
Abstract
Pseudomonas species are Gram-negative bacterial pathogens that affect a wide range of economically important crops, including almond. Increasing resistance of Pseudomonas syringae to conventional management strategies highlights the need for alternative disease control options. In this study, we isolated and characterized three lytic bacteriophages, including vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission, targeting almond-infecting Pseudomonas strains. Host-range analysis across 36 isolates revealed partially overlapping infectivity profiles, with strongest activity against phylogroup 2 (PG2) almond-associated P. syringae pv. syringae and reduced infectivity against phylogenetically distinct isolates, including PG7 P. viridiflava and other crop-associated pathovars. Efficiency-of-plating assays quantitatively supported these host-range patterns. Plaque morphology and transmission electron microscopy demonstrated icosahedral capsids with short, non-contractile tails consistent with tailed dsDNA bacteriophages of the class Caudoviricetes, with vB_PsyP_Mission producing halo-associated plaques consistent with predicted extracellular polysaccharide-modifying proteins. Genomic analyses revealed compact (~ 40 kb) genomes lacking integrases, tRNAs, lysogeny-associated genes, and known virulence or antimicrobial resistance determinants. Comparative genomics showed that vB_PsyP_Plaza and vB_PsyP_Mission likely represent novel isolates within previously described species-level groups, whereas vB_PsyP_Mobley is more genomically divergent. Collectively, these results define the phenotypic and genomic characteristics of three related but distinguishable Pseudomonas phages and provide a basis for future in planta evaluation of their potential utility against almond bacterial blast.
Similar content being viewed by others


Introduction
Pseudomonas species are Gram-negative bacteria with an exceptionally broad host range, which includes fruits, vegetables, cereals, ornamentals, and woody crops worldwide. These pathogens cause blights, cankers, leaf spots, wilts, and soft rots, resulting in substantial losses in global agricultural productivity1,2. Among this genus, Pseudomonas syringae is widely recognized as one of the most destructive plant pathogens worldwide. It encompasses numerous pathovars that collectively infect more than 180 host species, often causing severe disease outbreaks that can lead to yield losses approaching 80% in susceptible crops3,4. The ecological versatility of P. syringae, its ability to thrive on leaf surfaces, persist in water systems, and disperse via aerosols or rainfall, contributes to its widespread distribution and recurrent epidemic cycles5.
Within this diverse species, almond-infecting P. syringae pv. syringae (Pss) has emerged as a particularly serious threat. Almonds are California’s most valuable specialty crop with a $5 billion market value, and account for over 80% of global production6. Bacterial blast, caused by Pss, has intensified in recent years, leading to shoot dieback, blossom necrosis, and premature fruit loss, symptoms that are especially damaging to young orchards. Outbreaks are most severe during the cold, wet springs typical of California’s Central Valley7. Recent epidemics (2019–2021 and 2023) caused crop losses of up to 40% in affected orchards, highlighting the growing vulnerability of almond production systems8.
Current management strategies depend primarily on copper-based bactericides and kasugamycin (Kasumin), the latter of which is used under repeated Sect. 18 emergency exemptions7. However, copper resistance is now widespread, Kasumin efficacy is inconsistent, and tightening chemical regulations are reducing the availability of these tools9,10. In addition to declining performance, these products accumulate in soil, disrupt beneficial microbiota, and pose environmental and worker-safety concerns11,12. Collectively, these challenges highlight an urgent need for biologically based, sustainable alternatives that reduce pesticide reliance and enhance the long-term resilience of specialty crops worldwide.
Bacteriophages (phages) have re-emerged as promising biological control agents for plant pathogens13,14. Phage-based interventions offer several advantages: they exhibit high host specificity, minimizing damage to beneficial microbiota; they replicate in proportion to bacterial densities, enabling self-sustaining control; and they evolve alongside their bacterial hosts, potentially reducing the risk of long-term resistance15,16. Importantly, phage-based biocontrol aligns with integrated pest management and sustainable agriculture frameworks that prioritize reduced chemical input and the deployment of environmentally compatible disease management tools13,17. Previous studies have demonstrated the efficacy of lytic phages in suppressing P. syringae populations in vitro and in planta, delaying disease progression, and reducing symptom severity across multiple crops15,18,19,20,21,22.
Despite these advances, phages targeting almond-associated P. syringae pv. syringae remain poorly characterized. In this study, we isolated three distinct phages, including vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission, from sewage and soil samples and performed an integrated genomic, phylogenomic, and functional characterization to evaluate their potential as biocontrol agents against almond bacterial blast. We examined phage morphology, infection dynamics, environmental stability, and lytic activity to determine whether or not these phages could be viable tools for biocontrol efforts. We then quantified infectivity across a diverse panel of Pseudomonas isolates representing multiple crops and ecological backgrounds, including phylogenetically defined host groups (PG2 and PG7). We further conducted comparative genomic analyses to define their genomic features and identify traits relevant to biocontrol, including lytic modules, the absence of lysogeny-associated genes, and variation in predicted host-interaction proteins such as receptor-binding and depolymerase-associated candidates. Collectively, these findings expand current understanding of phage diversity, host specificity, and infection dynamics within the almond Pseudomonas pathosystem and provide a foundation for developing phage-based strategies to enhance sustainable disease management in perennial specialty crop systems.
Results
Phage selection and morphological characterization
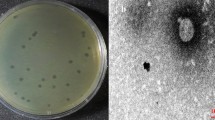
Representative phages were selected for detailed characterization based on distinct restriction digest profiles, with vB_PsyP_Mobley originating from soil-derived filtrates collected near UC Riverside and vB_PsyP_Plaza and vB_PsyP_Mission originating from sewage-derived filtrates. For mnemonic purposes, phages were named after street names within Riverside County, California, and candidate names were verified for uniqueness through literature searches and PhagesDB prior to assignment. All phages were prefixed with vB_PsyP in accordance with ICTV/BAVS nomenclature guidelines, reflecting their isolation and propagation on a Pseudomonas syringae host. Plaque assays revealed clear differences in plaque morphology among the three phages (Fig. 1A–C). vB_PsyP_Mobley produced small, well-defined plaques (Fig. 1A). In contrast, vB_PsyP_Plaza formed larger, uniformly clear plaques (Fig. 1B), whereas vB_PsyP_Mission produced large plaques surrounded by distinct halos (Fig. 1C), a phenotype commonly associated with extracellular polysaccharide–modifying activity. Transmission electron microscopy further showed that all three phages possess icosahedral capsids and short tails characteristic of the class Caudoviricetes (Fig. 1D–F). Capsid dimensions were comparable across phages, with mean widths of 58.47 ± 4.86 nm for vB_PsyP_Mobley, 53.87 ± 2.34 nm for vB_PsyP_Plaza, and 54.15 ± 1.20 nm for vB_PsyP_Mission (Table 1). Tail lengths differed modestly, with vB_PsyP_Mobley exhibiting the longest mean tail length (14.29 ± 3.64 nm), followed by vB_PsyP_Mission (10.60 ± 0.33 nm) and vB_PsyP_Plaza (9.23 ± 3.12 nm). Consistent with these measurements, all three phages exhibited short, non-contractile tails consistent with podovirus-like morphology.

Morphological characteristics of almond-infecting Pseudomonas phages propagated on host strain Pseudomonas syringae pv. syringae 2507. Plaque morphology of vB_PsyP_Mobley (A), vB_PsyP_Plaza (B), and vB_PsyP_Mission (C) following propagation on Pseudomonas syringae pv. syringae strain 2507. vB_PsyP_Mobley produces small, well-defined circular plaques lacking halos, vB_PsyP_Plaza forms larger, uniformly clear plaques, and vB_PsyP_Mission generates plaques surrounded by distinct turbid halos. Transmission electron micrographs (D–F) show representative virions of vB_PsyP_Mobley (D), vB_PsyP_Plaza (E), and vB_PsyP_Mission (F), each displaying icosahedral capsids and short, non-contractile tails characteristic of tailed dsDNA bacteriophages within the class Caudoviricetes. Scale bars are indicated in each panel.
Host-range and efficiency-of-plating analysis across almond-associated and phylogenetically distinct Pseudomonas isolates
Host-range and efficiency-of-plating (EOP) analyses using a panel of 36 Pseudomonas isolates revealed distinct yet partially overlapping infectivity profiles among the three phages (Fig. 2, Table S1). Almond-associated Pseudomonas syringae pv. syringae isolates included in this study were previously recovered from symptomatic almond tissues and extensively characterized, providing a biologically relevant host set7. These isolates belong primarily to phylogroup 2 (PG2), with additional representation from the phylogenetically distinct phylogroup 7 (PG7), enabling assessment of phage activity across related but divergent lineages. All three phages produced clear plaques and generally high EOP values across the majority of PG2 isolates, indicating efficient productive infection within this clade. In contrast, infectivity against PG7 P. viridiflava isolates was more variable and typically associated with lower EOP values, reflecting reduced infection efficiency. Among the three phages, vB_PsyP_Mission exhibited the broadest and most consistent activity across PG7 isolates and also infected several non-almond Pseudomonas pathovars, including strains associated with bean (pv. phaseolicola), tomato (pv. tomato DC3000), cauliflower (P. maculicola), oleander (pv. savastanoi), and radish (P. viridiflava), albeit at lower efficiencies. In contrast, vB_PsyP_Mobley and vB_PsyP_Plaza infected a narrower subset of non-almond isolates, primarily tomato- and cauliflower-associated strains, with both plaque formation and EOP values generally lower than those observed on almond-associated PG2 hosts. No productive infection (EOP ≈ 0) was detected for any phage against more phylogenetically distant Pseudomonas species, including P. syringae pv. glycinea, P. fluorescens, P. cepacia, P. corrugata, P. putida, P. syringae pv. apii, or P. syringae pv. atropurpurea. Collectively, these results demonstrate strong adaptation to PG2 almond-associated P. syringae pv. syringae, with reduced and often partial infectivity against phylogenetically distinct PG7 P. viridiflava and other non-almond Pseudomonas pathovars, consistent with clade-associated patterns of phage–host interaction.

Efficiency-of-plating profiles of three P. syringae phages across a diverse Pseudomonas panel. Efficiency of plating (EOP) was quantified for vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission across the indicated Pseudomonas isolates/pathovars. For each phage, plaque counts on each test host were normalized to the host yielding the highest plaque titer (set to 1.0), and EOP values for all other hosts were calculated relative to this maximum. Heatmap shading reflects relative plating efficiency (white = no detectable plaques; darker red = higher EOP). Isolates are grouped by phylogroup/pathovar, highlighting strong plating on PG2 P. syringae pv. syringae and more variable, generally reduced plating on PG7 P. viridiflava and other non-almond–associated Pseudomonas taxa.
Optimal multiplicity of infection
The optimal multiplicity of infection (MOI) for each phage was determined by infecting cultures of the almond-infecting Pseudomonas syringae pv. syringae strain 2057 with vB_PsyP_Mobley, vB_PsyP_Mission, and vB_PsyP_Plaza across a range of MOIs and monitoring phage titers over time. vB_PsyP_Mobley and vB_PsyP_Plaza exhibited maximal replication efficiency at an MOI of 10− 3 (Tables 2 and 3), whereas vB_PsyP_Mission reached peak propagation at an MOI of 10− 4 (Table 4). These MOI values corresponded to the conditions yielding the highest phage titers while maintaining effective bacterial suppression during the assay period.
Lytic activity against planktonic and biofilm cells
The bactericidal activity of phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission against planktonic Pseudomonas cells was assessed by monitoring optical density (OD600) over time. All three phages exhibited MOI-dependent reductions in bacterial density during the early phase of infection. vB_PsyP_Mobley reduced OD600 within 4–8 h across MOIs from 0.00001 to 1, with more rapid decline at higher MOIs; at an MOI of 10, OD600 dropped to near baseline within 2 h. However, bacterial regrowth consistent with the emergence of resistant cells was observed by 16 h (Fig. 3A, D). vB_PsyP_Plaza caused a rapid early decrease in OD600 (within ~ 2 h) across MOIs ranging from 10⁻⁹ to 10⁻², followed by regrowth at later time points (Fig. 3B, E). vB_PsyP_Mission showed a similar pattern, with a pronounced early reduction in OD600 within ~ 2 h across the same MOI range and subsequent regrowth at later time points (Fig. 3C, F). Both vB_PsyP_Plaza and vB_PsyP_Mission retained measurable activity at very low MOIs, whereas vB_PsyP_Mobley showed its strongest early activity at MOIs ≤ 1. All three phages also disrupted pre-formed biofilms. When applied at MOIs from 0.1 to 0.001, vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission reduced biofilm biomass by 63–88%, 76–78%, and 88–96%, respectively, as measured by crystal violet staining (Fig. 4A, B). vB_PsyP_Mission consistently produced the greatest reduction, consistent with its halo-forming plaque morphology, which suggests EPS-degrading activity. Together, these results indicate that vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission produce strong early bactericidal effects against planktonic cells and significantly reduce established biofilms, with differences in activity profiles across MOIs and in biofilm disruption.

Lytic activity of phages against Pseudomonas planktonic cells. Bacterial growth curves (OD₆₀₀) of Pseudomonas syringae pv. syringae 2507 treated with phages vB_PsyP_Mobley (A), vB_PsyP_Plaza (B), and vB_PsyP_Mission (C) at the indicated multiplicities of infection (MOIs). Corresponding areas under the growth curves (AUC) are shown in panels (D–F), respectively. Data represent the mean ± SD of three technical replicates from at least two independent experiments. Statistical differences among treatments were assessed using one-way ANOVA followed by Tukey’s multiple comparisons test. Bars sharing the same lowercase letter are not significantly different (p < 0.05).

Phage-mediated disruption of planktonic and biofilm cells of almond-infecting Pseudomonas syringae pv. syringae 2507. (A) Biofilm biomass following treatment with phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission at the indicated MOIs, quantified by 0.1% crystal violet staining and measured at 595 nm. (B) Percentage reduction in biofilm biomass relative to untreated controls. Data are presented as mean ± SD from three technical replicates and at least two independent experiments. Statistical significance was determined using one-way ANOVA followed by Tukey’s multiple comparisons test. Different lowercase letters indicate statistically significant differences among treatments (p < 0.05).
Stability and adsorption kinetics of phage
Temperature and pH stability are important considerations for field application of phages, as environmental conditions can influence infectivity. Thermal stability assays revealed that vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission remained viable after 24 h of incubation at between 4 °C and 25 °C (Fig. 5A–C). This stability is consistent with the cold spring conditions under which almond bacterial blast commonly develops3,7,8,23. Phage vB_PsyP_Mobley retained infectivity at 37 °C, whereas phages vB_PsyP_Plaza and vB_PsyP_Mission showed marked reductions in activity at this temperature. At 50 °C and above, infectivity of all three phages was minimal or undetectable. Across pH treatments, all phages maintained infectivity over a broad range (pH 3–11), although with phage-specific profiles (Fig. 5D–F). Extreme acidic (pH 1) or alkaline (pH 13) conditions eliminated detectable infectivity. Among the three phages, vB_PsyP_Mobley exhibited comparatively greater thermal tolerance, while vB_PsyP_Mission showed broader stability across the tested pH range, suggesting differences in capsid or virion structural resilience. Adsorption assays further distinguished the phages’ infection dynamics. vB_PsyP_Mobley adsorbed to Pseudomonas cells more rapidly than vB_PsyP_Plaza and vB_PsyP_Mission, which displayed similar but slower adsorption kinetics (Fig. 6). Together, these results indicate that the three phages differ in their stability and early infection traits, with vB_PsyP_Mobley combining faster host adsorption and greater thermal tolerance, and vB_PsyP_Plaza and vB_PsyP_Mission exhibiting moderate stability and slower adsorption.

Effect of temperature and pH on the viability of almond-infecting Pseudomonas syringae pv. syringae 2507 phages. (A–C) Viability of phages vB_PsyP_Mobley (A), vB_PsyP_Plaza (B), and vB_PsyP_Mission (C) following 24 h incubation at the indicated temperatures. (D–F) Viability of vB_PsyP_Mobley (D), vB_PsyP_Plaza (E), and vB_PsyP_Mission (F) following 1 h incubation at the indicated pH values. Phage viability is expressed as PFU/mL. Data represent mean ± SD from at least two independent experiments. Statistical comparisons were performed using one-way ANOVA with Tukey’s multiple comparisons test. Bars sharing the same lowercase letter are not significantly different (p < 0.05).

Adsorption kinetics of almond-infecting Pseudomonas syringae pv. syringae 2507 phages. Adsorption curves of phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission were determined by mixing each phage with host cells at multiplicities of infection (MOI) of 0.001 for vB_PsyP_Mobley and vB_PsyP_Plaza, and 0.0001 for vB_PsyP_Mission. At defined time intervals, aliquots were removed, and the number of unadsorbed phages was quantified by plaque assay.
Genomic features and assembly quality of vB_PsyP phages
The complete genomes of phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission were sequenced using the Illumina MiSeq platform and assembled into single, high-quality contigs. Genome sizes were 39,888 bp for vB_PsyP_Mobley, 41,879 bp for vB_PsyP_Plaza, and 40,099 bp for vB_PsyP_Mission, with GC contents of 63%, 57%, and 56%, respectively (Table 5). Assembly quality assessment using CheckV and MiUVIG criteria confirmed that all three genomes were complete, with 100% estimated completeness and average amino acid identity (AAI)–based confidence values exceeding 90%. All three genomes contained direct terminal repeats (DTRs) of identical length (87 bp; repeat count = 2), indicating a conserved DTR-mediated DNA packaging strategy. Functional annotation identified 58 open reading frames (ORFs) in vB_PsyP_Mobley, 54 in vB_PsyP_Plaza, and 51 in vB_PsyP_Mission (Table 6). Canonical modules for DNA replication, morphogenesis, and host lysis were present in all genomes, whereas integrases, recombination modules, tRNAs, CRISPR arrays, virulence-associated genes, and antimicrobial resistance determinants were absent, indicating that these phages do not establish lysogeny. vB_PsyP_Mobley encoded the highest proportion of hypothetical or uncharacterized ORFs (34), followed by vB_PsyP_Plaza (23) and vB_PsyP_Mission (20), suggesting differing degrees of functional novelty among the phages.
Genome-based classification and taxonomic placement
Genome-based classification using ICTV-recognized taxonomy frameworks placed all three phages within the realm Duplodnaviria, kingdom Heunggongvirae, phylum Uroviricota, class Caudoviricetes, and order Autographivirales. All were assigned to the family Autotranscriptaviridae (Table 7). Subfamily- and genus-level resolution differed among the phages. vB_PsyP_Plaza and vB_PsyP_Mission were placed within the subfamily Studiervirinae, clustering at the genus level within Ghunavirus and Pifdecavirus, respectively. In contrast, vB_PsyP_Mobley could not be confidently assigned to a defined subfamily or genus and is provisionally designated as unclassified within Autotranscriptaviridae. Whole-genome nucleotide comparisons supported these placements. Pairwise intergenomic identities among vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission ranged from 40.93% to 45.56% (Table 8), indicating that the three phages represent distinct evolutionary lineages relative to one another. Comparison with previously described phages (Table 9) showed that vB_PsyP_Mission shares 96.08% identity (98% coverage) with Pseudomonas phage vB_PpuP-Keila, and vB_PsyP_Plaza shares 97.91% identity (97% coverage) with Pseudomonas phage Pst_gh1. According to ICTV species demarcation thresholds (≥ 95% genome-wide nucleotide identity), these values indicate that vB_PsyP_Mission and vB_PsyP_Plaza represent novel isolates within previously established species-level groups rather than new species. In contrast, vB_PsyP_Mobley exhibits lower similarity to its closest reference, Pseudomonas phage KBC54 (83.87% identity; 80% coverage), consistent with greater genomic divergence.
Phylogenomic relationships and genome organization
Phylogenetic analyses based on amino acid sequences of two conserved structural markers, including the major capsid protein (MCP, Fig. S1) and terminase large subunit (TerL, Fig. S2), further resolved these relationships. Both trees showed congruent topologies. vB_PsyP_Plaza clustered within Group I alongside Pst_gh1-related phages, vB_PsyP_Mobley clustered within Group II with KBC54-related phages, and vB_PsyP_Mission clustered within Group III with vB_PpuP-Keila and related representatives. A separate Group IV comprised more distantly related Pseudomonas and EctoPseudomonas phages. In both trees, the Pectobacterium phages used as outgroups were clearly separated from the Pseudomonas clades. The consistent grouping across MCP and TerL phylogenies supports the lineage assignments inferred from whole-genome analyses. Phylogenomic clustering using ViPTree (Figs. 7, 8 A-C) produced similar relationships, with vB_PsyP_Plaza and vB_PsyP_Mission clustering closely with their respective reference phages, whereas vB_PsyP_Mobley formed a more distant branch. Whole-genome synteny analysis further reflected these patterns. vB_PsyP_Plaza and Pst_gh1, as well as vB_PsyP_Mission and vB_PpuP-Keila, exhibited extensive collinearity across structural, replication, and lysis modules. In contrast, vB_PsyP_Mobley shared only partial synteny with KBC54, with several divergent regions enriched in unique ORFs and hypothetical genes (Fig. 9). VIRIDIC intergenomic similarity analysis (Fig. 10) also showed that vB_PsyP_Mission and vB_PsyP_Plaza cluster with multiple closely related Pseudomonas phages, whereas vB_PsyP_Mobley clustered with only KBC54, further highlighting its relative genomic distinctness. Circular genome maps (Fig. 7A-C) showed densely packed coding regions and characteristic GC content and GC skew patterns corresponding to structural and replication modules, consistent with modular genome organization typical of tailed dsDNA phages.

Genomic architecture and whole-genome similarity of three almond-infecting vB_PsyP phages. (A–C) Circular genome maps of phages vB_PsyP_Mobley (A), vB_PsyP_Plaza (B), and vB_PsyP_Mission (C). Predicted coding sequences (CDS) are shown on the outer ring (color-coded by phage), with inner rings indicating GC content (black) and GC skew (green = positive; purple = negative). (D) BRIG-based whole-genome comparison of vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission, with shaded rings indicating nucleotide identity thresholds (50%, 70%, and 100%) relative to the reference genome.

Phylogenetic analysis of almond-infecting Pseudomonas syringae pv. syringae 2507 phages. Phylogenetic tree generated using ViPTree based on whole-genome proteomic alignments of phages vB_PsyP_Mobley (A), vB_PsyP_Plaza (B), and vB_PsyP_Mission (C) with related phages from established reference genomes.

Comparative genomic architecture of phages vB_PsyP_Mission, vB_PsyP_Plaza, and vB_PsyP_Mobley and closely-related phages. Gene synteny and amino acid identity are shown for each phage and its reference counterpart: (A) vB_PsyP_Mobley vs. KBC54, (B) vB_PsyP_Plaza vs. GIL1, and (C) vB_PsyP_Mission vs. Keila. Colored arrows represent predicted ORFs, and grey lines indicate homologous regions. The identity scale ranges from 0–100%.

VIRIDIC pairwise intergenomic similarities and distances among phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission. Color density represents aligned genome fraction, genome length ratio, and intergenomic similarity values, where darker shading corresponds to lower similarity or limited alignment. Gray bars indicate relative genome lengths. The names of the three focal phages are bolded and shown in red for emphasis.
Comparative gene content and predicted depolymerase repertoire
Comparative gene content analysis demonstrated that vB_PsyP_Plaza and vB_PsyP_Mission share a substantial proportion of orthologous genes, whereas vB_PsyP_Mobley shares fewer conserved genes with the other two phages (Fig. 7D; Fig. S3; Tables S2-S4). Although all three genomes retain conserved structural and replication backbones, each contains regions enriched in unique ORFs, with vB_PsyP_Mobley exhibiting the greatest divergence. Reciprocal BLASTP-based ortholog comparisons (Tables S5-S7) identified conserved structural and replication modules across the three genomes but revealed divergence in genes associated with host interaction and infection processes. Sequence variation was observed in tail fiber proteins, tail-associated proteins, head-tail adaptor components, and infection-related enzymes, including DNA polymerase, RNA polymerase, DNA ligase, and DNA primase/helicase. Differences were also detected in holin, Rz-like spanin, internal virion proteins, and a predicted host HNS inhibition protein, indicating variability in infection-related gene repertoires among the lineages. Depolymerase prediction analysis identified putative virion-associated enzymatic candidates in all three genomes (Table 10). Each phage encoded a predicted tail-associated depolymerase (YCJ12281.1_41 in vB_PsyP_Mobley; YCJ12353.1_55 in vB_PsyP_Plaza; YCJ12402.1_49 in vB_PsyP_Mission). In addition, vB_PsyP_Mobley encoded a second predicted depolymerase annotated as an internal virion protein, whereas vB_PsyP_Plaza and vB_PsyP_Mission each encoded an additional predicted depolymerase annotated as a tail fiber protein. These predicted candidates varied in sequence length and physicochemical properties among the genomes. All depolymerase assignments remain bioinformatic predictions pending biochemical validation.
Discussion
This study provides an integrated phenotypic, ecological, genomic, and phylogenomic characterization of three strictly lytic phages infecting P. syringae associated with almond bacterial blast. While in planta efficacy was not evaluated, the data define key attributes relevant to agricultural deployment, including phylogenetically structured host range, quantitative infectivity (EOP), infection dynamics, environmental stability, genome organization, and taxonomic placement. By comparing three evolutionarily distinct phages with overlapping but non-identical biological traits, this work illustrates how lineage-level diversity may be leveraged to address pathogen heterogeneity in perennial cropping systems.
A central finding is that all three phages exhibit their strongest infectivity against almond-associated P. syringae pv. syringae isolates belonging to phylogroup 2 (PG2), whereas activity against PG7 (P. viridiflava) is reduced and often partial. Importantly, host range conclusions are supported by quantitative EOP measurements in addition to plaque morphology. Framing host range in a phylogenetic context is particularly important in Pseudomonas, where phylogroups frequently reflect underlying differences in surface architecture, including lipopolysaccharide O-antigen composition, outer membrane proteins, and extracellular polysaccharides24,25. The PG-associated infectivity patterns observed here are therefore consistent with receptor conservation within PG2 and receptor divergence in PG7 affecting adsorption efficiency, post-entry compatibility, or replication dynamics. This aligns with broader evidence that phage host range often tracks receptor phylogeny more closely than formal species or pathovar boundaries26. From a disease management standpoint, this suggests that phage selection can be optimized for dominant outbreak lineages—such as PG2 almond-associated strains—while recognizing that coverage of more distant clades may require complementary isolates or engineered receptor-binding variants27.
Phylogenomic and whole-genome analyses further contextualize these ecological patterns. Despite low intergenomic nucleotide identity among vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission, all three infect PG2 populations, suggesting that distinct evolutionary lineages can independently access the same ecological niche through different structural or receptor-binding configurations28,29. Previous work has shown that infection outcomes are shaped by lineage-specific combinations of receptor-binding proteins, tail structures, and accessory genes that influence adsorption, intracellular compatibility, and lysis dynamic16,30. Whole-genome identity thresholds indicate that Plaza and Mission represent new isolates within previously defined species-level groups, whereas Mobley is more divergent and potentially represents a distinct lineage. Based on its comparatively low nucleotide identity to currently described relatives, limited clustering with reference phages, and concordant placement as a distinct lineage across MCP, TerL, synteny, and VIRIDIC analyses, vB_PsyP_Mobley may represent a member of a previously unrecognized genus. However, formal genus designation will require broader comparative analysis and validation under ICTV taxonomic criteria. Concordant placement across MCP and TerL phylogenies, genome synteny, and VIRIDIC clustering strengthens these assignments and supports their evolutionary distinctness. From an applied perspective, genomic distinctness combined with overlapping host coverage may be advantageous for cocktail design, as resistance often arises through receptor modification or intracellular defenses31,32,33,34. However, genomic divergence alone does not confirm independent receptor usage, as unrelated phages can converge on the same surface determinant35,36. Accordingly, while the present data support functional complementarity, definitive receptor mapping and cross-resistance testing are required to confirm distinct entry pathways.
The concordance between halo formation, biofilm disruption, and predicted depolymerase-like genes provides an additional functional dimension often lacking in phage biocontrol studies37,38. In this study, vB_PsyP_Mission consistently produced plaques surrounded by halos, whereas vB_PsyP_Mobley and vB_PsyP_Plaza did not. In Pseudomonas, extracellular matrices can include alginate-like polymers, levan, cellulose-associated components, and other strain- and environment-dependent EPS structures39,40,41,42. Virion-associated depolymerases facilitate penetration of these barriers and can improve access to surface receptors43,44. Interpreted conservatively, the concordance between halo formation in vB_PsyP_Mission and the presence of predicted depolymerase-like genes is consistent with a model in which matrix modification enhances access to bacterial cells embedded within structured environments, whereas vB_PsyP_Mobley and vB_PsyP_Plaza may rely primarily on lytic replication against more accessible targets45. From a biocontrol standpoint, this suggests potential ecological differentiation rather than superiority. Halo-forming phages may contribute to disruption of surface-associated microcolonies, whereas non-halo-forming phages may more efficiently target exposed populations46,47. In perennial systems such as almond orchards, where epiphytic persistence and microenvironmental heterogeneity are common, such complementary roles could be advantageous. Nonetheless, halo formation alone does not predict field efficacy, and biochemical validation of putative depolymerases remains necessary to confirm substrate specificity and functional relevance.
The shared presence of direct terminal repeats (DTRs) across all three phages, despite broader genomic divergence, highlights conservation of packaging architecture independent of lineage identity. DTR-mediated packaging imposes constraints on terminase function and capsid–genome interactions48,49. In this sense, DTR conservation represents an architecture-level similarity, indicating that distinct phage lineages infecting similar hosts can converge on comparable solutions for genome replication and packaging while differing substantially in host-recognition modules and accessory gene content50. It is noteworthy that, although this feature does not directly inform lytic efficacy, host range, or performance in combined applications, nor does it inherently imply interaction or interference among phages48, it nevertheless provides insight into conserved replication and packaging architectures shared across otherwise divergent phage lineages.
Differences in thermal tolerance among the phages have practical relevance for field deployment. The retention of infectivity by vB_PsyP_Mobley at 37 °C, compared with reduced stability of Plaza and Mission, suggests that environmental conditions and application timing may influence relative performance51,52. Almond bacterial blast typically develops under cool, wet conditions, but short-term temperature elevations above 30 °C are common in the Central Valley7,8. Under such conditions, Mobley may maintain activity more reliably, whereas Plaza and Mission are likely to perform best when applications are aligned with cooler windows, such as early morning or evening periods. In addition to temperature, pH is another field-relevant factor that may influence phage persistence. Although orchard conditions are not defined by extreme pH, phage exposure to variable pH can occur on leaf surfaces, particularly via irrigation water quality, tank mixes, and formulation additives. Consistent with our stability assays across a broad pH range, these results suggest that pH dynamics, together with temperature, should be considered when optimizing formulation and application conditions for orchard deployment.
In conclusion, this study provides an integrated characterization of three strictly lytic phages (vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission) infecting almond-associated P. syringae pv. syringae and establishes a foundation for developing phage-based management strategies. However, receptor identification and mechanistic analyses explaining cross-phylogroup infectivity remain priorities for future work. In addition, key assays were performed on a limited set of strains, and broader isolate-level robustness remains to be evaluated. Environmental stability was assessed under controlled conditions, and additional orchard-relevant stressors, including UV exposure and fluctuating humidity, were not tested. Finally, all experiments were conducted in vitro, and phage activity under epiphytic and in planta conditions remains to be determined. Collectively, these results define a genetically diverse set of strictly lytic phages with distinct infection traits and provide a strong platform for subsequent quantitative, mechanistic, and in planta/field-based evaluations. Notably, the degree of genomic and phylogenetic divergence observed for vB_PsyP_Mobley suggests that it may warrant future consideration as a representative of a previously unrecognized genus, pending formal taxonomic validation.
Materials and methods
Phage isolation and purification
Soil samples collected from the vicinity of Sproul Hall at UC Riverside and sewage samples obtained from the Riverside wastewater treatment facility were collected and processed for bacteriophage isolation. Samples were mixed with an equal volume of 2× LB medium and incubated at 28 °C with shaking for 4 h to facilitate the release of phages into solution. Following incubation, samples were clarified by centrifugation and passed through 0.22 μm syringe filters to remove bacterial debris. For phage enrichment, 10 mL of the filtered sample was combined with 20 mL of actively growing cultures of the target Pseudomonas strains and incubated overnight at 28 °C with shaking at 200 rpm. Enrichment cultures were then centrifuged at 3,500 rpm for 10 min, and the supernatants were filtered through 0.22 μm syringe filters. Phage activity was initially screened using spot assays. Briefly, 100 µL of each bacterial culture was mixed with 5 mL of molten LB soft agar (0.5% agar) and overlaid onto LB agar plates to form bacterial lawns. Aliquots (10 µL) of enriched phage suspensions were spotted onto the lawns, and plates were incubated overnight at 28 °C. Zones of lysis were identified and individual plaques were collected using sterile pipette tips and resuspended in SM buffer (50 mM Tris-HCl, 100 mM NaCl, 8 mM MgSO₄, 0.01% gelatin). To ensure clonal purity, phages were purified through three successive rounds of plaque isolation. High-titer phage stocks were prepared and stored at 4 °C for short-term use or at − 80 °C in SM buffer supplemented with 20% glycerol for long-term preservation.
Host range determination and efficiency of plating
The host range of phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission was evaluated using a panel of 36 Pseudomonas strains representing multiple species and pathovars associated with diverse plant hosts. Almond-associated P. syringae pv. syringae isolates were obtained from the culture collection of Dr. Florent Trouillas (University of California, Davis). These isolates were originally recovered from symptomatic almond tissues and were identified to the subspecies/pathovar level through whole-genome sequencing followed by BLAST-based comparative analyses. Additional Pseudomonas strains infecting soybean, bean, tomato, cauliflower, radish, oleander, celery, tobacco, onion, and cherry, as well as soil-associated isolates, were obtained from the culture collections of Dr. Donald Cooksey via Dr. Caroline Roper (University of California, Riverside). These additional strains were included to assess cross-species infectivity (Table 1). Overnight bacterial cultures were grown in LB broth at 28 °C with shaking and adjusted to an OD600 of 2.0. For each strain, 100 µL of the standardized culture was mixed with 5 mL of molten LB top agar (0.5% agar) and poured onto LB agar plates to generate uniform bacterial lawns. High-titer phage suspensions (~ 10⁸ PFU/mL) were spotted (15 µL per spot) onto the lawns. Each phage-host combination was tested using three technical replicates, and the entire assay was repeated using three independent biological replicates conducted on separate days. Plates were incubated at 28 °C for 18–24 h before scoring. Phage infection was defined by the reproducible formation of visible lysis zones at the spot site. Clear or turbid lysis zones were recorded as positive interactions, whereas the absence of visible lysis was recorded as resistance.
To quantitatively validate host susceptibility, efficiency of plating (EOP) was determined for strains that produced lysis in spot assays. For each phage-host pair, phage stocks were serially 10-fold diluted in SM buffer, and 100 µL of each dilution was plated using the double-layer agar method as described above (100 µL host culture in 5 mL top agar). Plates were incubated at 28 °C for 18–24 h, and plaque-forming units (PFU/mL) were calculated from countable dilutions. EOP was defined as the ratio of PFU/mL on the test strain to PFU/mL on the phage propagation host (primary isolation host) assayed in parallel on the same day. EOP values were averaged across technical replicates within each experiment, and the assay was repeated across three independent biological replicates.
Transmission electron microscopy
The morphology of phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission was examined using a Talos L120C transmission electron microscope (Thermo Fisher Scientific) operated at 100 kV. High-titer phage lysates (~ 10⁹ PFU/mL) were prepared from freshly amplified stocks by centrifugation at 10,000 × g for 10 min to remove bacterial debris, followed by filtration through 0.22 μm syringe filters. For grid preparation, 10 µL of each phage suspension was applied to 400-mesh carbon-coated Formvar copper grids and allowed to adsorb for 2 min. Excess liquid was removed with filter paper, and grids were negatively stained with 2% (wt/vol) uranyl acetate for 30 s. After air-drying, grids were briefly rinsed with sterile distilled water to remove excess stain. At least two grids per phage were prepared and imaged independently. Morphological parameters, including capsid diameter and tail length, were measured using ImageJ software (v 1.40) from at least three intact virions per grid (≥ 6 virions per phage). Representative micrographs were captured at multiple magnifications to document structural features.
Multiplicity of infection
The optimal multiplicity of infection (MOI) for phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission was determined following standard protocols with minor modifications. MOI optimization was conducted using Pseudomonas syringae pv. syringae strain 2057, which served as the isolation and propagation host for all three phages, providing a standardized and biologically relevant reference for comparing phage infection dynamics. Overnight bacterial cultures were grown in LB broth at 28 °C with shaking (200 rpm) and adjusted to an OD₆₀₀ of 0.2 (approximately 10⁸ CFU/mL). High-titer phage suspensions (≥ 10⁹ PFU/mL) were prepared, and phage–bacterium mixtures were established across a range of MOIs (10, 1, 10− 1, 10− 2, 10− 3, and 10− 4). Each 500 µL mixture was incubated in LB broth at 28 °C with shaking for 5 h to allow phage replication. Following incubation, cultures were centrifuged (10,000 × g for 5 min), and supernatants were filtered through 0.22 μm syringe filters to remove residual bacterial cells. Phage titers were quantified using the double-layer agar method, in which 100 µL of serially diluted phage suspension was mixed with 100 µL of mid-log-phase bacterial culture in 0.5% LB soft agar and overlaid onto LB agar plates. Plaques were enumerated after overnight incubation at 28 °C. The MOI yielding the highest phage titer (PFU/mL) was defined as the optimal MOI for each phage. All assays were performed in triplicate and repeated on at least two independent occasions to ensure reproducibility.
Thermal and pH viability assay
The stability of phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission was evaluated across a range of temperature and pH conditions. For thermal stability assays, high-titer phage suspensions (~ 10⁸ PFU/mL) were aliquoted into sterile 1.5-mL microcentrifuge tubes and incubated for 24 h at 4, 25, 37, 50, or 60 °C using calibrated temperature-controlled incubators (refrigerated incubator for 4 °C, benchtop incubator for 25 and 37 °C, and dry heat block for 50 and 60 °C). For pH stability assays, phage suspensions were diluted into SM buffer adjusted to pH 1, 3, 5, 7, 9, 11, and 13 using 1 M HCl or NaOH and incubated at room temperature for 24 h. Following incubation, samples were briefly centrifuged to remove precipitates, and residual phage infectivity was quantified using the double-layer agar method. Briefly, 100 µL of serially diluted phage suspension was mixed with 100 µL of mid-log-phase Pseudomonas syringae culture in 0.5% LB soft agar and overlaid onto LB agar plates. Plates were incubated at 28 °C for 18–24 h, after which plaques were enumerated to determine phage titers (PFU/mL). Phage survival under each condition was expressed as a percentage of the initial titer measured at time zero. All assays were performed in triplicate and repeated in at least two independent experiments, and results were averaged across replicates.
Phage adsorption
Adsorption kinetics of phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission were determined using their previously established optimal MOIs and a standardized culture of P. syringae pv. syringae, following established methods with minor modifications53. Exponentially growing bacterial cultures (OD600 ≈ 0.2; ~10⁸ CFU/mL) were mixed with each phage at the optimal MOI and incubated at 28 °C with gentle shaking. For adsorption assays, 100 µL aliquots were withdrawn at 2-min intervals over a 20-min period and immediately centrifuged (12,000 × g, 1 min) to pellet bacterial cells. Supernatants containing unadsorbed phages were serially diluted and quantified using the double-layer agar method. The proportion of unadsorbed phages at each time point was calculated relative to titers at time zero, and adsorption constants were estimated by linear regression. All assays were performed in triplicate and repeated in at least two independent experiments to ensure reproducibility.
Phage DNA extraction, sequencing, and genome assembly
Genomic DNA from purified phage particles was extracted using the Phage DNA Isolation Kit (Norgen Biotek, Thorold, Canada), following the manufacturer’s protocol. DNA concentration and purity were quantified using the Qubit Fluorometric Quantification System (Life Technologies, Carlsbad, CA, USA). Sequencing libraries were prepared and sequenced on the Illumina MiSeq platform using the MiSeq Reagent Kit v2 (500-cycle), generating paired-end reads of 250 base pairs54. To remove the adapters, N bases, and low-quality reads, the raw data were filtered using the Cutadapt program (http://code.google.com/p/cutadapt/) with the following parameters: −q 20 − m 20 -M 140. Quality of the processed reads was assessed with FastQC v0.11.7 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). De novo assembly was performed using SPAdes v3.15.5 with k-mer sizes of 21, 33, and 5555. Low-coverage contigs were discarded to retain a high-confidence assembly. Assembly quality and completeness were evaluated using QUAST v5.1.056 and CheckV vs. 1.0.157,58,59. Genome termini were analyzed with PhageTerm to determine genome packaging and structure48. Functional annotation of the assembled genome was conducted using Pharokka v1.3.060, with further manual curation. Protein-coding genes were predicted using PHANOTATE v1.5.161, and tRNA genes were identified with tRNAscan-SE v2.0.1162. Putative depolymerase genes were identified using the DePolymerase Predictor (DePP) tool63. Comparative genomic analyses were performed using BLASTn against the NCBI nt database to identify the closest reference phages. Pairwise nucleotide identity was calculated using the virus intergenomic distance calculator (VIRIDIC)64 and genomic similarity matrices were generated accordingly. Whole genome synteny among the phages and their closest relatives was examined using clinker and clustermap.js65. The complete genome sequences of the isolated phages are available in GenBank under accession numbers PX694326, PX694328, and PX694327, respectively.
Comparative genomics, phylogenomic, and taxonomic analyses
Comparative genomic analyses were performed using BLASTn searches against the NCBI nucleotide database to identify closely related reference phages. Pairwise nucleotide identity values were calculated using the Virus Intergenomic Distance Calculator (VIRIDIC), and similarity matrices were generated accordingly. Whole-genome comparisons and visualization of shared and unique genomic regions were conducted using BRIG and clinker with clustermap.js to assess gene content conservation and synteny among the three phages and their closest relatives. Reciprocal BLASTP analyses were performed to identify orthologous proteins shared between phage pairs and to quantify amino acid identity among structural, replication, and lysis-associated proteins. Phylogenomic relationships were further evaluated using ViPTree to place the phages within a broader viral proteomic context. To strengthen taxonomic inference, marker-gene phylogenies were constructed using the major capsid protein (MCP) and terminase large subunit (TerL). Homologous MCP and TerL sequences from closely related phages were retrieved from GenBank based on BLASTP searches. Protein sequences were aligned using MAFFT66, and maximum-likelihood phylogenetic trees were inferred using IQ-TREE (vs. 2.2.2.6) with automatic model selection and ultrafast bootstrap and SH-aLRT support67. Taxonomic classification was assigned according to current ICTV taxonomy based on genome-wide nucleotide identity, phylogenomic clustering, and marker-gene phylogenies. These combined analyses were used to evaluate genus- and species-level placement of the three phages within Autographiviridae.
Phage lytic activity against planktonic and biofilm cells
The bactericidal activity of phages vB_PsyP_Mobley, vB_PsyP_Plaza, and vB_PsyP_Mission against planktonic Pss was assessed using a microplate-based growth inhibition assay. Overnight bacterial cultures were grown in LB broth at 28 °C with shaking (200 rpm), adjusted to an OD₆₀₀ of 0.2 (~ 10⁸ CFU/mL), and diluted 1:100 in fresh LB medium. Phage suspensions were prepared at high titer (~ 10⁸ PFU/mL), and bacteria were mixed with phages at varying MOIs (10, 1, 10− 1, 10− 2, 10− 3, and 10− 4). A total volume of 200 µL per well was dispensed into sterile, flat-bottom 96-well microtiter plates. Control wells included bacteria-only (growth control), phage-only (sterility control), and media-only (blank). Plates were incubated at 28 °C in a SpectraMax iD3 microplate reader (Molecular Devices, San Jose, CA, USA) with continuous orbital shaking. Optical density at 600 nm (OD₆₀₀) was recorded every 30 min over a 24 h period. Each treatment was performed in triplicate and repeated in at least two independent experiments. Biofilm inhibition was evaluated using a crystal violet (CV) staining assay in 96-well microtiter plates. P. syringae cultures were grown in LB medium, adjusted to an OD₆₀₀ of 0.8, and 100 µL aliquots were added to each well. Plates were incubated at 28 °C under static conditions for 24 h to allow biofilm establishment. Phages were then added at varying MOIs, and plates were incubated for an additional 24 h. Following incubation, wells were gently washed three times with sterile PBS to remove planktonic cells, stained with 0.1% (w/v) crystal violet for 20 min, rinsed thoroughly, and air-dried. Bound dye was solubilized in 95% ethanol, and absorbance was measured at 595 nm68 using the SpectraMax iD3 microplate reader. Biofilm biomass reduction was expressed relative to untreated controls. All treatments were conducted in triplicate, and assays were repeated independently at least twice.
Statistical analysis
All experiments were performed with three technical replicates and repeated in at least two independent biological experiments unless otherwise stated. Data are presented as mean ± standard deviation (SD). Statistical analyses were conducted using GraphPad Prism (version 9.3.1). Depending on the experimental design, differences among groups were evaluated using one-way or two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons test. Statistical significance was determined at p < 0.05, and significant differences among treatments are indicated by different lowercase letters in the figures.
Data availability
The complete genome sequences of phages vB_PsyP_Mobley, vB_PsyP_Mission, and vB_PsyP_Plaza are available in GenBank (NCBI) under accession numbers PX694326, PX694328, and PX694327, respectively.
References
Alattas, H., Glick, B. R., Murphy, D. V. & Scott, C. Harnessing Pseudomonas spp. for sustainable plant crop protection. Front. Microbiol. 15, 1485197. https://doi.org/10.3389/fmicb.2024.1485197 (2024).
Bisht, N., Dixit, R. & Chauhan, P. S. Pseudomonas. In Compendium of Phytopathogenic Microbes in Agro-Ecology: Vol. 3, Bacteria, Protozoa, Algae and Nematodes (eds Amaresan, N. & Kumar, K.) 125–142 (Springer Nature Switzerland, 2025).
López, M., Roselló Pérez, M. & Palacio-Bielsa, A. Diagnosis and detection of the main bacterial pathogens of stone fruit and almond. Journal of Plant Pathology 92 (2010).
Bultreys, A. & Kałużna, M. Bacterial cankers caused by Pseudomonas syringae on stone fruit species with special emphasis on the pathovars syringae and morsprunorum race 1 and race 2. J. Plant Pathol. 92, 21–33 (2010).
Donati, I. et al. Pseudomonas syringae pv. actinidiae: Ecology, infection dynamics and disease epidemiology. Microb. Ecol. 80, 81–102. https://doi.org/10.1007/s00248-019-01459-8 (2020).
Ajibade, A. & Saghaian, S. Us almond exports and retaliatory trade tariffs. Sustainability 14, 15. https://doi.org/10.3390/su14116409 (2022).
Maguvu, T. E. et al. Phylogenomic analyses and comparative genomics of Pseudomonas syringae associated with almond (Prunus dulcis) in California. PLoS One 19, 24. https://doi.org/10.1371/journal.pone.0297867 (2024).
Maguvu, T. et al. Comprehensive phylogenomic and comparative genomic analysis of Pseudomonas syringae associated with almond in California. Phytopathology 113, 1 (2023).
Kaur, A., Minsavage, G., Potnis, N., Jones, J. & Goss, E. Evolution of copper resistance in Xanthomonas euvesicatoria pv. perforans population. mSystems 9, 16. https://doi.org/10.1128/msystems.01427-24 (2024).
Lamichhane, J. et al. Thirteen decades of antimicrobial copper compounds applied in agriculture. A review. Agron. Sustain. Dev. 38, 18. https://doi.org/10.1007/s13593-018-0503-9 (2018).
Mehmood, M. et al. Impact of antibacterial agents in horticulture: Risks to non-target organisms and sustainable alternatives. Horticulturae 11, 28. https://doi.org/10.3390/horticulturae11070753 (2025).
Tegenaw, A., Sorial, G. & Sahle-Demessie, E. Potential environmental implications of select copper-based fungicide/bactericide employed in world markets. Abstr. Pap. Am. Chem. Soc. 254, 2 (2017).
Kortright, K. E., Chan, B. K., Koff, J. L. & Turner, P. E. Phage therapy: A renewed approach to combat antibiotic-resistant bacteria. Cell Host Microbe 25, 219–232. https://doi.org/10.1016/j.chom.2019.01.014 (2019).
Haq, I. U., Rahim, K. & Paker, N. P. Exploring the historical roots, advantages and efficacy of phage therapy in plant diseases management. Plant Sci. 346, 112164. https://doi.org/10.1016/j.plantsci.2024.112164 (2024).
Pinheiro, L., Pereira, C., Frazão, C., Balcão, V. & Almeida, A. Efficiency of phage φ6 for biocontrol of Pseudomonas syringae pv. syringae: An in vitro preliminary study. Microorganisms https://doi.org/10.3390/microorganisms7090286 (2019).
Cui, L. Z. et al. A comprehensive review on phage therapy and phage-based drug development. Antibiotics 13, 47. https://doi.org/10.3390/antibiotics13090870 (2024).
Buttimer, C. et al. Bacteriophages and bacterial plant diseases. Front. Microbiol. 8, 34. https://doi.org/10.3389/fmicb.2017.00034 (2017).
Fiorillo, A., Frezza, D., Di Lallo, G. & Visconti, S. A phage therapy model for the prevention of Pseudomonas syringae pv. actinidiae infection of kiwifruit plants. Plant Dis. 107, 267–271. https://doi.org/10.1094/pdis-02-22-0348-sc (2023).
Papp-Rupar, M. et al. Impact of phage therapy on Pseudomonas syringae pv. syringae and plant microbiome dynamics through coevolution and field experiments. Environ. Microbiol. 27, e70076. https://doi.org/10.1111/1462-2920.70076 (2025).
Liu, L. et al. Biological characteristics of the bacteriophage LDT325 and its potential application against the plant pathogen Pseudomonas syringae. Front. Microbiol. https://doi.org/10.3389/fmicb.2024.1370332 (2024).
Akbaba, M. & Ozaktan, H. Evaluation of bacteriophages in the biocontrol of Pseudomonas syringae pv. syringae isolated from cankers on sweet cherry (Prunus avium L.) in Turkey. Egypt. J. Biol. Pest Control 31(1), 35. https://doi.org/10.1186/s41938-021-00385-7 (2021).
Skliros, D. et al. In planta interactions of a novel bacteriophage against Pseudomonas syringae pv. tomato. Appl. Microbiol. Biotechnol. 107, 3801–3815. https://doi.org/10.1007/s00253-023-12493-5 (2023).
Samavatian, H. in 4th International Symposium on Pistachios and Almonds. 667–671 (International Society Horticultural Science, 2006).
Lam, J., Taylor, V., Islam, S., Hao, Y. & Kocíncová, D. Genetic and functional diversity of Pseudomonas aeruginosa lipopolysaccharide. Front. Microbiol. 2, 25. https://doi.org/10.3389/fmicb.2011.00118 (2011).
Quiroz-Morales, S., García-Reyes, S., Ponce-Soto, G., Servín-González, L. & Soberón-Chávez, G. Tracking the origins of Pseudomonas aeruginosa phylogroups by diversity and evolutionary analysis of important pathogenic marker genes. Diversity 14(5), 18. https://doi.org/10.3390/d14050345 (2022).
Camejo, P. et al. A machine learning approach to predict strain-specific phage-host interactions. Sci. Rep. 15, 14. https://doi.org/10.1038/s41598-025-22075-2 (2025).
Dunne, M. et al. Reprogramming bacteriophage host range through structure-guided design of chimeric receptor binding proteins. Cell Rep. 29, 1336. https://doi.org/10.1016/j.celrep.2019.09.062 (2019).
McInerney, J. Classifying convergences in the light of horizontal gene transfer: Epaktovars and xenotypes. Mol. Biol. Evol. 42, 7. https://doi.org/10.1093/molbev/msaf279 (2025).
Yebra, G. et al. Bacteriophage-driven emergence and expansion of Staphylococcus aureus in rodent populations. Plos Pathogens 20, 21. https://doi.org/10.1371/journal.ppat.1012378 (2024).
Witte, S. et al. Structural and functional characterization of the receptor binding proteins of Escherichia coli O157 phages EP75 and EP335. Comput. Struct. Biotechnol. J. 19, 3416–3426. https://doi.org/10.1016/j.csbj.2021.06.001 (2021).
Farooq, T. et al. Deploying viruses against phytobacteria: Potential use of phage cocktails as a multifaceted approach to combat resistant bacterial plant pathogens. Viruses 14, 27. https://doi.org/10.3390/v14020171 (2022).
Silva, E. C. et al. Control of coffee canker associated with Pseudomonas coronafaciens pv. garcae using a cocktail integrating two virulent polyvalent bacteriophages encapsulated in nanoparticles: In planta studies. J. Appl. Microbiol. 136, 18. https://doi.org/10.1093/jambio/lxae310 (2025).
Silva, E. C., Rodrigues, L. M. R., Vila, M. & Balca, V. M. Newly isolated phages preying on Pseudomonas syringae pv. garcae: In vitro and ex vivo inactivation studies in coffee plant leafs. Enzyme Microb. Technol. 171, 16. https://doi.org/10.1016/j.enzmictec.2023.110325 (2023).
Ul Haq, I., Khan, M. & Khan, I. Phytopathological management through bacteriophages: Enhancing food security amidst climate change. J. Ind. Microbiol. Biotechnol. 51, 12. https://doi.org/10.1093/jimb/kuae031 (2024).
Kaneko, T. et al. From phenotype to receptor: Validating physiological clustering of Escherichia coli phages through comprehensive receptor analysis. J. Virol. 99, 21. https://doi.org/10.1128/jvi.01061-25 (2025).
Selpiev, Z., Leptihn, S., Müsken, M. & Loh, B. Deciphering shared receptor usage in genomically unrelated bacteriophages infecting hypervirulent Klebsiella pneumoniae K1 ST23. FEMS Microbes https://doi.org/10.1093/femsmc/xtaf014 (2025).
Nga, N. et al. Phage biocontrol of bacterial leaf blight disease on Welsh onion caused by Xanthomonas axonopodis pv. allii. Antibiotics 10, 14. https://doi.org/10.3390/antibiotics10050517 (2021).
Vanneste, J. & Paulin, J. in 5th International Workshop on Fireblight. 95–98 (Int Soc Horticultural Science, 1992).
Colvin, K. et al. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ. Microbiol. 14, 1913–1928. https://doi.org/10.1111/j.1462-2920.2011.02657.x (2012).
Molina-Santiago, C., de Vicente, A. & Romero, D. Bacterial extracellular matrix as a natural source of biotechnologically multivalent materials. Comput. Struct. Biotechnol. J. 19, 2796–2805. https://doi.org/10.1016/j.csbj.2021.05.008 (2021).
Muthuraman, S., Flint, S. & Palmer, J. Characterization of the extracellular polymeric substances matrix of Pseudomonas biofilms formed at the air-liquid interface. Food Biosci. 64, 13. https://doi.org/10.1016/j.fbio.2025.105918 (2025).
Yang, Y. et al. Inactivation of Pseudomonas aeruginosa biofilms by 405-nanometer-light-emitting diode illumination. Appl. Environ. Microbiol. 86, 13. https://doi.org/10.1128/aem.00092-20 (2020).
Cornelissen, A. et al. The T7-related Pseudomonas putida phage φ15 displays virion-associated biofilm degradation properties. Plos One https://doi.org/10.1371/journal.pone.0018597 (2011).
Hughes, K. A., Sutherland, I. W., Clark, J. & Jones, M. V. Bacteriophage and associated polysaccharide depolymerases - novel tools for study of bacterial biofilms. J. Appl. Microbiol. 85, 583–590. https://doi.org/10.1046/j.1365-2672.1998.853541.x (1998).
Latka, A., Maciejewska, B., Majkowska-Skrobek, G., Briers, Y. & Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 101, 3103–3119. https://doi.org/10.1007/s00253-017-8224-6 (2017).
Hirano, S. & Upper, C. Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae - a pathogen, ice nucleus, and epiphyte. Microbiol. Mol. Biol. Rev. 64, 624. https://doi.org/10.1128/mmbr.64.3.624-653.2000 (2000).
Monier, J. & Lindow, S. Differential survival of solitary and aggregated bacterial cells promotes aggregate formation on leaf surfaces. Proc. Natl. Acad. Sci. U. S. A. 100, 15977–15982. https://doi.org/10.1073/pnas.2436560100 (2003).
Garneau, J., Depardieu, F., Fortier, L., Bikard, D. & Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. https://doi.org/10.1038/s41598-017-07910-5 (2017).
Hyde, J. et al. Diversity and conservation of the genome architecture of phages infecting the Alphaproteobacteria. Microbiol. Spectr. https://doi.org/10.1128/spectrum.02827-23 (2024).
Lokareddy, R., Hou, C., Li, F., Yang, R. & Cingolani, G. Viral small terminase: A divergent structural framework for a conserved biological function. Viruses 14, 18. https://doi.org/10.3390/v14102215 (2022).
Jones, J. B., Obradovic, A. & Balogh, B. Considerations for Using Bacteriophages in Plant Pathosystems. (2014).
Ke, D. et al. Advancements in bacteriophages for the fire blight pathogen Erwinia amylovora. Viruses-Basel 16, 26. https://doi.org/10.3390/v16101619 (2024).
Summer, E. J. et al. Genomic and biological analysis of phage Xfas53 and related prophages of Xylella fastidiosa. J. Bacteriol. 192, 179–190. https://doi.org/10.1128/jb.01174-09 (2010).
Steemers, F. J. & Gunderson, K. L. Illumina, Inc. Pharmacogenomics 6, 777–782 (2005). https://doi.org/10.2217/14622416.6.7.777
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19(5), 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086 (2013).
Nayfach, S. et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39(5), 578–585. https://doi.org/10.1038/s41587-020-00774-7 (2021).
Ghanaim, A. M. et al. Bacteriophage therapy as an alternative technique for treatment of multidrug-resistant bacteria causing diabetic foot infection. Int. Microbiol. 26, 343–359. https://doi.org/10.1007/s10123-022-00293-2 (2023).
Aiewsakun, P., Adriaenssens, E. M., Lavigne, R., Kropinski, A. M. & Simmonds, P. Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: Steps towards a unified taxonomy. J. Gen. Virol. 99(9), 1331–1343. https://doi.org/10.1099/jgv.0.001110 (2018).
Bouras, G. et al. Pharokka: A fast scalable bacteriophage annotation tool. Bioinformatics https://doi.org/10.1093/bioinformatics/btac776 (2023).
McNair, K., Zhou, C., Dinsdale, E. A., Souza, B. & Edwards, R. A. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 35, 4537–4542. https://doi.org/10.1093/bioinformatics/btz265 (2019).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. TRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. https://doi.org/10.1093/nar/gkab688 (2021).
Magill, D. J. & Skvortsov, T. A. DePolymerase predictor (DePP): A machine learning tool for the targeted identification of phage depolymerases. Bmc Bioinformatics 24, 11. https://doi.org/10.1186/s12859-023-05341-w (2023).
Moraru, C., Varsani, A. & Kropinski, A. M. VIRIDIC-a novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses-Basel 12, 10. https://doi.org/10.3390/v12111268 (2020).
Gilchrist, C. L. M. & Chooi, Y. H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 37, 2473–2475. https://doi.org/10.1093/bioinformatics/btab007 (2021).
Katoh, K. & Standley, D. MAFFT: Iterative Refinement and Additional Methods In (ed. Russell, D. J.) (2014).
Minh, B. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. https://doi.org/10.1093/molbev/msaa015 (2020).
Fong, S. A. et al. Activity of bacteriophages in removing biofilms of Pseudomonas aeruginosa isolates from chronic rhinosinusitis patients. Front. Cell. Infect. Microbiol. 7, 11. https://doi.org/10.3389/fcimb.2017.00418 (2017).
Acknowledgements
We thank Dr. Caroline Roper (Department of Microbiology and Plant Pathology, University of California, Riverside) for generously providing the non-almond Pseudomonas isolates used in this study.
Funding
This study was supported by an initial complement package provided to O.I.O. by the College of Natural and Agricultural Sciences, University of California, Riverside.
Author information
Authors and Affiliations
Contributions
O.I.O. conceived and supervised the study, designed the experimental framework, and provided oversight throughout the project. C.V.H., J.F., H.L., L.B., D.N., A.D., and S.G. carried out the laboratory experiments. C.V.H. and O.I.O. performed data processing, analysis, and interpretation. C.V.H., O.I.O., J.F., L.B., D.N., A.D., R.Q., S.G., and L.S. contributed to manuscript drafting and revisions. J.E.A., T.E.M., and F.P.T. supplied materials, provided guidance with *Pseudomonas* cultivation and maintenance, and provided critical feedback on experimental design and manuscript development. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hoang, C.V., Fan, J., Lee, H. et al. Genomic and biological characterization of lytic phages infecting Pseudomonas syringae associated with almond bacterial blast. Sci Rep 16, 11657 (2026). https://doi.org/10.1038/s41598-026-47496-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-47496-5
