogen phosphate (NaH2PO4), disodium hydrogen phosphate (Na2HPO4), dimethyl sulfoxide (DMSO), phenylbutazone (PBZ; MW: 308.4 Da), biliverdin hydrochloride (BVD; MW: 619.12 Da), teicoplanin (TEICO; MW: 1880 Da), sodium chloride (NaCl), sodium acetate and protease inhibitor cocktail were all purchased from Sigma–Aldrich Millipore (Milan, Italy). Dansyl-L-phenylalanine (DPA; MW: 398.5 Da) was purchased from Tokyo Chemical Industry (Tokyo, Japan). Galantamine hydrobromide (GAL; MW: 368.3 Da) was obtained from Tocris (Cookson, UK). Research-grade CM5 sensor chips and an amine coupling kit consisting of N-ethyl-N-(3-dimethyl-aminopropyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS) and ethanolamine hydrochloride (pH 8.5; 1 M) were all purchased from Cytiva. Bromocresol green (BCG; MW: 698.1 Da) was purchased from Fluka Honeywell (Milan, Italy), and succinic acid was purchased from Carlo Erba (Milan, Italy). Thermo Scientific Nunc Microwell 96-well plates were purchased from Fisher Scientific Italia (Rodano, Milan, Italy). HPLC-grade (≥ 99.9%) acetonitrile (ACN) was obtained from Honeywell (Milan, Italy). Deionized water was obtained with a Milli-Q system (Millipore, Milford, MA, USA), and all aqueous solutions were filtered through 0.22 μm membrane filters before use.
Preparation and validation of an HA-sensing surface
SPR analyses were performed on a Biacore X100 system (Cytiva, Uppsala, Sweden) equipped with an inline degasser, thermostated at 25 °C. The data were analyzed and processed using Biacore X100 evaluation software v. 4.1. Software information is available at:
Preparation of the anti-HA sensing surface for reversible capture of HA
Phosphate-buffered saline (PBS) (20 mM, pH 7.4 + 0.05% (v/v)) with Tween-20 (running buffer A) was used for the immobilization procedure, while running buffer A + DMSO (98:2, v/v) (pH 7.4) (running buffer B) was used for binding studies. All buffer solutions were freshly prepared every day and filtered through a 0.22 μm cellulose nitrate membrane before use.
To determine the optimal pH for immobilization, a pH-scouting investigation was performed by sequentially injecting 50 µg/mL anti-HA antibody solution in sodium acetate buffer (10 mM) at various pH values (pH 4.00, 4.35, 4.50, 4.76, 5.00, 5.22, and 5.50) for 120 s at 10 µL/min using running buffer A. After each injection, the baseline was re-established by injecting a NaOH solution (50 mM). The best preconcentration was obtained using a pH 5.0 solution. Therefore, these conditions were used to immobilize antibodies covalently on the test flow cell (FC2) of a carboxymethyl-dextran (CM) 5 sensor chip via amine coupling, according to the standard Biacore procedure. Briefly, the chip was equilibrated at room temperature for 30 min before docking, and the system was primed with running buffer A three times. The sensor-chip test surface was activated by flushing a freshly prepared mixture of 0.4 M EDC and 0.1 M NHS (final concentrations) for 420 s at 10 µL/min. Then, 50 µg/mL anti-HA antibody solution was injected over the activated flow cell(s) at a flow rate of 10 µL/min for 120 s to achieve the desired immobilization level, i.e., 18,000 RU, corresponding to a surface density of approximately 18 ng/mm2. The remaining active esters were quenched by injecting a 1 M solution of ethanolamine hydrochloride (pH 8.5) for 420 s at the same flow rate. Finally, the system was left to equilibrate for at least 12 h to achieve a steady baseline.
Two chips were prepared according to the described procedure: a sensor chip 1 (SC-1), in which the anti-HA antibody was immobilized only on the test flow cell, and a sensor chip 2 (SC-2), in which it was immobilized in both the test and reference flow cells. The sensor chips functionalized with the anti-HA antibody were used to immunocapture HA.
Preparation of a reversibly functionalized HA-sensing surface by immunocapture
Optimal conditions for HA immunocapture were achieved by injecting a 50 µM HA solution in 10 mM sodium acetate buffer (pH 7.4) at 5 µL/min for 840 s.
Validation of the sensing surface
To confirm the correctness of the capture procedure and validate the binding capacity of the chip surface, the affinities of three well-known HA markers, PBZ, DPA, and BVD, were evaluated as site I, site II, and site III HA binders, respectively. The steady-state dissociation constant (KD) was measured for each sample by multiple-cycle analysis. Stock solutions of 10 mM PBZ, DPA, and BVD in DMSO were further diluted with running buffer B to obtain the desired final concentrations: PBZ (0.620–50.0 µM), DPA (0.940–30.0 µM), and BVD (3.13–50.0 µM). The analytes were injected into both flow cells at a flow rate of 75 µL/min, with a contact time of 40 s, followed by a 40 s dissociation time.
Due to the high bulk response of DMSO relative to the intrinsically low response of small molecules, a solvent correction procedure was employed to account for the observed responses to DMSO and improve the robustness.
Furthermore, based on binding data available in the literature27, galantamine was chosen as a negative control to assess the absence of possible artifacts. To determine the binding of galantamine, starting from a 3 mM stock solution in running buffer A, test solutions of increasing concentrations of galantamine, namely, 0.370, 1.11, 3.33, 10.0, and 30.0 µM, were prepared in running buffer B and injected at a flow rate of 10 µL/min for 60 s.
Data analysis
Responses from the test flow cell were double-referenced against those from the reference flow cell and against the average of all the blank injections taken at the beginning of each multicycle of analysis. Finally, the solvent refractive index correction was applied55. The equilibrium dissociation constant (KD) of the ligand-analyte complex was determined by fitting the responses at steady state to a 1:1 isotherm binding model, defined in Eq. (1):
where KD is expressed in M, C is the analyte concentration (in M), Req is the SPR response of the binding complex at equilibrium (in RU), Rmax is the maximum response upon saturation of the analyte (in RU), and the offset is the response at zero analyte concentration (in RU).
Each dataset was fitted separately in the binding model, using at least two independent measurements. The resulting parameters (n = 3) were averaged and reported with their corresponding standard deviation.
Patients
Two study populations were selected for the pilot studies conducted in this work. The first involved six cirrhotic patients admitted to the IRCCS Azienda Ospedaliero-Universitaria di Bologna in Bologna (Italy) due to acute-on-chronic liver failure (ACLF) between January 2014 and March 2016. The selection of those patients was driven by the evaluation of the relative amount of the native form of the protein (nHA): selected patients had the lowest amount of nHA. Six age-matched healthy volunteers were considered the reference population.
The second study involved 20 patients attending the outpatient clinic of the Metabolic Diseases & Clinical Dietetics Unit and the Nephrology, Dialysis and Transplantation Unit of the IRCCS Azienda Ospedaliero-Universitaria di Bologna (Italy). The inclusion criteria were a diagnosis of T2DM for at least one year without renal impairment (n = 10) or with renal impairment at the “very high risk” stage (n = 10) according to the guidelines for the evaluation and management of chronic kidney disease50. Vital parameters, weight, height, BMI, and systolic and diastolic blood pressure were assessed in all patients. A medical history was also collected to document current drug therapy.
Blood samples were collected from all subjects after fasting in EDTA tubes (Becton Dickinson Italia, Milan, Italy) and were centrifuged at 3000 × g for 10 min; the plasma was aliquoted into cryotubes (Corning, Inc., Corning BV, Amsterdam, The Netherlands) and stored at − 80 °C until analysis. The study protocol was approved by the local institutional review board, and written informed consent was obtained from patients or legal surrogates before enrollment, in accordance with the Declaration of Helsinki and later amendments.
Ethics approval
The study protocols were approved by the Ethics Committee of the Sant’Orsola Malpighi University Hospital (protocol codes 88_2017U\Sper, 2017, and 75/2012/U/OSS) and conducted in accordance with the declaration of Helsinki and later amendments. Informed consent was obtained from all participants before their enrollment in the study.
Evaluation of albumin microheterogeneity before and after immunoextraction from plasma samples
Plasma from ten T2DM patients with (n = 5) or without (n = 5) renal impairment was divided into two aliquots and stored at − 80 °C before use. One aliquot was diluted 1:100 with 10 mM phosphate buffer (pH 7.4), filtered through 0.22 μm syringe filters, and directly analyzed via LC‒MS analysis following the methods described in the following section. The second aliquot was diluted with phosphate-buffered saline (PBS), filtered through 0.22 μm syringe filters, and subjected to albumin immunoextraction using a monolithic affinity column that contained the selected anti-HA polyclonal antibody immobilized on the monolithic stationary phase (Sartorius BIA Separations, Slovenia, beta version affinity column). Extraction was performed according to the vendor protocol. The collected HA eluate was concentrated, and the elution buffer was replaced with 10 mM phosphate buffer (pH 7.4) by ultrafiltration (Amicon Ultra tubes; 0.5 mL; cutoff: 10 kDa). The HA concentration in the collected samples was assessed via spectrophotometric analysis. Before LC-ESI-MS analysis, the samples were diluted to a final protein concentration of 100 µg/mL.
Liquid chromatography‒mass spectrometry (LC–MS) analyses
For the relative quantitation of HA structural alterations, the LC‒MS method previously reported by Naldi et al. was slightly adapted45. The plasma samples were diluted 100 times with ultrapure water and filtered through a 0.22 μm syringe filter (Merck KGaA, Darmstadt, Germany). HPLC analyses were conducted on an Agilent 1200 HPLC System (Walbronn, Germany) using a Phenomenex Jupiter C4 column (5 μm, 300 Å, 150 mm × 2.0 mm i.d.) to achieve chromatographic separation from other plasma proteins. A gradient composed of mobile phases A [water/acetonitrile/formic acid (99/1/0.1, v/v/v)] and B [acetonitrile/water/formic acid (98/2/0.1, v/v/v)] was applied: 20–70% B for 5 min, followed by 70% B for 1 min. Between injections, the column was equilibrated for 5 min. The flow rate was 0.4 mL/min, and the injection volume was 3 µL.
A quadrupole-time of flight hybrid mass analyzer (Q-ToF Micro, Micromass, Manchester, UK) with a Z-spray electrospray ionization (ESI) source was used for mass spectrometry analysis. The capillary and cone voltages were set at 3.0 kV and 40 V, respectively. The ESI-Q-ToF source temperature was 150 °C, and the desolvation temperature was 300 °C. The scan and interscan times were set at 2.4 s and 0.1 s, respectively. The desolvation gas flow rate was 1000 L/h, and the cone gas flow rate was 120 L/h. Total ion current (TIC) chromatograms were acquired in positive polarity within the 1000–1800 m/z range. Using MassLynx software v. 4.2 (Waters Corporation, Milford, MA, USA) with the maximum entropy (MaxEnt1)-based tool, the HA baseline-subtracted spectrum (m/z 1084–1534) was deconvoluted into a genuine mass scale, with parameters set at a mass range of 61,500–71,500 Da and a resolution of 2 Da/channel. The relative abundance of HA forms was determined by dividing the intensity of each form (from the deconvoluted spectrum) by the sum of the intensities of all the forms multiplied by 100. Microsoft Excel software (Microsoft Corporation, 2016) was used for the data analysis. MassLynx software information is available at:
https://help.waters.com/help/en/support/library-details.html?documentid=715010225.
Bromocresol green (BCG) colorimetric method
The HA concentration in the plasma samples under investigation was determined using a BCG colorimetric assay, adapting the well-established method currently used in the clinic on a smaller scale56. The BCG reagent contained 0.2 mM BCG, 0.1 mM succinate buffer (pH 4.2), and 0.8% v/v Tween 20. The plasma samples were diluted 5-fold in ultrapure water. A 5 µL aliquot of diluted plasma sample was added to 200 µL of BCG reagent and gently mixed. The samples were incubated at room temperature for 5 min. Blank solutions were prepared in parallel and contained all the components except for plasma. Two hundred microliters of each sample and blank solution were transferred to a well of a clear 96-well flat-bottom microplate, and the absorbance in the range of 570–670 nm (at 620 nm) was measured using a Spark multimode microplate reader (Tecan, Austria). HA quantitation was performed by interpolating the absorbance at 620 nm in a calibration curve, which was built using HA standard solutions at increasing concentrations (5.0, 7.5, 10, 15, and 20 mg/mL). A standard curve was generated with each set of assays. All assays were performed in triplicate.
Ex vivo SPR-based assessment of HA binding functions in cirrhotic patients and T2DM + DKD patients
Prior to SPR analysis, the samples were diluted to match the analysis conditions, HA degradation by proteases was inhibited, and the HA concentration was normalized. In detail, based on the HA concentration (as determined by the BCG method), each plasma sample was diluted with PBS (pH 7.4) containing 2% (v/v) protease inhibitor mixture to obtain a final HA concentration of 100 µM. The samples were then filtered through a 0.22 μm syringe filter and further diluted 1 to 2 in 1× PBS (pH 7.4) + 0.1% (v/v) Tween20 + 4% (v/v) DMSO to obtain a final concentration of 50 µM HA and to match running buffer B.
HA immunocapture was performed by injecting a diluted plasma solution (HA concentration = 50 µM) over the functionalized sensor chip (SC1 or SC2) at 5 µL/min for 840 s. The surface was allowed to stabilize for 420 s. Association and dissociation profiles of the selected site-specific binders, namely, PBZ, DPA, and BVD, were monitored for 40 s at a flow rate of 75 µL/min. For each subject-specific HA-sensing surface, the steady-state dissociation constants for the three site-specific markers, PBZ, DPA, and BVD, were determined using the same concentration range used for sensor chip validation. Furthermore, the affinity of teicoplanin (TEICO), a non-site-specific HA binder, was also assessed for each patient. To this end, solutions of TEICO with final concentrations ranging from 0.880 to 550 µM in running buffer B were screened. At the end of each multicycle analysis, two subsequent regeneration steps were performed by injecting 100 mM Gly-HCl (pH 2) and 50 mM NaOH at a flow rate of 10 µL/min for 40 s. All the assays were performed at 25 °C.
Statistical analysis
The data are reported as the mean and standard deviation or the median and interquartile range. For each parameter, the assumption of normality was checked using the Shapiro‒Wilk test. To compare the mean difference between plasma samples and extracted samples for each form to an expected mean of zero, the one-sample t-test was used.
The Mann‒Whitney U test was used to compare the relative amounts of HA forms (as determined by LC–MS) between groups. Student’s t-test was used to assess differences in SPR binding.
All tests were two-sided, and p values less than 0.05 were used to indicate statistical significance. The data were analyzed using the Statistical Package for Social Sciences (SPSS) version 28 (IBM) and GraphPad Prism software v. 8.4.2 (GraphPad Software, San Diego, CA, USA). Software information is available at: https://www.graphpad.com/scientific-software/prism/.
Conclusions
We developed a robust, cost-effective SPR-based method for analyzing albumin binding capacity in clinically relevant settings. By combining single-step selective immunocapture with label-free, real-time detection, this approach enables the cost-effective assessment of HA-drug interactions, preserving the intrinsic and clinically relevant HA microheterogeneity. This enables the evaluation of HA binding properties under clinically meaningful conditions, without the need for prior purification or protein modification. The method requires only minimal sample volumes, is highly reproducible, and supports extended reusability of the sensing surface for several hundred analytical cycles without loss of performance. These features make it a practical and scalable tool for the reliable investigation of HA functional alterations associated with pathological states or therapeutic interventions. Overall, this analytical approach provides a solid foundation for the future implementation of personalized and condition-specific assessments of albumin functionality in both research and clinical settings.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Quinlan, G. J., Martin, G. S. & Evans, T. W. Albumin: Biochemical properties and therapeutic potential. Hepatology 41, 1211–1219 (2005).
Ascenzi, P., Fanali, G., Fasano, M., Pallottini, V. & Trezza, V. Clinical relevance of drug binding to plasma proteins q. J. Mol. Struct. 1077, 4–13 (2014).
Fasano, M. et al. The extraordinary ligand binding properties of human serum albumin. IUBMB Life. 57, 787–796 (2005).
Sudlow, G., Birkett, D. J. & Wade, D. N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 12, 1052–1061 (1976).
Sudlow, G., Birkett, D. J. & Wade, D. N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 11, 824–832 (1975).
Zsila, F. Subdomain IB is the third major drug binding region of human serum albumin: Toward the three-sites model. Mol. Pharm. 10, 1668–1682 (2013).
Ghuman, J. et al. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 353, 38–52 (2005).
Fanali, G. et al. Human serum albumin: From bench to bedside. Mol. Aspects Med. 33, 209–290 (2012).
Paramasivan, S. et al. Serum albumin cysteine trioxidation is a potential oxidative stress biomarker of type 2 diabetes mellitus. Sci. Rep. 10, 1–12 (2020).
Watanabe, H. Oxidized albumin: Evaluation of oxidative stress as a marker for the progression of kidney disease. Biol. Pharm. Bull. 45, 1728–1732 (2022).
Domenicali, M. et al. Posttranscriptional changes of serum albumin: Clinical and prognostic significance in hospitalized patients with cirrhosis. Hepatology 60, 1851–1860 (2014).
Nakajou, K., Watanabe, H., Kragh-Hansen, U., Maruyama, T. & Otagiri, M. The effect of glycation on the structure, function and biological fate of human serum albumin as revealed by recombinant mutants. Biochim. Biophys. Acta - Gen. Subj. 1623, 88–97 (2003).
Qiu, H. et al. Comprehensive glycomic analysis reveals that human serum albumin glycation specifically affects the pharmacokinetics and efficacy of different anticoagulant drugs in diabetes. Diabetes 69, 760–770 (2020).
Fender, A. C. & Dobrev, D. Bound to bleed: How altered albumin binding may dictate warfarin treatment outcome. IJC Hear. Vasc. 22, 214–215 (2019).
Bertozo, L., de Neto, C. T., de Oliveira, E., Ximenes, V. F. & L. C. & Oxidative alteration of Trp-214 and Lys-199 in human serum albumin increases binding affinity with phenylbutazone: A combined experimental and computational investigation. Int. J. Mol. Sci. 2018. 19, 2868 (2018).
Michalcová, L. & Glatz, Z. Study on the interactions of sulfonylurea antidiabetic drugs with normal and glycated human serum albumin by capillary electrophoresis-frontal analysis. J. Sep. Sci. 39, 3631–3637 (2016).
Joseph, K. S. S. & Hage, D. S. The effects of glycation on the binding of human serum albumin to warfarin and l-tryptophan. J. Pharm. Biomed. Anal. 53, 811–818 (2010).
Wilting, J. et al. The effect of albumin conformation on the binding of warfarin to human serum albumin. The dependence of the binding of warfarin to human serum albumin on the hydrogen, calcium, and chloride ion concentrations as studied by circular dichroism, fluorescence. J. Biol. Chem. 255, 3032–3037 (1980).
Fredrickson, D. S. & Gordon, R. S. The metabolism of albumin-bound C14-labeled unesterified fatty acids in normal human subjects. J. Clin. Invest. 37, 1504–1515 (1958).
Paar, M. et al. Albumin in patients with liver disease shows an altered conformation. Commun. Biol. 4, 1–9 (2021).
Baraka-Vidot, J., Guerin-Dubourg, A., Bourdon, E. & Rondeau, P. Impaired drug-binding capacities of in vitro and in vivo glycated albumin. Biochimie 94, 1960–1967 (2012).
Oettl, K. et al. Oxidative albumin damage in chronic liver failure: Relation to albumin binding capacity, liver dysfunction and survival. J. Hepatol. 59, 978–983 (2013).
Nagumo, K. et al. Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS One. 9, e85216 (2014).
Blache, D. et al. Glycated albumin with loss of fatty acid binding capacity contributes to enhanced arachidonate oxygenation and platelet hyperactivity: Relevance in patients with type 2 diabetes. Diabetes 64, 960–972 (2015).
Masson, J. F. Surface plasmon resonance clinical biosensors for medical diagnostics. ACS Sens. 2, 16–30 (2017).
Lai, T., Hou, Q., Yang, H., Luo, X. & Xi, M. Clinical application of a novel sliver nanoparticles biosensor based on localized surface plasmon resonance for detecting the microalbuminuria. Acta Biochim. Biophys. Sin (Shanghai). 42, 787–792 (2010).
Fabini, E., Tramarin, A. & Bartolini, M. Combination of human acetylcholinesterase and serum albumin sensing surfaces as highly informative analytical tool for inhibitor screening. J. Pharm. Biomed. Anal. 155, 177–184 (2018).
Fabini, E., Fiori, G. M. L., Tedesco, D., Lopes, N. P. & Bertucci, C. Surface plasmon resonance and circular dichroism characterization of cucurbitacins binding to serum albumins for early pharmacokinetic profiling. J. Pharm. Biomed. Anal. 122, 166–172 (2016).
Sharifi, M. et al. Surface plasmon resonance and molecular docking studies of bovine serum albumin interaction with neomycin: Kinetic and thermodynamic analysis. BioImpacts 7, 91–97 (2017).
Onishi, R. et al. Surface plasmon resonance assay of binding properties of antisense oligonucleotides to serum albumins and lipoproteins. Anal. Sci. 31, 1255–1260 (2015).
Fabini, E. & Danielson, U. H. Monitoring drug–serum protein interactions for early ADME prediction through Surface Plasmon Resonance technology. J. Pharm. Biomed. Anal. 144, 188–194 (2017).
Baldassarre, M. et al. Albumin Homodimers in Patients with Cirrhosis: Clinical and Prognostic Relevance of a Novel Identified Structural Alteration of the Molecule. Sci. Rep. 6, 1–9 (2016).
Jha, J. C., Banal, C., Chow, B. S. M., Cooper, M. E. & Jandeleit-Dahm, K. Diabetes and kidney disease: Role of oxidative stress. Antioxid. Redox Signal. 25, 657–684 (2016).
Naldi, M., Baldassarre, M., Domenicali, M., Bartolini, M. & Caraceni, P. Structural and functional integrity of human serum albumin: Analytical approaches and clinical relevance in patients with liver cirrhosis. J Pharm. Biomed. Anal 144 (2017).
de Mol, N. J. & Fischer, M. J. E. Surface plasmon resonance: Methods and protocols. Life Sci. 55–73. https://doi.org/10.1007/978-1-60761-670-2 (2010).
Naldi, M. et al. A fast and validated mass spectrometry method for the evaluation of human serum albumin structural modifications in the clinical field. Eur. J. Mass. Spectrom. 19, 491–496 (2013).
Kuroda, Y., Saito, M., Sakai, H. & Yamaoka, T. Rapid characterization of drug-drug interaction in plasma protein binding using a surface plasmon resonance biosensor. Drug Metab. Pharmacokinet. 23, 120–127 (2008).
Wang, Y. et al. A fluorescent fatty acid probe, DAUDA, selectively displaces two myristates bound in human serum albumin. Protein Sci. 20, 2095–2101 (2011).
Brodersen, R. Competitive binding of bilirubin and drugs to human serum albumin studied by enzymatic oxidation. J. Clin. Invest. 54, 1353–1364 (1974).
Day, Y. S. N. & Myszka, D. G. Characterizing a drug’s primary binding site on albumin. J. Pharm. Sci. 9, 333–343 (2003).
Bakar, K. A. & Feroz, S. R. A critical view on the analysis of fluorescence quenching data for determining ligand–protein binding affinity. Spectrochim Acta - Part. Mol. Biomol. Spectrosc. 223, 1–5 (2019).
Goncharova, I. & Urbanová, M. Stereoselective bile pigment binding to polypeptides and albumins: A circular dichroism study. Anal. Bioanal Chem. 392, 1355–1365 (2008).
Clària, J. et al. Systemic inflammation in decompensated cirrhosis: Characterization and role in acute-on-chronic liver failure. Hepatology 64, 1249–1264 (2016).
Baldassarre, M. et al. Determination of effective albumin in patients with decompensated cirrhosis: Clinical and prognostic implications. Hepatology 74, 2058–2073 (2021).
Naldi, M. et al. Mass spectrometry characterization of circulating human serum albumin microheterogeneity in patients with alcoholic hepatitis. J. Pharm. Biomed. Anal. 122, 141–147 (2016).
Assandri, A. & Bernareggi, A. Binding of teicoplanin to human serum albumin. Eur. J. Clin. Pharmacol. 33, 191–195 (1987).
Zoratti, C. et al. Antibiotics and liver cirrhosis: What the physicians need to know. Antibiotics 11, 1–19 (2022).
Ulldemolins, M., Roberts, J. A., Rello, J., Paterson, D. L. & Lipman, J. The effects of hypoalbuminaemia on optimizing antibacterial dosing in critically ill patients. Clin. Pharmacokinet. 50, 99–110 (2011).
Prakash, S. Role of human serum albumin and oxidative stress in diabetes. J. Appl. Biotechnol. Bioeng. 3, 281–285 (2017).
International, K. KDIGO 2020 clinical practice guideline for diabetes management in chronic kidney disease. Kidney Int. 98, S1–S115 (2020).
Babu Kondaveeti, S., Kumaraswamy, D., Mishra, S. & Aravind Kumar, R. Anand Shaker, I. Evaluation of glycated albumin and microalbuminuria as early risk markers of nephropathy in type 2 diabetes mellitus. J. Clin. Diagn. Res. 7, 1280–1283 (2013).
Copeland, R. A. A Practical Introduction to Structure, Mechanism, and Data Analysis (Wiley-VCH, 2000).
Segel, I. H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems (York, Wiley-Interscience: New, 1975).
Cantor, C. R. & Schimmel, P. R. Biophysical Chemistry; Part III: The Behavior of Biological Macromolecules (W. H. Freeman & Co., 1980).
Myszka, D. G. Improving biosensor analysis. J. Mol. Recognit. 12, 279–284 (1999).
Beng, C. G., Rasanayagam, L. J., Lim, K. L. & Lau, K. S. Solubility and absorption spectra of complexes resulting from interaction among human albumin, bromcresol green and detergents. Clin. Chim. Acta. 52, 257–269 (1974).
Acknowledgements
MBar and MN would like to thank Drs Francesca Marchignoli and Chiara Carrisi from the Unit of Clinical Nutrition, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy, for collecting and handling the plasma samples from DM patients, which were used in this study. Miss Martina Chimisso, Miss Alessia Cavaliere, and Miss Claudia Dal Monte are also acknowledged for technical support.
Funding
This work was financially supported by the University of Bologna (RFO funding scheme).
Author information
Authors and Affiliations
Contributions
Marta Nugnes: Investigation, validation, data curation, writing—original draft preparation; Maurizio Baldassarre: Visualization, formal analysis, resources; Paolo Caraceni: Resources, writing—review & editing; Marina Naldi: Visualization, methodology, supervision, writing—review & editing; Manuela Bartolini: Conceptualization, project administration, writing—review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nugnes, M., Baldassarre, M., Caraceni, P. et al. Albumin-on-a-chip: binding profiling of circulating human albumin via selective immunocapture and real-time SPR analysis. Sci Rep 16, 10920 (2026). https://doi.org/10.1038/s41598-026-44934-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-44934-2
