
Abstract
Management of advanced hormone receptor-positive, HER2-negative breast cancer after progression on endocrine therapy plus CDK4/6 inhibitors is challenging due to mutational heterogeneity. Current therapies yield limited efficacy, achieving 4–6 months PFS. FGFR signaling is implicated in resistance to endocrine plus CDK4/6 inhibitors, but FGFR inhibitors have shown limited activity in unselected populations. Co-clinical trials bridge preclinical and clinical findings, optimize resources, and enable biomarker identification. Using patient-derived organoids (PDOs), we demonstrated that FGFR-amplified PDOs respond to fulvestrant, palbociclib, and rogaratinib only when PIK3CA and ESR1 are wild-type. In a dose-escalation trial pre-screening 66 patients with FGFR1/2-amplification (FISH) and/or overexpression (RNAScope) patients, >40% harbored FGFR alterations. Nine patients were enrolled; the combination showed activity specifically in PIK3CA- and ESR1-wild type patients (9.1 vs. 1.9 months PFS; P = 0.0005). Toxicity was manageable and consistent with prior data. Our findings highlight biomarker-driven approaches as essential for refining FGFR-targeted strategies in this resistant population.
Similar content being viewed by others

Introduction
The standard of care for advanced or metastatic hormone receptor-positive breast cancer (HR + MBC) in the first-line metastatic setting has been transformed by the introduction of CDK4/6 inhibitors, which, in combination with endocrine therapy, benefit the majority of patients1,2,3. However, in the second-line setting the disease exhibits a more heterogeneous behavior4. The drug development landscape in this setting is currently focused on novel inhibitors targeting endocrine or CDK4/6-inhibitors resistance mechanisms5, including mutant ESR16 or PIK3CA7, and on drugs aimed at cell surface targets, such as HER28 or TROP29. Here, the challenge is to extend progression-free survival (PFS) intervals to 6–12 months in a broad patient population using oral therapies.
FGFR1/2 amplification is a frequently observed genomic aberration in this clinical context10. Progress in developing effective therapies for this target in breast cancer has been limited. Studies, including our own, have reported over a 10% incidence of FGFR1/2 amplification—primarily FGFR1—in early hormone-receptor-positive breast cancer11,12,13. This amplification correlates with a two- to fourfold increased risk of metastatic relapse and resistance to endocrine therapy, suggesting that such amplifications are more prevalent among metastatic cases11,13,14,15. Additionally, our research indicates that patients with elevated FGFR1/2 expression, as measured by RNAScope, exhibit a comparable long-term prognosis to those with amplification, irrespective of actual amplification status12. A significant concordance has been observed between FGFR amplification as determined by FISH and overexpression identified by RNAScope, although many cases display only one or the other alteration12. Approximately 25% of early hormone-receptor positive patients exhibit either amplification or overexpression of FGFR1/2, each correlating with poorer clinical outcomes12. In the metastatic setting, sub-analyses of the BOLERO216 or MONALEESA-217 trials suggest that FGFR amplification decreases the PFS expectancy of standard-of-care treatment combinations with MTOR or CDK4/6 inhibitors respectively, highlighting this patient sub-population as a difficult-to-treat breast cancer subtype.
So far, FGFR inhibitors have been used alone or in combination with endocrine therapy in breast cancer, leading to modest clinical outcomes18,19,20,21. Our preclinical research suggests several critical insights that could explain this situation: 1) FGFR1 amplification is directly linked to resistance to endocrine therapy; 2) extended treatment with endocrine therapy favors the emergence of FGFR1 amplification; 3) the presence of FGFR1 amplification diminishes the effectiveness of endocrine therapy when combined with CDK4/6 inhibitors; 4) resistance developed from endocrine therapy combined with CDK4/6 inhibitors, involves FGFR1 amplification; and 5) complete inhibition of RB phosphorylation in presence of FGFR1 amplification is attainable exclusively with the combined use of CDK4/6, ER, and FGFR inhibitors12.
Considering the significant incidence of FGFR amplification and overexpression in HR + MBC, and its association with resistance to endocrine therapy and CDK4/6 inhibitors, we hypothesized that a triple blockade of CDK4/6, estrogen receptor (ER), and FGFR could be beneficial. This approach could potentially address a substantial segment of the HR + MBC population. Inhibition of RB phosphorylation, completely achieved through this combination, may result in sustained disease control in this challenging clinical context. With this hypothesis, we set out to investigate rogaratinib—a pan-FGFR inhibitor initially developed for FGFR-amplified or mutant urothelial cancers22—in FGFR1/2 amplified or overexpressed HR + MBC patients who progressed after first-line therapy with aromatase inhibitors and CDK4/6 inhibitors. Given the diverse mutational landscape and the influence of mutations like ESR1 or PIK3CA on treatment response to different drug combos, we initiated a co-clinical trial combining patients and patient-derived organoids (PDOs). This research design, involving concurrent patient treatment and model testing, aims to expedite and enhance the translation of laboratory discoveries to clinical practice. By employing the same therapeutic strategies in clinical and model environments, we can swiftly assess treatment efficacy and safety, modify protocols based on real-time data, and make informed predictions about the success of these therapies in a broader patient cohort, thus conserving resources and minimizing patient exposure to suboptimal treatments. Our objectives included: 1) determining the prevalence of FGFR1/2 amplification or overexpression in HR + MBC after CDK4/6 inhibitor and endocrine therapy; 2) delineating the mutational landscape of HR + MBC that is responsive to the triple-drug regimen using PDOs; 3) evaluating the initial efficacy and tolerability in patients of the combination of rogaratinib, fulvestrant and palbociclib based on the underlying mutational profile.
Results
Patient-derived organoids description
Four PDOs (H12_5, H12_7, H12_28 and H12_31) were used for this study, all of them established from HR + MBC patients right after disease progression to a CDK4/6 inhibitor in combination with endocrine therapy administered in the metastatic setting. H12_5 and H12_7 PDOs were established from breast lesions; H12_28 was established from a liver metastasis, and H12_31 was established from a pleural metastasis. Patients 5 and 7 were de novo metastatic patients, whereas 28 and 31 were metastatic relapses from a stage IIA and a stage IA primary tumors, respectively. Treatment regimens along the patients’ medical history are summarized in Fig. 1A; all PDOs were obtained from patients progressing to first-line endocrine therapy combined with a CDK4/6i, with the exception of H12_31, who received the CDK4/6 inhibitor after multiple endocrine therapy lines. The tumor biopsy obtained at the moment of PDO establishment revealed that H12_5, H12_7, H12_28 were Luminal-B tumors, whereas H12_31 was a Luminal-A tumor (Fig. 1B). Three of them (H12_5, H12_7 and H12_31) displayed FGFR1 amplification, whereas H12_28 was not amplified (for either FGFR1 or FGFR2; Fig. 1B, C). ER, PR, Ki67 immunohistochemistry and hematoxylin/eosin staining for the 4 PDOs are shown in Fig. 1B as well. The panel shown in Fig. 1C displays the molecular profiling regarding the FGFR1/2 status and top-20 mutated genes in advanced breast cancer4. As it can be observed, among the 3 amplified PDOs, one each harbored one PIK3CA activating mutation and ESR1 activating mutation, respectively; the third one was wild type for both genes. Finally, the FGFR-non amplified PDO was as well wild-type for both genes, but displayed two AKT mutations and one deleterious mutation in NF1, a mutation previously associated with acquired resistance to endocrine therapy23.

A Schematic representation of the clinical course of the patients from whom the CDKi- and endocrine-resistant PDOs were established, including the treatment lines and biopsy site, as well as basic demographic data. B Hematoxylin & Eosin and immunohistochemical staining (ER, PR and Ki67) of the four PDOs. FGFR1/2 status (amplification, demonstrated by FISH, and/or overexpression, demonstrated by RNAScope) is also shown. C FGFR1/2 and mutational status of the top-20 mutated genes in advanced breast cancer among the amplified and non-amplified PDOs. Those genes that were wild-type in all PDOs are not shown.
Patient-derived organoids’ response to single, double, and triple-therapy combinations by genomic profile
Figure 2A, B show the effect on viability of fulvestrant and palbociclib, respectively, over the 3 FGFR-amplified/overexpressed and the one non-amplified/overexpressed PDOs, in accordance with their endocrine/CDK4/6-resistant clinical profile. As expected, rogaratinib monotherapy was devoid of meaningful activity as well (Fig. 2C). The effects of the triple blockade, fixing fulvestrant and palbociclib dose at 1 μM each, and exposing the PDOs to increasing rogaratinib doses, are shown in Fig. 2D. It can be observed that the IC50 for H12_7, significantly lower than for the other organoids (FGFR-amplified and wild-type for ESR1 and PIK3CA). The control, non-amplified PDO (H12_28) did not display specific sensitivity to any drug or combination, suggesting that FGFR amplification/overexpression is a key determinant for drug efficacy.

FGFR-amplified and non-amplified PDOs were exposed to increasing concentrations of fulvestrant (A), palbociclib (B), and rogaratinib (C) monotherapy; IC50 was not reached (NR) for palbociclib or fulvestrant monotherapy in any organoid. H12-7 was moderately sensitive to rogaratinib monotherapy. D PDOs were exposed to fixed concentrations of fulvestrant and palbociclib (1 μM each) and increasing concentrations of rogaratinib; IC50s are shown for rogaratinib in the presence of fulvestrant+palbociclib. E Efficacy of increasing concentrations of camizestrant over a backbone of fixed palbociclib and rogaratinib in the ESR1-mutant PDO. F Efficacy of increasing concentrations of alpelisib over a backbone of fixed fulvestrant and rogaratinib in the PIK3CA-mutant PDO. IC50 values displayed in (E) and (F) correspond to the IC50 of camizestrant and alpelisib, respectively, over fixed backbone doublets. Error bars: standard error.
If, instead of fulvestrant, a drug targeting the ESR1 Y573S activating mutation (camizestrant) is used, H12_31 (ESR1-mutant) becomes sensitized (Fig. 2E) to the combo. Similarly, if palbociclib is substituted by PIK3CA H1047R-targeting drug alpelisib, the same effect is observed in the PIK3CA-mutant PDO (H12_5; Fig. 2F).
The former data highlighting the role of ESR1 and PIK3CA mutations in treatment resistance, suggesting that an FGFR inhibitor-based combo is only active in case both ESR1 and PIK3CA are wild-type.
Clinical trial and patients
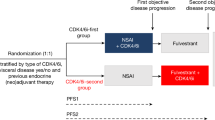
The ROGABREAST trial was designed in light of the findings observed in the PDOs. Candidate patients could be pre-screened during their first-line treatment or screened right after experiencing disease progression to first-line treatment with the combination of a CDK4/6 inhibitor and an aromatase inhibitor. Patients displaying FGFR1 and/or FGFR2 amplification (by FISH) and/or mRNA overexpression (by RNAScope) were eligible for the trial. The screening results are shown in Fig. 3A. Twenty-nine out of 67 screened patients (43%) were positive for at least one of the four tests. Among screening-positive patients, 15 patients were positive in more than one test, and the most frequent phenotype was FGFR1-amplified/FGFR1-overexpressed (12/29 cases, 41.4%; Fig. 3B). A representative FISH/RNAScope image for each test result combination is shown in Fig. 3B as well. The concordance between FISH and RNAScope positivity was far from 1:1, as expected from our previous results12, with several patients testing positive in only one assay. This highlights the importance of performing both tests to detect all potentially responsive patients. The trial schedule is depicted in Fig. 3C, and consisted in treatment with rogaratinib twice per day on days 1–28, palbociclib once per day on days 1–21, and fulvestrant q28 days (with a second dose on day 15 in the first cycle). Fulvestrant and palbociclib doses were fixed at 500 ug and 100 mg/day, respectively (scalable to 125 in the second cycle in case of good tolerance). Rogaratinib dosage started at 400 mg/bid (Level 1). The first enrolled patient presented with grade 2 diarrhea in the urgent care room the 8th treatment day at night; urgent care room physicians discharged the patient with a loperamide prescription without interrupting treatment drugs. Three days later the patient was seen by the site IP, and had to be admitted for I.V. hydration because of the increasing diarrhea symptoms. Qualifying as a grade 3 event, a potential DLT event could have not been discarded since the drug was not interrupted as per protocol during the urgent care visit. Accordingly, Level 1 was expanded to 6 patients. No further patient experienced a DLT, and Level 2 was opened (rogaratinib 600 mg bid). When 3 patients were enrolled, and despite no DLTs had been observed, Bayer decided to discontinue the development of rogaratinib and the study was closed; patients continued on medication until disease progression. Table 1 describes the main clinical and demographic characteristics of the 9 enrolled patients, and Table 2 gathers the toxicity summary observed in the trial during the 49 administered treatment cycles. No grade 4 toxicities were registered, and, in general, the toxicities were manageable with support measures and dose interruptions and/or reduction. The most frequent toxicity attributed to rogaratinib was hyperphosphatemia, whereas the one most frequently attributed to palbociclib was neutropenia. No distinction was made by investigators between palbociclib and rogaratinib as a cause for incident diarrhea. The individual dose trajectories for rogaratinib and palbociclib are shown in Fig. S1 (no dose modifications or interruptions were undertaken for fulvestrant). Except for one patient, no one was escalated to 125mg palbociclib, which suggests that the RP2D is 600 mg bid rogaratinib (days 1–28) plus 100 mg/d palbociclib (days 1–21) and 500 µg fulvestrant q28 days (two injections on the first cycle).

A CONSORT diagram. B Status of FGFR1 and FGFR2 amplification and overexpression assessed by FISH and RNAScope, respectively, in the patients pre-screened for the trial. The upper table lists the frequency of each test result combination. Representative images for each pattern are shown below. Two contingency tables on the right show the concordance between FISH and RNAScope results for FGFR1 (top) and FGFR2 (bottom), based on patients with valid results for both assays (n = 61 for FGFR1; n = 58 for FGFR2). C Trial treatment schedule.
Efficacy and mutational profiles
The Kaplan–Meier plot in Fig. 4A shows the PFS time registered for the nine patients. Median PFS was 3.5 months (95% C.I.: 0.11–6.8 months). No RECIST V.1.1 responses were observed; six patients (66.7%) experienced stable disease, and 2 (22.2%) progressive disease as best response. One patient was non-evaluable (premature withdrawal because of DLT, at day +11). The mutational landscape of the nine patients is shown in Fig. 4B. Overall, five patients displayed PIK3CA and/or ESR1 mutations in the tumor or plasma sample. The swimmers plot in Fig. 4C depicts the disease control duration of patients with or without mutations in those two genes, evidencing that only those that were wild-type for both made it beyond the 6-month mark. In addition, the panel grid on the left-hand side depicts the FGFR1 and FGFR2 status (by FISH and RNAScope) of the nine patients. The Kaplan–Meier curve in 4D compares the PFS time between patients with and without PIK3CA and/or ESR1 mutations, with wild-type patients experiencing a statistically significant, almost fivefold advantage in PFS (274 versus 58 days median PFS; P = 0.005; 95% C.I.: 183.8–364.2 versus 51.6–64.4 days). ERBB2 mutations, although present in several patients, were not associated with PFS (Fig. S2). We did not observe any correlation, however, between the level of amplification and/or overexpression of FGFR1/2 and overall PFS (Fig. S3).

A Kaplan–Meier curve showing the progression-free survival time of all the patients enrolled in the trial. B Mutational status of the top-20 genes most-frequently mutated in breast cancer in the nine enrolled patients. C Swimmers plot. Green: patients wild-type both for ESR1 and PIK3CA. Red: patients with mutation in either PIK3CA and/or ESR1. The status of FGFR1 and FGFR2 (by FISH or RNAScope) is shown for each patient in the left-hand side. D Kaplan–Meier curve comparing the progression-free survival of PIK3CA/ESR1 wild-type versus mutant patients.
Discussion
FGFR1 amplification has been associated with tumor progression features in breast cancer and other malignancies24. Multiple studies, including our own, have demonstrated its link to increased metastatic relapse in early hormone-receptor positive breast cancer, irrespective of treatment type (endocrine therapy alone or combined with chemotherapy) or other prognostic factors (grade, Ki67, nodal status)11,12,13. The identification of FGFR1 as an amplified kinase, coupled with the efficacy of FGFR inhibitors in FGFR-mutated or rearranged bladder25 and cholangiocarcinomas26, initially raised expectations for a new targetable oncogene addiction driver in breast cancer. However, despite promising preclinical data, clinical results have been modest at best. First-generation multi-kinase FGFR inhibitors, such as dovitinib, showed limited efficacy alone or combined with endocrine therapy, accompanied by substantial toxicity18,20. Novel, selective FGFR inhibitors have demonstrated better tolerability but still limited activity in breast cancer19,21. We previously demonstrated that in FGFR1/2-amplified/overexpressed breast cancer, complete cell cycle arrest requires simultaneous inhibition of the ER, CDK4/6, and FGFR12. The post-CDK4/6i clinical setting is highly heterogeneous4, with acquired mutations potentially modulating response to triple-targeted therapies. Conducting trials in this scenario poses challenges, including patient attrition, high resource consumption, and uncertain clinical activity despite biomarker selection. Furthermore, combining three targeted agents raises concerns about toxicity. To address these limitations, we implemented a co-clinical trial design using pPDOs to precisely define the patient population most likely to benefit from FGFR inhibition in combination with ER and CDK4/6 blockade. Co-clinical trials enhance translational relevance by enabling direct patient correlation, real-time protocol adaptation based on emerging data, and improved biomarker-driven patient selection.
To generate a PDO collection relevant for this co-clinical study, we adopted a pragmatic approach, focusing on the most frequent resistance mechanisms and available therapies for future studies. PIK3CA activating mutations, present in ~30% of HR + MBC4, are associated with resistance to endocrine therapy and CDK4/6 inhibitors. Similarly, ESR1 activating mutations, found in up to 50% of cases27, predict failure of aromatase inhibitors and fulvestrant. Given that FGFR1-amplified breast cancer cells require endocrine therapy and CDK4/6 inhibition for effective proliferation suppression, it is plausible that PIK3CA- and/or ESR1-mutant tumors would not respond to the triple combination therapy. From a collection of >15 breast cancer PDOs established in our study (data not shown), we selected representative models of distinct mutational combinations: H12-5 (PIK3CA-mutant, ESR1-wild type), H12-31 (PIK3CA-wild type, ESR1-mutant), and H12-7 (PIK3CA-wild type, ESR1-wild type), all of which were FGFR1-amplified/overexpressed. H12-28, a non-amplified, PIK3CA-wild type, ESR1-wild type model, served as a control. As anticipated, the therapeutic results aligned with our predictions, with only H12-7 demonstrating sensitivity to the triple combination (Fig. 2D). Also, targeting mutant ESR1 or PIK3CA, alongside FGFR inhibition for mutant tumors, could represent alternative strategies for FGFR-amplified patients following progression on CDK4/6 inhibitors. This approach merits further investigation in clinical settings (Fig. 2E, F).
As expected, the clinical trial included a diverse mix of patients based on the mutational status of PIK3CA and ESR1 (Fig. 4B). FGFR-amplified patients are known to have a poor prognosis even in the CDKi+AI first-line setting17. In our screened population, patients with FGFR1/2 amplification or overexpression had a median PFS of only 14.9 months in the first line (Table 1). In the second-line setting, current standards of care for non-FGFR-amplified patients have historically failed to surpass the 6-month PFS threshold. The recent EMBER trial with imlunestrant achieved promising results, but no subset analysis has been provided for FGFR-amplified/overexpressed patients28. Consequently, the median PFS for this subgroup in the second line, with or without imlunestrant, remains unknown but likely low, as reflected in our data (Fig. 4A). The 3.5-month median PFS observed in our trial cohort is far from remarkable. However, exploratory analysis of the cohort using PDO co-clinical data revealed an improved outcome in the PIK3CA-wild type, ESR1-wild type subgroup (Fig. 4C, D; 9.1 vs. 1.9 months; P = 0.005). Although these results are based on a limited sample size and should be interpreted with caution, they suggest a potentially actionable biomarker signature that may help refine patient selection in future trials in the post-CDK4/6 inhibitor setting. These results should not be interpreted as definitive evidence for the predictive role of PIK3CA and ESR1 mutations, but rather as a hypothesis-generating observation that warrants further prospective validation. Critics may argue that the PDO collection was limited, and we fully acknowledge this limitation. Nonetheless, the concordance between preclinical and clinical data, together with a statistically significant association in an internally consistent dataset, provides preliminary support for the biological relevance of these findings. Still, larger, independent cohorts will be required to confirm these results and formally establish the predictive value of these mutations. Finally, other mutations potentially impacting the therapeutic regimen include AKT (observed in two patients). However, the specific variants (R121W and R406C) were not among the well-characterized activating mutations (e.g., E17K), and their functional impact remains uncertain. These findings highlight the complexity of signaling cross-talk and underscore the need for further studies to delineate the role of less frequent mutations in modulating therapeutic response.
Of note, our study revealed partial concordance between FGFR1/2 amplification (FISH) and mRNA overexpression (RNAScope), a finding consistent with our prior observations in early breast cancer12. This reflects the known biological divergence between genomic amplification and transcriptional output, and mirrors similar observations in other oncogenes such as HER2, where mRNA and FISH correlate imperfectly29,30,31. Importantly, both alterations were associated with poor prognosis and predicted response to FGFR inhibition in our previous study. We therefore propose that FISH and RNAScope serve as complementary biomarkers for patient selection, maximizing the likelihood of therapeutic benefit in FGFR-driven tumors.
Triple-targeted therapies have historically been only moderately feasible in clinical settings. To the best of our knowledge, this study represents the first published trial combining endocrine therapy and CDK4/6 inhibition with FGFR blockade. Importantly, no new emergent toxicities from potential drug-drug interactions were observed (Table 2). The most frequent adverse events, neutropenia and hyperphosphatemia, were consistent with known class effects of CDK4/6 and FGFR inhibitors, respectively. These findings suggest that a triple combination of a SERD, CDK4/6 inhibitor, and FGFR inhibitor is feasible and well-tolerated in principle, supporting its consideration for future trials.
This study has several limitations. First, the small sample size of both the clinical trial cohort and the PDO collection may restrict the generalizability of our results. Larger datasets will be required to confirm these observations. Second, the trial was discontinued prematurely due to sponsor-driven strategic decisions, precluding further validation of the observed clinical trends. Third, although the co-clinical trial design offers significant advantages, it does not fully recapitulate the tumor microenvironment, which could influence treatment responses beyond the capabilities of PDO models.
Our findings underscore the relevance of the FGFR-amplified/overexpressed population, which comprises 40–50% of HR + MBC cases (Fig. 3A). This subgroup faces a particularly poor prognosis16. By leveraging a co-clinical trial design, we have identified and preliminarily characterized a biomarker-defined subset—FGFR1/2-amplified/overexpressed, PIK3CA-wild type, ESR1-wild type—that is most likely to benefit from triple-targeted therapies. This approach may serve as a rationale for biomarker-enriched trial design rather than as a definitive patient stratification at this stage. Future prospective trials could prioritize this population to validate the efficacy of combining FGFR inhibitors with SERDs and CDK4/6 inhibitors. Additionally, the development of next-generation FGFR inhibitors with improved selectivity and tolerability could further enhance the feasibility of triple combinations. The co-clinical trial framework demonstrated here provides a compelling model for accelerating drug development in breast cancer. Collectively, our findings establish a rationale for future registration trials, ultimately aiming to advance FGFR-targeted therapies toward clinical implementation and improve outcomes for this difficult-to-treat breast cancer population.
Methods
Clinical trial design
This trial was a phase I, open-label, multicenter trial (ClinicalTrials.gov: NCT04483505) conducted at eight centers in Spain between November 2020 and May 2023. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice standards. Ethics approval was obtained from the Madrid Regional Government Ethics Board for Drug Research and the Spanish Agency for Medicine and Health Products (AEMPS; protocol number 20/392). Patients were pre-screened for the trial; those patients eligible for trial inclusion were then tested for FGFR1/2 amplification and overexpression. Patients could be screened during their previous treatment line. The main inclusion criteria for pre-screening were as follows: 1) Women >18 years-old; 2) diagnostic of metastatic or locally advanced non-resectable breast cancer; 3) hormone-receptor positivity defined by at least 5% positivity of ER and/orPR (no central laboratory testing was required) and HER2-negativity (Herceptest 0+, 1+ or 2+ with negative FISH/CISH/SISH); 4) patients must have undergone a previous aromatase inhibitor treatment line for metastatic disease, with anastrozole, letrozole or exemestane, plus a cell cycle inhibitor (palbociclib, ribociclib or abemaciclib); 5) recovery of toxicities from previous regimens to equal or below tolerable grade 2. 6) ECOG performance status of 0/1 and life expectancy of >24 weeks; 7) adequate organ function according to usual criteria. Patients fulfilling the inclusion criteria were tested for FGFR1/2 amplification (by FISH) or overexpression (RNAScope) in a recent tumor sample. Positivity in either of the 4 tests allowed full screening and trial inclusion; demonstration of disease progression (RECIST 1.1) on a CDK inhibitor in combination with a CDK inhibitor was mandatory prior to trial enrollment.
Treatment consisted of a fulvestrant injection (500 ug IM) every 4 weeks (two injections on days 1 and 15 in cycle 1) combined with continuous rogaratinib and palbociclib from day 1 to 21 in 28-day cycles. Rogaratinib dosage started at 400 mg twice per day, orally (Level 1); palbociclib started at 100 mg/day. Rogaratinib escalation took place in 200 mg/bid increments following a classic 3 + 3 schedule. Intra-patient palbociclib dose-escalation was allowed to 125 mg/day in cycle 2 in case no significant tolerance problems were observed according to investigators’ criteria. Toxicity was graded according to the NCI CTC AE V. 5.0. Two and one dose-reductions were allowed for rogaratinib and palbociclib, respectively; no dose-reductions were planned for fulvestrant. Treatment continued until disease progression, incidence of significant toxicity or intercurrent disease, or investigators’ decision. RECIST V.1.1 evaluation took place every 8 weeks.
Patient-derived organoids establishment and culture
Patient-derived organoids were established and maintained as described previously32. In summary, fresh tumor biopsies were placed in 10 mL sterile ice-cold base media (Advanced DMEM-F12 (Gibco, 12634010) supplemented with 5% FBS (Biowest, S1810), 1× Pen-Strep (Gibco, 15140-122) and 10 mM HEPES (Gibco, 15630056) and kept on ice until processing. For reliable organoid generation, it was imperative that the tissue was processed immediately, as a prolonged time between surgical removal and tissue processing reduced the reliability of PDO generation. Tissue was washed with ADF+++ media (Advanced DMEM-F12 containing 1× Glutamax (Gibco, 35050061), 10 mM HEPES and 1× Pen-Strep), manually cut into smaller pieces (1–3 mm3) and digested with 1.5 mg/ml collagenase from Clostridium histolyticum (Sigma C9407) on an orbital shaker at 37 °C for 90 mins. Intermittent vortexing every 15 min is recommended to ensure proper digestion of the tissue. The digested tissue suspension was strained over a 100 μM filter and centrifuged at 3505 × g for 5 mins at room temperature (RT). In case of a visible red pellet, erythrocytes were lysed in 2 mL red blood cell lysis buffer (Invitrogen, 00-4333-57) for 5 min at room temperature before the addition of 10 ml of ADF+++ and centrifugation at 350 g for 5 min at RT. The pellet was resuspended in 100% cold Matrigel (Cultrex, RND-3533-010-02) and 45μL drops were plated in a pre-warmed 24-well culture plate (Corning, 3524) and allowed to solidify at 37°C for 30 min. Upon completed gelation, 530 μl of pre-warmed completed-medium (10% R-Spondin 3 (Peprotech, 120-44), 5 nM Neuregulin 1 (Pepotech 100-03), 5 ng/ml FGF7 (Peprotech, 100-19), 20 ng/ml FGF10 (Peprotech, 100-26), 5 ng/ml EGF (Stemcell 78006.1), 100 ng/ml Noggin (Stemcell, 78060), 500 nM A83–01 (Biogen, 2939), 5 μM Y-27632 (Biogen, TO-1254), 1.2 μM SB202190 (Sigma-Aldrich S7067), 1× B27 supplement (Gibco 17504-44), 1.25 mM N-Acetylcysteine (Merck, A9165), 5 mM Nicotinamide (Merck, N0636), 50μg/ml, Primocin (InvivoGen, ant-pm-1) in ADF+++) was added to each well and plates were transferred to humidified 37 °C/ 5% CO2 incubator. Medium was changed every 4 days, and organoids were passaged every 15–30 days.
For passaging, dense organoids were dissociated in 1 mL of TrypLe Express (Gibco 12604-013) by vigorous pipetting and incubated for 5 min at 37 °C. Cells were resuspended in 10 mL of ADF+++ media and centrifugated at 350 × g for 5 min at 4 °C. Cell pellet was resuspended in fresh cold Matrigel and reseeded as above at ratios (1:1 to 1:6) allowing the formation of new organoids.
Organoids treatments and cell viability assays
For cell viability assays, organoids were dissociated in 1 mL of pre-warm TrypLE™, resuspended in 10 mL of cold ADF +++ media and centrifugated at 350 × g for 5 min at 4 °C. Cell pellet was resuspended in 1–2 mL of complete medium, and single cells were counted in a Neubauer chamber. Cells were seeded at a density of 7500 to 10,000 single cells per well in a clear-bottom 96-well plate (Corning) in 50% Matrigel+ complete growth medium. After Matrigel gelation (37 °C for 1 h), 100 μl of completed media was added to each well. Fresh complete growth medium was applied two times a week, and organoids were allowed to form for 2–4 weeks. Then, each organoid line was evaluated for multiple anticancer agents, including fulvestrant (MedChemExpress, HY-13636), palbociclib (Selleckchem, S1116), rogaratinib (MedChemExpress, HY-100019), alpelisib (MedChemExpress, HY-15244) and camizestrant (MedChemExpress, HY-136255). To determine the inhibitory concentration of 50% (IC50) in monotherapy, organoids were exposed to a concentration range of seven drug concentrations of each compound: fulvestrant (0.25 μM–10 μM), palbociclib (0.25 μM–10 μM), rogaratinib (0.75 μM–50 μM), alpelisib (0.25–10 μM) and camizestrant (0.25 μM–10 μM). On the same plate, a medium control and a DMSO control were included. For the triple-therapy combination fulvestrant, palbociclib and rogaratinib, doses of fulvestrant and palbociclib were fixed at 1 μM each and the PDOs to increasing rogaratinib doses (0.75 μM -50 μM). If fulvestrant is substituted by camizestrant, camizestrant was fixed at 10 μM. Similarly, if palbociclib is substituted by alpelisib, it was fixed at 10 μM. After six days, the number of living cells was assessed using CellTiter-Glo® assay (Promega, G9683) according to the modified manufacturer´s instructions for tumor spheroids. The IC50 values were derived by a sigmoidal dose-response (variable slope) curve using GraphPad Prims software version 5.04. All experiments were performed at least in triplicate.
Fluorescence in situ hybridization (FISH) determination and RNAScope
Chromosome enumeration probes for FGFR1 and FGFR2 were acquired from ZytoVision (ZytoLight SPEC FGFR1/CEN8 and ZytoLight SPEC FGFR2/CEN 10 dual color probes). FISH analyses on 5 μm TMA sections mounted on positively charged slides (SuperFrost, Thermo Scientific) were performed as outlined in the manufacturer’s instructions. Briefly, slides were deparaffined in xylene, rehydrated in a graded ethanol series, and treated using the Histology FISH Accessory Kit (DAKO). For paraffin-embedded tissues, pre-treatment involved 2-[N-morpholino]ethanesulphonic acid (MES), whereas organoid samples received no pre-treatment. Protein digestion was performed using a pepsin solution in both cases. After dehydration, samples were denatured with the specific probes, followed by overnight hybridization. Post-hybridizations, slides were washed and mounted using DAPI-containing fluorescence mounting medium. FISH signals were manually enumerated within nuclei all over the tissue. Images were captured using a Photometrics SenSys CCD camera connected to a PC running the Zytovision image analysis system (Applied Imaging Ltd., UK). WE analyzed signals in a minimum of 200 cells with the appropriate filters. Results were quantified as the ratio of gene signal to the centromere signal. Interpretation of these ratios was as follows: a FISH ratio below 1.8 indicated no gene amplification (negative); a ratio above 2.2 suggested gene amplification (positive); and ratios between 1.8 and 2.2 were classified as equivocal. Additionally, gene/chromosome copy number alterations were categorized based on signal counts, with four gene and centromere signals indicating moderate polysomy, and counts exceeding four suggesting high polysomy.
Regarding RNAScope, tissue samples were fixed in 10% neutral buffered formalin (4% formaldehyde in solution), paraffin-embedded and cut at 4μm, mounted in superfrost®plus slides and dried overnight. RNAScope staining method was performed in an automated immunostaining platform (Ventana Discovery ULTRA, Roche). Antigen retrieval was first performed with the appropriate buffer and protease (RNAScope VS Universal Sample Prep ReagentV2, 323740, ACD), and endogenous peroxidase was blocked (peroxide hydrogen at 3%). Then, slides were incubated with the human FGFR1 probe, transcript variant 1, RNAScope™ 2.5 VS Probe- Hs-FGFR1 (ACD, 310079) or human FGFR2 probe, transcript variant 2, RNAScope™ 2.5 VS Probe- Hs-FGFR2 (ACD, 311179). Slides were then incubated with the corresponding Probe Amplification kit (RNAScope VS Universal HRP Detection Reagent, 323210, ACD), conjugated with horseradish peroxidase, and the reaction was developed using 3, -3'-diaminobenzidinetetrahydrochloride (DAB Detection Kit, 760-224, Ventana, Roche); nuclei were counterstained with Hematoxylin II and slides were mounted. Positive control sections were included for each staining run using Positive Control Probe_Hs-PPIB (313909, ACD). Samples were acquired and digitized using the AxioScan.Z1 system (Zeiss). Digitalized images were analyzed with the ZEN 2.3 lite software (Zeiss) and tumoral areas were categorized in the different scores: score 0 (no staining or <1 dot/10 cells), score 1 (1–3 dots/cell), score 2 (4–9 dots/cell and none or very few are in clusters), score 3 (10–15 dots/cell and <10% dots are in clusters) and score 4 (>15 dots/cell and >10% dots are in clusters). Scores of 3 and 4 were considered RNAScope-positive.
Genomic DNA extraction
Genomic DNA from paraffin tissue sections was extracted using the High Pure FFPET DNA Isolation Kit (Roche, 06650767001) following manufacturer instructions. Briefly, 8–10 paraffin tissue sections were deparaffinized in xylenes followed by ethanol washes. The deparaffinized tissue pellet is incubated with lysis buffer and proteinase K for 60 minutes at 56 °C. After inactivation of the enzyme at 90 °C, tissue lysates were placed into a high-pure filter tube, previously adding DNA-binding buffer and isopropanol. After washing the column with buffers I and II, DNA was eluted in a fresh 1.5 ml reaction tube. Genomic DNA from organoid cells was extracted using the DNeasy Blood and Tissue kit (Qiagen, 69504) following the manufacturer's protocol.
Whole-exome sequencing
Whole-exome sequencing (WES) was performed on genomic DNA samples by Macrogen Inc. (Seoul, Korea) using the SureSelectV6 capture kit and the NovaSeq6000 System (Illumina, San Diego, CA, USA). DNA was quantified using the Qubit 1× dsDNA HS Assay Kit (ThermoFisher Scientific) in the Qubit 4 Florometer (ThermoFisher Scientific). DNA integrity evaluation was performed using the Genomic DNA Assay kit (Revvity) on the LabChip GX Touch (Revvity). Libraries were prepared using the Illumina DNA Prep with Enrichment v.2.5 kit as recommended by the manufacturer. A set of 12-plex pre-hybridization pools was created and included for their hybridization with the Twist Bioscience for Illumina Exome 2.5 Panel (Illumina). Library pools were quantified using Qubit 1× dsDNA HS Assay Kit (ThermoFisher Scientific) following the manufacturer’s suggested protocol in the Qubit 4 Florometer (ThermoFisher Scientific). The final library size was determined using High Sensitivity DNA Assay (Revvity) on the LabChip GX Touch (Revvity). Libraries were sequenced using NovaSeq6000 System (Illumina) at a loading concentration of 150pM in a 10B Flowcell (2x150bp). Illumina BCL Convert software was used for generating fastq files.
Sequencing data processing
Variant calls were generated from the raw data using the Varca pipeline (https://github.com/cnio-bu/varca) in tumor-only mode, which implements the GATK´s best practices. For this purpose, raw reads were trimmed to remove adapters and low-quality bases using Trimmomatic version 0.36 (with the following parameters: ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10:2:True; LEADING:3; TRAILING:3; SLIDINGWINDOW:4:15; MINLEN:36. Residual polynucleotide ends were trimmed using fastp version 0.23.4, with the parameters: –trim_poly_x –trim_poly_g -Q -A -l 36.
The processed data were aligned to the human reference genome GRCh38 with BWA-MEM2 version 2.2.1. Subsequent variant calling was performed using the Genome Analysis Toolkit (GATK) version 4.4.0.0 with HaplotypeCaller and MuTect2. Variants were annotated with the Ensembl Variant Effect Predictor (VEP) version 109.
Putative variants were then filtered to retain only somatic variants, keeping only those passing the pipeline quality control filters, with a frequency lower than 1% in the general population, affecting primary transcripts identified by APPRIS, or any transcripts if principal isoforms were not identified; and labeled as moderate or high impact variants by Ensembl’s VEP.
Cell-free DNA analysis
Total circulating free nucleic acids were isolated from 5 ml of plasma using QIAamp Circulating Nucleic Acid Kit (Qiagen). Low-size fragments were purified with the Agencourt® AMPure® XP magnetic beads (Beckman Coulter, Life Science). Quantification as well as DNA quality assessment were carried out Cell-free DNA ScreenTape Assay for TapeStation Systems (Agilent Technologies). NGS was performed using Oncomine Pan-Cancer Cell-Free Assay (Thermofisher Scientific, SL), which is a targeted next-generation sequencing, multi-biomarker assay that enables the detection of genomic alterations across 52 cancer-relevant genes. This assay allows detection of SNVs, Indels and CNVs. Library preparation is performed manually, and the chip template is prepared using the Ion Chef System (Thermofisher Scientific, SL). Sequencing is performed using the Ion S5™ System (ThermoFisher Scientific). Sequence alignment to the hg19 human reference genome was performed using Torrent Suite version 5.18 (ThermoFisher Scientific). For variant calling and annotation, Ion Reporter Software 5.20 was used (workflow: Oncomine TagSeq Pan-Cancer Liquid Biopsy - w2.6 - Single Sample, ThermoFisher Scientific). The mean sequencing depth recommended to detect a somatic variant is 20000x. The LOD of the test is ≥ 0.1% when cfDNA used is between 20–50 ng (recommended for this assay). Sensitivity to detect Hotspot mutations (SNV and indels) is >80% and specificity is >98%. The nomenclature follows the guidelines of the Human Genome Variant Society (HGVS) v21.02.
The hotspots detected included SNVs and indels in the following gene exons and flanking intronic regions: AKT1 (3), ALK(21, 22, 23, 24, 25), AR(6, 8), ARAF(7), BRAF(11, 15), CHEK2(11), CTNNB1(3), DDR2(8, 15, 17), EGFR (3, 7, 12, 18, 19, 20, 21, 22), ERBB2(8, 17, 19, 20, 21, 22), ERBB3(3, 7, 12, 18, 19), ESR1(6, 8, 9), FGFR1(5, 14, 15), FGFR2(7, 9, 12, 13, 14, 15), FGFR3(7, 9, 13, 14, 16), FGFR4(13), FLT3(20), GNA11(5), GNAQ(5), GNAS(8, 9), HRAS(2, 3), IDH1(4), IDH2(4), KIT(8, 9, 10, 11, 13, 14, 17, 18), KRAS(2, 3, 4), MAP2K1(2, 3, 6),MAP2K2(2, 6), MET(14, 16, 19), MTOR(4, 30. 39, 40, 43, 44, 45, 47, 48, 50), NRAS(2, 3, 4), NTRK1(14, 15), NTRK3(16), PDGFRA(7, 10, 11, 12, 14, 16, 18), PIK3CA(2, 3, 5, 8, 10, 14, 21), RAF1(7), RET(10, 11, 13, 14, 15, 16), ROS1(36, 37, 38, 39, 42), SF3B1(15), SMAD4(3, 9, 10, 12), SMO(3, 5, 6, 8, 9, 11), APC(16), FBXW7(9, 10, 11, 12), PTEN(1, 2, 5, 6, 7, 8, 9), TP53(4, 5, 6, 7, 8).
CNV genes: CCND1, CCND2, CCND3, CDK4, CDK6, EGFR, ERBB2, FGFR1, FGFR2, FGFR3, MET, MYC.
Fusions: ALK, BRAF, ERG, ETV1, FGFR1, FGFR2, FGFR3, MET, NTRK1, NTRK3, RET, ROS1.
Statistical analysis
Sample size was not pre-specified due to the dose-escalation nature of the trial. The primary and secondary objectives of the study were to assess toxicity and efficacy (measured according to the NCI CTC AE V.5.0 and determining PFS time, respectively). The efficacy and toxicity population was defined as those patients receiving at least one dose of the study drugs. Kaplan–Meier survival analyses were conducted to estimate PFS, and comparisons between groups were assessed using the log-rank test. Median PFS and 95% confidence intervals were calculated with statistical significance set at P < 0.005 (two-sided). Statistical tests were performed using Python (version 3.8) with statistical packages.
Data availability
WES and NGS data presented in this study has been deposited on the public repository The Sequence Read Archive (NCBI SRA) with the following accession codes PRJNA1241528 and PRJNA1242686 respectively.
Code availability
Not applicable.
References
Finn, R. S. et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 375, 1925–1936 (2016).
Goetz, M. P. et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 35, 3638–3646 (2017).
Hortobagyi, G. N. et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med. 375, 1738–1748 (2016).
Bertucci, F. et al. Genomic characterization of metastatic breast cancers. Nature 569, 560–564 (2019).
Li, Z. et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the Hippo pathway. Cancer Cell 34, 893–905.e898 (2018).
Robinson, D. R. et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 45, 1446–1451 (2013).
Lloyd, M. R., Jhaveri, K., Kalinsky, K., Bardia, A. & Wander, S. A. Precision therapeutics and emerging strategies for HR-positive metastatic breast cancer. Nat. Rev. Clin. Oncol. 21, 743–761 (2024).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022).
Rugo, H. S. et al. Sacituzumab govitecan in hormone receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 40, 3365–3376 (2022).
Touat, M., Ileana, E., Postel-Vinay, S., Andre, F. & Soria, J. C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 21, 2684–2694 (2015).
Turner, N. et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 70, 2085–2094 (2010).
Mouron, S. et al. FGFR1 amplification or overexpression and hormonal resistance in luminal breast cancer: rationale for a triple blockade of ER, CDK4/6, and FGFR1. Breast Cancer Res. 23, 21 (2021).
Elbauomy Elsheikh, S. et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res. 9, R23 (2007).
Drago, J. Z. et al. FGFR1 amplification mediates endocrine resistance but retains TORC sensitivity in metastatic hormone receptor-positive (HR(+)) breast cancer. Clin. Cancer Res. 25, 6443–6451 (2019).
Formisano, L. et al. Association of FGFR1 with ERalpha maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER(+) breast cancer. Clin. Cancer Res. 23, 6138–6150 (2017).
Hortobagyi, G. N. et al. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J. Clin. Oncol. 34, 419–426 (2016).
Formisano, L. et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 10, 1373 (2019).
Andre, F. et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin. Cancer Res. 19, 3693–3702 (2013).
Chae, Y. K. et al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the FGFR pathway: results from the NCI-MATCH trial (EAY131) subprotocol W. J. Clin. Oncol. 38, 2407–2417 (2020).
Musolino, A. et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR(+), HER2(-) breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res. 19, 18 (2017).
Coombes, R. C. et al. Results of the phase IIa RADICAL trial of the FGFR inhibitor AZD4547 in endocrine-resistant breast cancer. Nat. Commun. 13, 3246 (2022).
Schuler, M. et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 20, 1454–1466 (2019).
Pearson, A. et al. Inactivating NF1 mutations are enriched in advanced breast cancer and contribute to endocrine therapy resistance. Clin. Cancer Res. 26, 608–622 (2020).
Katoh, M. et al. FGFR-targeted therapeutics: clinical activity, mechanisms of resistance and new directions. Nat. Rev. Clin. Oncol. 21, 312–329 (2024).
Loriot, Y. et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 381, 338–348 (2019).
Goyal, L. et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N. Engl. J. Med. 388, 228–239 (2023).
Bidard, F. C. et al. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 23, 1367–1377 (2022).
Jhaveri, K. L. et al. Imlunestrant with or without abemaciclib in advanced breast cancer. N. Engl. J. Med. 392, 1189–1202 (2024).
Vassilakopoulou, M. et al. In situ quantitative measurement of HER2mRNA predicts benefit from trastuzumab-containing chemotherapy in a cohort of metastatic breast cancer patients. PLoS One 9, e99131 (2014).
Lien, H. C. et al. In situ HER2 RNA expression as a predictor of pathologic complete response of HER2-positive breast cancer patients receiving neoadjuvant chemotherapy and anti-HER2 targeted treatment. Breast Cancer Res. 26, 100 (2024).
Tseng, Y. F. et al. Correlation of in situ HER2 RNA expression with HER2 immunohistochemistry and fluorescence in situ hybridization categories in breast cancer. Arch. Pathol. Lab. Med. 148, e48–e56 (2024).
Dekkers, J. F. et al. Long-term culture, genetic manipulation and xenotransplantation of human normal and breast cancer organoids. Nat. Protoc. 16, 1936–1965 (2021).
Acknowledgements
This study was funded with the following grants: Bayer Inc. (research contract); CRIS Contra El Cancer Foundation (donation); Proyectos de Investigación en Salud PI22/00317, funded by Instituto de Salud Carlos III and the European Union (European Regional Development fund) and Proyectos de investigación de Medicina Personalizada PMP22/00032, funded by the Instituto de Salud Carlos III and the European Union (NextGeneration EU/PRTR) (awarded to M.Q.F). We thank Ana Moreno, Susana Vara and Juan Luis Sanz (Apices, Madrid; Spain) for their support with the study setup, coordination and project management, monitoring and data management.
Author information
Authors and Affiliations
Contributions
N.M.J., J.A.G.S., S.P., B.B., S.M., J.G., J.S., L.M., E.C., P.T., R.S.B. and M.A. contribute to the acquisition of samples. S.C.F., S.G., L. M.R., E.P.Y., E.C., M.J.B., S.M. and M.Q.F. contribute to methodology, formal analysis, and interpretation of data. M.J.B., S.M. and M.Q.F. contributed to writing the original draft and generating figures. M.Q.F. contributes to the conception and design of the work, funding acquisition and supervision of the project. All authors contributed to the refinement of the study protocol and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Martinez-Jañez, N., García-Saenz, J.Á., Pernas, S. et al. Co-clinical trial targeting ER, FGFR and CDK4/6 in resistant hormone-positive breast cancer with FGFR alterations. npj Precis. Onc. 9, 343 (2025). https://doi.org/10.1038/s41698-025-01106-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01106-1
