
Abstract
Advancements in next-generation sequencing have facilitated tumour-agnostic approaches for cancer therapy. Here, we demonstrate the clinical utility of molecularly guided tumour-agnostic precision medicine in an Asian cohort, leveraging an Asian-centric DNA/RNA comprehensive genomic profiling (CGP) panel. A total of 1166 tissue samples encompassing 29 cancer types underwent real-world CGP testing. Actionable biomarkers were identified in 62.3% of samples, including 1291 (4.7%) somatic variants potentially targetable by regulatory-approved therapies. At least one tumour-agnostic biomarker, including high tumour mutation burden (TMB-high), microsatellite instability (MSI-high), NTRK/RET fusions, and BRAF V600E was identified in 98 samples across 26 cancer types (8.4%). ERBB2 amplification was identified in 42 samples (3.6%) and was most frequently detected in breast (15.0%), followed by endometrial (11.8%) and ovarian tumours (8.9%). Homologous recombination deficiency (HRD) was observed in 407 samples (34.9%). The high prevalence of actionable biomarkers underscores the significance of CGP in facilitating precision medicine in an Asian setting.
Similar content being viewed by others

Introduction
Over the past decade, technological advances in genomic diagnostics have led to a rapid increase in their clinical adoption as an essential component of the modern cancer management strategy1,2,3. Next-generation sequencing (NGS) technologies have enabled high-throughput interrogation of the cancer genome with increasing fidelity while maintaining accessible cost. Simultaneously, there has been a steady increase in the number of actionable diagnostic and therapeutic targets at the molecular level that leverage on our ever-expanding knowledge of genomic alterations that drive cancer development4, thereby paving the way for precision oncology clinical trials.
More recently, tumour-agnostic approaches, which prioritise molecular alterations over tumour tissue origin and histopathology for personalised treatment, have come to the forefront with the regulatory approval of several immunotherapies and targeted therapies. These genomic alterations include NTRK fusion, RET fusion, BRAF V600E mutation, microsatellite instability (MSI-high), and the presence of high tumour mutation burden (TMB-high)4, resulting in eight drugs to date being approved for the treatment of patients with advanced solid tumours harbouring these specific molecular profiles. There is currently a knowledge gap in the tumour-agnostic biomarker landscape across the diversity of Asian cancers.
Emerging targets such as ERBB2 amplification5,6, FGFR fusion/mutation7,8, NRG1 fusion9, MTAP loss10, ALK fusion, KRAS G12C, TP53 Y220C, and homologous recombination deficiency (HRD)11, amongst others, are expected to further transform the entire drug approval landscape12. Consequently, cancer diagnostics that enable broad profiling of the actionable genome will continue to play an increasingly important role in molecularly-guided therapies.
Given the importance of comprehensive tumour genomic testing, major oncology societies including the European Society of Medical Oncology (ESMO) and the American Society of Clinical Oncology (ASCO) have published professional recommendations for the clinical implementation of multigene panel tests in the setting of metastatic and advanced cancers13. However, the potential clinical utility of such tests, particularly in Asia, has not been well-described. Therefore, we report here for the first time a range of actionable outcomes from a comprehensive DNA/RNA tissue assay designed specifically to probe Asian-prevalent cancers and biomarkers in an international multi-centre real-world study in Asia.
Results
Characteristics of study cohort
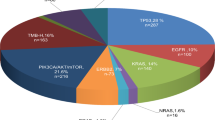
A total of 1166 patient samples were included in our study and underwent CGP with multiplex NGS testing using the UNITED DNA/RNA multigene panel from 2021 to 2024. This consisted of samples from 592 (50.8%) male and 574 (49.2%) female patients, with a median age of 62 years (range, 3–96 years) (Table 1). In terms of geographical distribution, samples were derived largely from patient cohorts from Singapore (SG, n = 777) (66.7%) and Hong Kong (HK, n = 326) (27.9%), with others from Taiwan (TW, n = 46), Macau (n = 13), Philippines (n = 2), Thailand (n = 1) and Malaysia (n = 1). In terms of ethnic distribution, a vast majority were ethnic Chinese (77.2%), while the rest include Vietnamese (3.9%), Indonesian (3.9%), Indian (3.3%), Malay (2.8%), Filipino (1.5%), Caucasian (1.4%) and others (e.g. Myanmese, Arab, Bangladeshi, Cambodian, Eurasian, Thai, Sri Lankan, Korean, Pakistani, Sikh). A spectrum of 29 cancer types were included, consisting of subtypes beyond the original multigene panel design14. Major tumour types included lung (17.8%), colorectal (12.8%), prostate (11.1%), pancreas (8.7%), breast (8.6%), sarcoma (5.1%), and others (Fig. 1).

A A total of 1166 cases were included, derived from patient cohorts from Singapore (SG, n = 777), Hong Kong (HK, n = 326), Taiwan (TW, n = 46) and other countries/territories (n = 17). B Ethnicity distribution of the study cohort. C A spectrum of 29 cancer types was included, consisting of subtypes beyond the original multigene panel design.
Somatic mutational landscape of Asian cancers
The genomic mutational landscape across all the cancer types is shown in Fig. 2A. The majority (clinically actionable %) of samples contained at least a single somatic alteration that was broadly deemed actionable. The commonest actionable gene alterations were TP53, KRAS, EGFR and PIK3CA. However, only 1299 out of 27,552 (4.7%) of somatic variants identified were potentially targetable by regulatory-approved therapies as per FDA. The relative contribution from DNA (SNV, indel and CNV), RNA (gene fusion) information was 4.6% and 0.1%, respectively. The likelihood of identifying at least one actionable molecular alteration was highest in CNS tumours (83.6%), followed by lung cancer (81.2%), and breast cancer (79.0%) (Fig. 2B).

A 62.3% of samples harboured at least 1 alteration that were deemed broadly actionable. The most common actionable gene alterations were TP53, KRAS, EGFR and PIK3CA. B 4.7% of somatic variants were potentially targetable by regulatory-approved therapies. The likelihood of identifying at least one actionable mutation was highest in CNS tumours (83.6%), followed by lung cancer (81.2%), breast cancer (79.0%), and others.
Clinical actionability of genomic alterations based on ESMO Scale for Clinical Actionability of molecular Targets (ESCAT) classification
The clinical actionability of genomic alterations as per ESCAT classification is summarised in Table 2. In our cohort, 148 (12.7%) samples harboured Tier I alterations, which denotes targets linked to approved standard-of-care therapies supported by robust clinical evidence. These include PIK3CA mutations in breast cancer (n = 39), EGFR exon 19 mutations in non-small cell lung cancer (NSCLC) (n = 29), BRCA1/2 somatic mutations or deletions in prostate cancer (n = 22). Meanwhile, 70 samples (6.0%) harboured Tier II alterations, which include targets with evidence of benefit in clinical trials but without established standard-of-care status in a particular cancer type. These include BRCA1/2 somatic mutations in breast cancer (n = 23), ERBB2 mutations (n = 21), PTEN mutations or deletions in prostate cancer (n = 17). The most prevalent Tier IA genomic alterations observed across cancer types included KIT mutations in all gastrointestinal stromal tumours (100%), HRD in nearly half of ovarian tumours (40.0%), and PIK3CA mutations in breast cancer (39%) (Table 2).
Landscape of established tumour-agnostic biomarkers
Amongst the 29 cancer types studied, at least a single tumour-agnostic biomarker was detected in 26 cancer types (89.7%), across 98 samples (8.4%), including in 30% of thyroid, 22.7% of melanoma, 16.8% of lung, 12% of unknown primary origin, 11.8% of endometrial tumours, and others (Figs. 3 and 4). These alterations included RET fusions, NTRK fusions, BRAF V600E mutations, TMB-high and MSI-high statuses. RET fusions were only detected in lung cancers (n = 2), while NTRK fusions were detected in pancreatic, gastric and colorectal cancers (n = 1, each). BRAF V600E mutation was found in colorectal cancer (n = 5), melanoma (n = 3), thyroid cancer (n = 3), CNS tumours (n = 2), cancer of unknown primary, lung cancer, and biliary tract cancer (n = 1, each). A total of 16 cases with MSI-high status were observed, including prostate cancer (n = 5), colorectal cancer (n = 3), cancer of unknown primary (n = 2), pancreatic cancer (n = 2), gastric cancer (n = 2), endometrial cancer (n = 1), and sarcoma (n = 1) (Figs. 3B and 4A). By proportion, endometrial tumours (5.9%), gastric tumours (4.7%) and cancer of unknown primary (4%) had the highest proportions of MSI-high tumours (Fig. 5A). TMB was significantly higher in MSI-high compared to microsatellite stable (MSS) tumours (median TMB 23.0 vs 5.15, Mann-Whitney U p = 3.303-11) (Fig. 5B).

A At least 1 established tumour agnostic marker was detected in 19 out of the 29 cancer types, across 98 samples (8.4%), including in 30% of thyroid, 22.7% of melanoma, 16.8% of lung, 12% of unknown primary origin, 11.8% of endometrial tumours, and others. B Distribution of tumour-agnostic biomarkers except TMB-high across tumour types with at least 1 tumour-agnostic marker detected.

A 77/1166 (6.6%) of samples were TMB-high, with a median TMB of 5.2 mutations/Mb. Tumours of the lung (15.4%), endometrium (11.8%) and oesophagus (11.1%) had the highest proportions of TMB-high tumours. B Range of TMB scores (mutations/Mb) in the top 10 cancer types with highest proportion of TMB-high cases are shown.

A 16/1166 (1.4%) of samples were MSI-high, with endometrial tumours (5.9%), gastric tumours (4.7%) and unknown primary tumours (4%) having highest proportions of MSI-high tumours. B TMB was significantly higher in MSI-high compared to MSS tumours (median TMB 23.0 vs 5.15, p = 3.303-11).
In terms of TMB status, 77 out of the 1166 (6.6%) samples tested were TMB-high, with a median TMB of 5.2 mutations/Mb. Tumours of the lung (15.4%), endometrium (11.8%) and oesophagus (11.1%) had the highest proportions of TMB-high tumours (Fig. 4A). The range of TMB scores in the top 10 cancer types with highest proportion of TMB-high cases are summarised in Fig. 4B. Including TMB-high samples, a total of 114 (9.8%) of all samples tested harboured at least one tumour-agnostic biomarker. The majority of MSI-high tumours (87.5%) except for two cases (sarcoma and colorectal cancer) were also TMB-high. On the other hand, most of the TMB-high tumours (81.8%) were MSS, altogether indicating that 79 out of 1166 samples (6.8%) evaluated are either MSI-high or TMB-high (or both), and thus eligible for immune checkpoint therapy via a tumour-agnostic approach.
HRD, ERBB2 amplification and other emerging tumour-agnostic biomarkers
Potential tumour-agnostic biomarkers were curated from recent literature4,5,7,8,9,10,11,12,15. HRD was observed in 407 samples (34.9%) and was present in about half of the tumours of the breast (50%), colon (49.0%), lung (44.2%), ovary (42.2%), and gastric (39.5%) (Fig. 6A). Notably, HRD-positive tumours exhibited significantly higher TMB compared to HRD-negative tumours (median TMB 5.58 vs. 5.15, respectively, Mann-Whitney U p = 0.000455) (Fig. 6B). ERBB2 amplification (copy number fold-change ≥ 2X), a potential surrogate marker for HER2 overexpression, was identified in 42 samples (3.6%) and was most frequently detected in breast tumours (15%), endometrial tumours (11.8%), ovarian tumours (8.9%), and small intestinal tumours (8.3%) (Fig. 6C). Interestingly, ERBB2-amplified tumours exhibited significantly higher proportion of HRD-positive cases (22 of 41, 53.7%) compared to ERBB2 non-amplified tumours (385 of 1097, 35.1%; p = 0.023) (Fig. 6D).

A HRD was observed in 407 samples (34.9%) and were present in about half of the tumours of the breast (50%), colon (49.0%), lung (44.2%), ovary (42.2%) and gastric (39.5%). B Notably, HRD-positive tumours exhibited significantly higher TMB compared to HRD-negative tumours (median TMB 5.58 vs. 5.15, respectively, p = 0.000455). C ERBB2 amplification, a potential surrogate marker for HER2 overexpression, was identified in 42 samples (3.6%) and was most frequently detected in breast tumours (15%), followed by endometrial tumours (11.8%), ovarian tumours (8.9%), small intestinal tumours (8.3%), and others. Shown are cancers with ≥ 1 ERBB2-amplified case only. D ERBB2-amplified tumours exhibited significantly higher proportion of HRD-positive cases compared to ERBB2 non-amplified tumours (p = 0.023).
ALK fusions were detected in 12 cases (1.0%), including mostly NSCLC (n = 10), but also a case of cancer of unknown primary (n = 1) and breast carcinoma (n = 1). BRAF fusions were detected in a case of breast cancer and thyroid cancer. FGFR2 fusions (n = 2) were detected in leiomyosarcoma (n = 1) and biliary cancer (n = 1), while a case of oesophageal cancer harboured an FGFR3 fusion. FGFR1-4 amplifications (n = 35, 3.0%) were found in cancers of the breast (n = 15), stomach (n = 5), colorectal (n = 4), lung (n = 3), pancreas (n = 2), ovary, prostate, endometrium, oesophagus, sarcoma and fallopian tube (n = 1, each). Other FGFR1-4 alterations identified included copy number deletions (n = 7), indels (n = 9), and SNVs (n = 126). KRAS G12C mutations were found in 14 cases (1.2%), including lung cancer (n = 9), pancreatic cancer (n = 3), biliary cancer (n = 1), and colorectal cancer (n = 1). TP53 Y220C mutations were found in 12 cases (1.0%), including colorectal cancer (n = 3), and cancers of the pancreas, oesophagus, ovary, lung, breast, prostate, as well as astrocytoma, melanoma, and leiomyosarcoma (n = 1, each). MTAP deletions were identified in 59 cases (5.1%), including CNS (n = 12), lung (n = 11), pancreas (n = 6), breast/sarcoma (n = 5, each), melanoma/biliary (n = 3, each), gastric/oesophagus/unknown primary/urinary tract (n = 2, each), colorectal/haematologic/head and neck/ovary/prostate/other (n = 1, each). NRG1 fusions were not identified in our cohort.
A summary of the potentially actionable biomarker landscape in our current study cohort is provided in Fig. 7.

A Established and emerging tumour agnostic biomarkers detected across all cancer types. B Proportion of clinically actionable biomarkers by ESCAT Tiers or otherwise. Inner circle: actionable and non-actionable. Outer circle: ESCAT tier proportions within each group.
Discussion
Comprehensive molecular profiling with combined DNA/RNA NGS on tumour tissue is now increasingly being used as standard-of-care for cancer therapy selection16,17. Combined DNA/RNA NGS is also considered standard-of-care for central nervous system tumour classification18. Despite the genetic heterogeneity and diversity of cancers, most combined DNA/RNA NGS assays are validated in non-Asian populations, resulting in knowledge gaps in Asian patients. In our current study, we have presented real-world Asian data of a DNA-RNA multigene panel of over 1000 tumour samples across multiple cancer types to identify genomic variants (SNV/indels), copy number alterations, and gene fusions. The patient phenotypes and genomic profiles of cancers are known to be different in Asian as compared with Western populations. East Asians, for example, are known to harbour high prevalence of oncogene-driven lung cancers, characterized by EGFR mutations and ALK fusions, amongst others19. On a similar note, breast cancers in East Asian women tend to occur at a younger age, with a more frequent incidence of luminal B subtype, ERBB2 amplification and TP53 mutations, as compared with to the West20. Our findings are generally similar in this regard with a higher frequency of ERBB2 amplification (15%) and TP53 mutations (62%) in our breast cancer cohort, in keeping with a recently reported Korean study (20% and 48%, respectively), as compared to known TCGA data (32% and 9%, respectively)21. Regardless of these differences, our observations on the genomic actionability of cancers are consistent with previous studies17,22,23, our results showed that ~60% of cancers harboured at least one potentially-actionable target. In a recent meta-analysis of 144 real-world studies investigating the potential impact of comprehensive genomic profiling on clinical decision-making in patients with metastatic cancers, close to three-fifths of patients were identified to harbour actionable alterations, though only 15.6% eventually received molecularly-directed therapy and objective responses observed only in 23.9%23. Indeed, successful therapeutic matching can only occur if targeted therapies or molecularly-driven clinical trials are readily accessible. Our data showed that only 4.7% of the genomic alterations identified were potentially targetable by any FDA-approved therapies—even then, real-world clinical use of these drugs may be hampered by issues such as high costs, lack of drug access, or local constraints for off-label usage24.
Despite emerging evidence that comprehensive genomic profiling and a precision medicine approach may confer improved survival outcomes in patients with metastatic cancers23,25, particularly in those with “hard-to-treat” relapsed cancers26 or rare cancers such as sarcomas27,28, the widespread adoption and successful implementation of such a strategy in clinical practice remains limited. While these figures are clearly modest, the real-world impact is anticipated to rise as we gain new knowledge on the molecular target landscape of cancers, alongside the development of novel therapies, drug repurposing efforts, as well as the rapid growth of tumour-agnostic drug approvals. Our data demonstrated a significant prevalence of established tumour-agnostic biomarkers (9.8%) such as TMB-high (6.6%), MSI-high (1.4%), BRAF V600E mutation (1.4%), as well as NTRK and RET fusions. Other potential tumour-agnostic biomarkers identified include HRD (34.9%), FGFR alterations (14.8%), MTAP deletions (5.1%), ERBB2 amplification (3.6%), KRAS G12C mutations (1.2%), ALK fusions (1.0%), and TP53 Y220C mutations (1.0%). While formal approvals will require validation via further clinical trials, early reports suggested promising activity5,7,8,11,12,15,26,29,30,31,32. Notably, we identified ERBB2 amplification in 3.6% of the cases, including several tumour types apart from breast cancer. Together with recent studies demonstrating strong correlation between ERBB2 copy number status by NGS and HER2 status standard immunohistochemistry or fluorescence in situ hybridization assays, this further supports the clinical utility of an NGS approach for therapeutic target identification6.
Building upon these findings, our analyses reinforce the clinical relevance of molecular profiling by applying the ESCAT framework to stratify genomic alterations by levels of actionability. In our cohort, nearly one-fifth of tumours harboured Tier I alterations, reflecting the presence of well-established, targetable drivers with approved standard-of-care therapies. These include PIK3CA mutations in breast cancer, EGFR exon 19 deletions in non-small cell lung cancer, and BRCA1/2 alterations in prostate cancer, amongst others. Importantly, tumour types with characteristic molecular drivers were well captured, such as KIT mutations in all gastrointestinal stromal tumours and HRD in almost half of ovarian tumours—both of which have immediate therapeutic implications26,29,30. Additionally, Tier II alterations were detected in nearly 10% of cases, highlighting how genomic profiling may springboard discovery of emerging therapeutic targets and the value of genomic profiling in enabling access to clinical trials. When integrated into real-world practice, such frameworks can guide treatment selection and broaden access to precision oncology.
While a DNA/RNA-based assay can identify all actionable mutations, its general use remains hindered by high costs, complex data interpretation24,31, and a range of practical implementation barriers, particularly in Asian healthcare systems32,33. These barriers include the lack of reimbursement, limited access to testing, regulatory constraints, and inadequate clinician education. Consequently, this may result in missed opportunities for optimal therapeutic matching and improved patient outcomes. As our data suggests, a more targeted DNA/RNA-based CGP approach will detect more or comparable actionable variants compared to whole transcriptome/exome approach, with deeper coverage while lowering cost. Addressing the other challenges may require adaptation to local clinical and resource environments, as well as efficient molecular multidisciplinary teams3, and improved infrastructure to ensure timely, practical, and equitable delivery of precision oncology. In Asian healthcare systems, the need for sustainability-driven political will, greater interdisciplinary collaboration, improved computational infrastructure, and an expanded bioinformatic workforce have been suggested in order to drive clinical implementation32. Recently, Stackland et al. evaluated the rigour of the peer-reviewed literature cited in the Centers for Medicare & Medicaid Services (CMS) National Coverage Determination Memorandum for FoundationOne CDx (F1CDx), a hitherto DNA-only NGS companion diagnostic. F1CDx interrogates 324 genes in addition to a few genomic signatures and TMB; it was one of the in vitro diagnostic products to undergo parallel review by FDA and CMS in a novel programme intended to reduce time to completion of premarket clearance and CMS coverage decisions. The findings indicate gaps in the supporting evidence for broad comprehensive genomic profiling use in patients with solid tumours. Certainly, more rigorous studies that assess clinical utility would better inform the approval process for novel diagnostic tests34.
Our study is limited by the retrospective nature of data collection and inherent selection biases in the population under study. The genomic information is also specific to that obtainable by the hybrid-capture panel used. Nevertheless, this study provides valuable real-world data of the therapeutic biomarker landscape from a large multi-country Asian cohort. Future studies will be needed to validate the impact on patient outcomes through a molecular-guided targeted therapy approach in Asian populations, as well as to explore additional novel biomarkers (e.g. proteomics, single-cell spatial omics profiling)35. In conclusion, we demonstrate the potential clinical utility of molecularly guided tumour-agnostic precision medicine in an Asian cohort, leveraging an Asian-centric CGP panel tailored to address the unique genetic landscape of the region.
Methods
Patient cohort
Patient samples of various cancer types (n = 29) which underwent comprehensive genomic testing using an Asian-centric pan-cancer combined DNA/RNA hybrid-capture panel, UNITED, were included in the study. The panel profiles 572 cancer-related genes and 71 RNA fusion partners. Patient information such as age at time of genomic profiling, sex, and tumour type were provided by the managing physician. Clinical characteristics of all samples included in the study are summarised in Table 1. A total of 1166 formalin-fixed paraffin-embedded (FFPE) tissue samples from 1149 Asian cancer patients across >70 centres in seven countries and territories underwent real-world CGP testing in a CAP-accredited CLIA-certified laboratory from August 2021 to June 2024 (Lucence Diagnostics, Singapore). The study was approved by the relevant institutional review boards. All samples were obtained following written informed consent in accordance with the Declaration of Helsinki. Participants and/or their legal guardians provided informed consent for their data to be used in this research.
DNA/RNA extraction and quantification
FFPE samples were processed as part of routine clinical care. For each sample, simultaneous DNA and RNA extraction was performed using Allprep DNA/RNA FFPE Kit (Cat No. 80234) (Qiagen, Germany), following the manufacturer’s recommendations. Extracted nucleic acids were quantified using Qubit dsDNA HS kit (Cat No. Q32851) and Qubit RNA HS Kit (Cat No. Q32855) (Thermo Fisher Scientific, MA, USA). Nucleic acid was quality controlled using Agilent Genomic DNA ScreenTape and Agilent High Sensitivity RNA ScreenTape on a TapeStation 4200 (Agilent Technologies, CA, USA), following the manufacturer’s protocol.
Library preparation, sequencing, and bioinformatics analysis
The protocol for library preparation, next generation sequencing, and bioinformatics analysis is as previously described14. Briefly, a customised probe panel based on the Agilent SureSelect XTHS Target Enrichment System (Agilent Technologies, CA, USA) was deployed for Illumina paired-end multiplex sequencing, using an Illumina NextSeq 550 or NextSeq 2000 system (Illumina, CA, USA). The panel used includes the full protein coding DNA regions from the main isoform of the majority of 572 genes (except for MED12, TERT, RARA where only certain exons/hotspots are covered), and all exons in the main isoform of RNA gene partners. Genomic alterations investigated included single nucleotide variations (SNVs), insertions/deletions (indels), gene fusions, copy number alterations, tumour mutation burden (TMB), microsatellite instability (MSI), and homologous recombination deficiency (HRD). A validated in-house DNA and RNA sequencing analytic pipeline was used to call somatic variants of interest including SNVs, indels, and gene fusions. Copy number changes were determined using a read count-based approach. TMB and MSI were determined using in-house methods as described previously14. HRD was assessed based on global loss of heterozygosity (LOH) using an in-house algorithm. A continuous LOH score was calculated based on the percentage of chromosome arms exhibiting LOH in the tumor using B-allele frequencies of >6000 SNPs targeted by the panel. Cases with an LOH score ≥ 35 were considered HRD positive. Definitions for TMB-high (>10 mutations/Mb) and MSI-high were described previously14.
Definitions for actionability
Biomarkers were broadly classified as clinically actionable if therapeutic indications in solid tumours or haematological malignancies were FDA-approved for any indication36, guideline-recommended, and/or supported by robust phase III clinical trials by manual curation. References used included primarily National Comprehensive Cancer Network (NCCN) and National Medical Products Administration (NMPA) guidelines. In addition, we also included the ESCAT (ESMO Scale for Clinical Actionability of molecular Targets) framework to rank genomic alterations according to the level of clinical evidence for molecular targets in a 6-tier scale, from targets ready for implementation (tier I) to preclinical-only or those with lack of evidence37.
Statistical analyses
Continuous variables were represented by box and whisker plots, and their associations with categorical variables were evaluated by Mann-Whitney U tests. Comparisons between categorical variables were evaluated by Chi-squared tests. All analyses were conducted using R version 4.4.0, including packages maftools v2.20.0, ggplot2 v3.5.1, ComplexHeatmap v2.20.0 and RColorBrewer v1.1–3. The threshold for statistical significance was taken as two-tailed p < 0.05 for all analyses.
Data Availability
All data are available upon reasonable request.
References
Mosele, F. et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO precision medicine working group. Ann. Oncol. 31, 1491–1505 (2020).
Chakravarty, D. et al. Somatic genomic testing in patients with metastatic or advanced cancer: ASCO provisional clinical opinion. J. Clin. Oncol. 40, 1231–1258 (2022).
Westphalen, C. B. et al. ESMO precision oncology working group recommendations on the structure and quality indicators for molecular tumour boards in clinical practice. Ann. Oncol. 36, 614–625 (2025).
Mosele, M. F. et al. Recommendations for the use of next-generation sequencing (NGS) for patients with advanced cancer in 2024: a report from the ESMO precision medicine working group. Ann. Oncol. 35, 588–606 (2024).
Liu, D. et al. Diverse ERBB2/ERBB3 activating alterations and coalterations have implications for HER2/3-targeted therapies across solid tumors. Cancer Res. Commun. 5, 680–693 (2025).
Xia, D. et al. HER2/ERBB2 copy number analysis by targeted next-generation sequencing in breast cancer. Am. J. Clin. Pathol. 161, 436–442 (2024).
Ellis, H. & Goyal, L. Are FGFR fusions and mutations the next tumor-agnostic targets in oncology?. JCO Precis. Oncol. 8, e2400113 (2024).
Pant, S. et al. Erdafitinib in patients with advanced solid tumours with FGFR alterations (RAGNAR): an international, single-arm, phase 2 study. Lancet Oncol. 24, 925–935 (2023).
Schram, A. M. et al. Efficacy of zenocutuzumab in NRG1 fusion–positive cancer. N. Engl. J. Med. 392, 566–576 (2025).
Kryukov, G. V. et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science (1979) 351, 1214–1218 (2016).
Nguyen, L. et al. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 11, 5584 (2020).
Bhamidipati, D. & Schram, A. M. Emerging tumor-agnostic molecular targets. Mol. Cancer Ther. 23, 1544–1554 (2024).
Schilsky, R. L. & Longo, D. L. Closing the gap in cancer genomic testing. N. Engl. J. Med. 387, 2107–2110 (2022).
Ng, C. C.-Y. et al. A comprehensive next generation sequencing tissue assay for Asian-prevalent cancers—Analytical validation and performance evaluation with clinical samples. Front. Mol. Biosci. 9, 963243 (2022).
Thavaneswaran, S. et al. A signal-seeking phase 2 study of trastuzumab emtansine in tumours harbouring HER2 amplification or mutation. NPJ Precis. Oncol 8, 195 (2024).
Rodon, J. et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat. Med. 25, 751–758 (2019).
Pleasance, E. et al. Whole-genome and transcriptome analysis enhances precision cancer treatment options. Ann. Oncol. 33, 939–949 (2022).
Sahm, F. et al. Molecular diagnostic tools for the World Health Organization (WHO) 2021 classification of gliomas, glioneuronal and neuronal tumors; an EANO guideline. Neuro Oncol. 25, 1731–1749 (2023).
Zhou, W. & Christiani, D. C. East meets West: ethnic differences in epidemiology and clinical behaviors of lung cancer between East Asians and Caucasians. Chin. J. Cancer 30, 287–292 (2011).
Yap, Y.-S. et al. Insights into breast cancer in the east vs the west. JAMA Oncol. 5, 1489 (2019).
Kan, Z. et al. Multi-omics profiling of younger Asian breast cancers reveals distinctive molecular signatures. Nat. Commun. 9, 1725 (2018).
Lee, S.-H. et al. Landscape of actionable genetic alterations profiled from 1,071 tumor samples in Korean cancer patients. Cancer Res. Treat. 51, 211–222 (2019).
Zerdes, I. et al. Comprehensive genome profiling for treatment decisions in patients with metastatic tumors: real-world evidence meta-analysis and registry data implementation. JNCI: J. Natl. Cancer Inst. 117, 1117–1124 (2025).
Lawler, M. et al. Empowering effective biomarker-driven precision oncology: a call to action. Eur. J. Cancer 209, 114225 (2024).
Schwaederle, M. et al. Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J. Clin. Oncol. 33, 38173825 (2015).
Mirza, M. R. et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 375, 2154–2164 (2016).
Horak, P. et al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov. 11, 2780–2795 (2021).
Tay, T. K. Y. et al. Soft tissue leiomyosarcoma with microsatellite instability, high tumor mutational burden, and programmed death ligand-1 expression showing pathologic complete response to pembrolizumab: a case report. JCO Precis. Oncol. 6, e2200068 (2022).
Heinrich, M. C. et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 21, 4342–4349 (2003).
Coleman, R. L. et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1949–1961 (2017).
van de Haar, J. et al. ESMO Recommendations on clinical reporting of genomic test results for solid cancers. Ann. Oncol. 35, 954–967 (2024).
Phanthunane, C. et al. Precision medicine in Asia enhanced by next-generation sequencing: implications for Thailand through a scoping review and interview study.Clin. Transl. Sci 17, e13868 (2024).
Chong, H. Y., Allotey, P. A. & Chaiyakunapruk, N. Current landscape of personalized medicine adoption and implementation in Southeast Asia. BMC Med Genomics 11, 94 (2018).
Stackland, S., Schnabel, D., Dinan, M. A., Presley, C. J. & Gross, C. P. Strength of evidence underlying the CMS-FDA parallel review of comprehensive genomic profiling tests in the cancer setting. JNCI: J. Natl. Cancer Inst. 117, 144–151 (2025).
Lee, J. Y. et al. The multi-dimensional biomarker landscape in cancer immunotherapy. Int J. Mol. Sci. 23, 7839 (2022).
U.S. Food & Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling. https://www.fda.gov/media/124784 (2024).
Mateo, J. et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO scale for clinical actionability of molecular targets (ESCAT). Ann. Oncol. 29, 1895–1902 (2018).
Acknowledgements
This work was supported by the Singapore Ministry of Health’s National Medical Research Council under its Transition Award (TA21jun-0005), Clinician Scientist Individual Research Grant (CIRG25jan-0007), Large Collaborative Grant (OFLCG18May-0028 and OFLCG23May-0039), and TETRAD II Collaborative Centre Grant (CG21APR2002), SingHealth Duke-NUS AM/ACP-Designated Philanthropic Fund Grant Award (08/FY2023/EX/27-A65), as well as the Khoo Bridge Funding Award provided by Duke-NUS Medical School and the “Estate of Tan Sri Khoo Teck Puat” (Duke-NUS-KBrFA/2025/0090).
Author information
Authors and Affiliations
Contributions
J.Y.L. and J.Y.C. analysed the data and drafted the manuscript; Z.Y.W., T.Y., M.P., R.W., T.B.T., M.H.T. and J.P. provided technical expertise and performed various assays. D.J.T. contributed to manuscript writing. A.E.H., D.P., J.S., T.H.S., S.P.C., R.K., C.V.H., J.S.A. and R.S. provided clinical input; J.Y.L. and J.Y.C. conceived the study, interpreted the results, and revised the manuscript; and all authors read and approved the final version of the manuscript. All authors confirm that they had full access to all the data in the study and accept responsibility to submit for publication.
Corresponding author
Ethics declarations
Competing interests
M-H.T., Z.Y.W., T.Y., M.P. and J.P. are employees of and/or hold stock ownership in Lucence Diagnostics Pte Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, J.Y., El Helali, A., Tan, D.J.J. et al. High clinical actionability of a pan-cancer tissue-based combined DNA and RNA next generation sequencing assay in a diverse Asian population. npj Precis. Onc. 9, 347 (2025). https://doi.org/10.1038/s41698-025-01126-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01126-x
