
Abstract
Van der Waals crystals offer opportunities to engineer material properties by stacking diverse 2D layers into heterostructures. Charge redistribution at these interfaces, governed by band alignment and Fermi levels, enables control over optical, electronic, and magnetic behavior. In this study, we investigated the band alignment and excitation spectra of exfoliated MPS3 (M = Mn, Fe, Co, Ni) using X-ray and UV photoelectron spectroscopy, optical absorption, and DFT + U calculations. Ionization potentials from 5.4 eV (FePS3) to 6.2 eV (NiPS3) were determined. The resulting band diagrams differentiate localized d states from hybridized p-d states, offering insights for designing functional heterostructures. Selective occupation of unoccupied 3d states provides a pathway to tune magnetic order. Theoretical results show that MnPS3 is well-aligned for both hydrogen and oxygen evolution reactions (HER and OER), while other MPS3 are promising for OER. MnPS3/NiPS3 heterostructure exhibits optimal band alignment for efficient water splitting across a broad pH range.

Similar content being viewed by others

Introduction
The study of magnetic ordering in two-dimensional (2D) systems has been a central focus of condensed matter physics for several decades1. Recently, magnetism in 2D van der Waals (vdW) materials has gained prominence as a promising avenue for exploring this phenomenon2,3,4,5. These materials exhibit intriguing properties and hold significant potential for applications in fields such as topological magnonics, low-power spintronics, quantum computing, and optical communications6,7,8,9,10,11,12,13,14,15,16,17. Metal phosphorus trichalcogenides (MPX3), where M represents a transition metal, P is phosphorus, and X is sulfur or selenium, exhibit MII, MI, and MIII states that are stably located within octahedral sulfur environments18,19,20. The strong ionic bonds between the metal cations and the [P2X6]4− anions provide MPX3 materials with a wide range of functionalities18. Significantly, MPS3 displays magnetic properties for M = Mn, Fe, Co, and Ni, characterized by Curie-Weiss susceptibility at high temperatures and antiferromagnetic order at low temperatures3,6,20,21,22. The magnetic behavior arises from a competition between direct M-M exchange and indirect M-S-M super-exchange interactions within the layers, along with interlayer exchange interactions3,20,21,22. Furthermore, theoretical and experimental studies indicate that the band gaps within this family range from 1.3 to 3.5 eV, significantly expanding the potential applications of 2D materials across a broad wavelength spectrum23,24. These properties of MPX3 nanocrystals are particularly promising for applications in catalysis and optoelectronics18,24. Moreover, these materials exhibit unusual behaviors such as intercalation-substitution or intercalation-reduction, along with notable ionic conductivity, making them suitable for use in Li-ion batteries25,26, hydrogen storage27, photodetectors28, and photoelectrochemical reactions29,30,31.
A crucial aspect of vdW materials is the limitless possibility for combining and modifying their properties by stacking different types of 2D materials into heterostructures with atomically sharp interfaces19,32,33,34. Studying band alignment and charge transfer across such heterostructures is a powerful method for tailoring their optical and electronic properties35. Designing heterostructures with magnetic vdW materials offers the potential to modulate the intrinsic magnetic order within the layers, enabling the creation of materials with elevated critical temperatures or the development of magneto-tunnel junctions36.
Despite the recent surge of interest in MPS3 as functional materials, the understanding of their fundamental physical quantities, such as work function or ionization potential, remains limited18,19,37,38. These properties are crucial in determining the characteristics and performance of potential devices based on heterojunctions, where an electron barrier might form. In this report, the electronic properties of exfoliated MPS3, such as MnPS3, FePS3, CoPS3, and NiPS3, were determined by using X-ray and UV photoelectron spectroscopies (XPS and UPS). Additionally, the band gap widths for all studied MPS3 were determined using the optical absorption technique. The obtained data allowed us to construct band energy diagrams for MPS3 relative to the vacuum level, which can be advantageous for tailoring the optical and electronic properties of devices based on 2D material heterostructures. To interpret the experimentally measured ionization potentials, theoretical calculations were conducted in the framework of density functional theory (DFT) to determine the valence band position relative to the vacuum level. The resulting band alignment, with distinguished localized d states from purely ligand or strongly hybridized states, serves as a crucial tool for designing heterostructures with customized optoelectronic properties and offers a pathway to manipulate magnetic behavior. Additionally, the layered structures of MPX3 materials provide a high surface area and active sites for catalytic reactions. The ability to intercalate ions between layers can further improve catalytic efficiency, while magnetic interactions in these materials can influence reaction selectivity. By selectively filling the localized, unoccupied 3d states of transition metals, distinct from ligand states, it becomes feasible to regulate the intrinsic magnetic order within the layer. Additionally, the localized d-orbitals can enhance the adsorption and activation of reactants in water-splitting reactions.
Results
XPS measurements
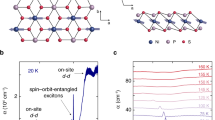
To examine the physicochemical properties and evaluate the purity of the obtained MPS3 samples, XPS analysis was performed. Each sample was exfoliated under ultra-high vacuum and then measured (see Fig. 1a, b), ensuring the removal of air-contaminated layers to obtain pristine surfaces.

a Mounting of NiPS3 sample on a molybdenum plate using silver paste. b The sample is exfoliated under ultra-high vacuum conditions. Adhesive tape is affixed to the side of the preparation chamber, and the forward motion of the transfer arm allows for the exfoliation process. c Wide XPS spectrum and d high-resolution XPS spectra of S-2p, Ni-2p, and P-2p core level lines for exfoliated NiPS3 relative to the valence band maximum.
Figure 1 shows a wide XPS spectrum and high-resolution XPS spectra of Ni-2p, S-2p, and P-2p core level lines for exfoliated NiPS3. For a more accurate analysis, the binding energies of the main core level lines are presented relative to the valence band maximum (VBM), as the Fermi level may lie within the sample’s band gap, potentially affecting the measured absolute values. The survey XPS spectrum obtained for NiPS3, Fig. 1c, shows a series of characteristic emission peaks with no additional lines from possible oxygen and carbon contaminants. In Fig. 1d, six fitted peaks observed at 854.4 and 871.7 eV, 859.1 and 876.0 eV, 864.5 and 881.7 eV in the Ni-2p spectrum are attributed to the characteristic multiplet structure of typical Ni2+ species, along with the expected satellite peaks39,40,41. In the S-2p spectrum, two fitted peaks at 161.9 and 163.3 eV correspond to the spin-orbit 2p3/2 and 2p1/2 split. The P-2p spectrum shows two distinct fitted peaks at 131.4 and 132.4 eV, corresponding to a single spin-orbit split doublet. The XPS spectra for other studied MPS3 are presented in Fig. S1 in Supporting Information. The obtained results indicate the high quality of the analyzed MPS3 layers.
UPS measurements
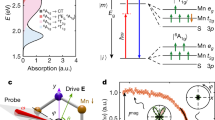
To investigate fundamental physical quantities such as the work function and ionization potential of the examined MPS3 samples, UPS analysis was conducted. Measurements of parameters such as the VBM position and the work function can be affected by charging effects, as both are referenced to the Fermi level, which may shift due to surface charging. Thus, even if the XPS/UPS measurements are performed correctly, the Fermi level may be located within the sample’s band gap. However, in such cases, the ionization potential - defined as the energy difference between the VBM and the vacuum level - remains unaffected, since both the VBM and the vacuum level shift together. As a result, their relative positions stay constant. Figure 2a shows UPS spectra of all studied MPS3 measured using He I photons (hν = 21.2 eV), referenced to the vacuum level. As a result, the cutoff energy (Ecutoff) for all samples was set to 21.2 eV. The ionization potential can be determined by linearly extrapolating the onset of the spectrum and identifying the point where it intersects with the background in the valence band region. The lowest ionization potential of 5.4 eV was observed for FePS3. Subsequently, values of 6.0 and 6.1 eV were determined for materials MnPS3 and CoPS3, respectively. The highest value of ionization potential was observed for NiPS3, equal to 6.2 eV. It can also be observed that the valence band shapes of the studied MPS3 samples differ from those of typical semiconductors, such as GaN42, where the electron density of states begins below the Fermi level, forming a distinct valence band edge. In contrast, the states associated with high kinetic energy electrons in MPS3 samples exhibit low photoelectron emissivity. Among the samples, the most well-defined valence band edge was observed for FePS3. The experiment on exfoliated samples following air exposure was also conducted. The UPS results for NiPS3, shown in Fig. S2 in Supporting Information, reveal that air exposure shifts the ionization energy only by 0.1 eV. This result suggests that although surface chemistry may change upon exposure, the electronic levels relevant for band alignment remain relatively stable within the timeframe and conditions tested.

a UPS spectra relative to the vacuum level, measured using He I photons (hν = 21.2 eV) for all studied and exfoliated MPS3. Gray dashed lines represent experimentally determined values of ionization potential. b Room temperature optical absorption spectra. c The excitation scheme of d–d and charge transfer (CT) transitions for all employed systems. d Schematic band alignment diagram for all studied MPS3 constructed based on a UPS and b absorption results.
Absorption measurements
The next step of our experimental study involved determining the electron affinity (EA), defined as the difference between the vacuum level and the conduction band minimum, which corresponds to the difference between the ionization potential and the optical band gap. To achieve this, optical absorption measurements were conducted. Figure 2b presents room temperature optical absorption spectra of all studied MPS3. These transitions primarily take place between the metal ion states and its surrounding sulfur ligand p states (CT transitions) or occur between 3d states of the same ion (d–d transitions). Ligand arrangement yields a nearly octahedral (trigonal antiprismatic) crystal field that splits the 3d states into even and odd levels, as reported previously43,44. Due to the different electronic configuration in the 3d shell of the ion states, excited states represent different symmetries, typically named using Mullikan notation (see Fig. 2c)45. In general, in a centrosymmetric environment within metal complexes, d–d transitions are typically forbidden in the electric-dipole transition regime due to the parity rule. However, while this restriction does not generally apply to magnetic-dipole transitions, they are usually several orders of magnitude less intense46. Additionally, according to ref. 47, even a slight change in parity can cause electric-dipole transitions to dominate. Moreover, in the case of MPS3, the occupied 3d states are not entirely isolated from the rest of the states but hybridize with ligand states, a topic that will be discussed later. For these reasons, such transitions can occur and are observed in absorption spectra, particularly in systems containing transition metals48,49,50, including MPS3. The spectrum of a bulk MnPS3 crystal reveals three distinct absorption bands associated with electronic transitions within the 3d shell of the Mn2+ (3d5) configuration51. For MnPS3, the Mn2+ ground state is 6A1g (S), characterized by the orbital configuration (t2g3 eg2) with five unpaired spins. In the presence of an octahedral field, the free-ion 4G state is split into several levels: two triply degenerate states, 4T1g (G, t2g4 eg1) and 4T2g (G, t2g4 eg1); a doubly degenerate state, 4Eg (G, t2g3 eg2); and a nondegenerate state, 4A1g (G, t2g3 eg2)52. These represent the first four excited states of Mn2+. Hence, the observed transitions correspond to 6A1g (6S)→4T1g, {4A1g, 4Eg (4G)}, and P,S 3pxpy → 6A1g (6S) with respective energies of 1.91, 2.7, and 2.85 eV51,52. The optical absorption spectrum of a bulk FePS3 crystal exhibits a broad, symmetric absorption feature centered around 1.03 eV, followed by a steep absorption edge at 1.35 eV. The 1.03 eV absorption peak corresponds to a 3d–3d transition in Fe²⁺, specifically from the ground 5T2g state to the first excited 5Eg state (hereafter referred to as the d–d transition), within the forbidden energy region38. Similarly, for CoPS3, an on-site d–d transition is visible around 0.94 eV (4T1g → 4T2g), followed by a steep absorption edge at 1.3 eV53, while NiPS3 displays an on-site d–d transition near 1.05 eV (3A2g → 3T2g), with a subsequent absorption edge at 1.4 eV54. To evaluate the practical relevance of the optical absorption properties of the investigated materials, we compared the calculated absorption spectra with the AM1.5G solar spectrum, as shown in Fig. S3 (Supporting Information). This comparison highlights the spectral range in which the materials can effectively harvest solar energy. While the absorption characteristics suggest promising light-harvesting potential, we note that experimental determination of photocatalytic spectral efficiency lies beyond the scope of this work and is not quantitatively assessed here.
The determined parameters allow us to construct band diagrams for all studied MPS3 samples, as presented in Fig. 2d, where the electron affinity for all materials refers to the position of the vacuum level. The conduction and valence band offsets between the two materials were estimated using a first-approximation method, calculated as the differences in their electron affinities and ionization potentials, respectively. The results indicate that an interface consisting solely of MnPS3 and FePS3 forms a type I (straddling gap) heterojunction, while other heterostructure configurations result in type II (staggered gap) heterojunctions.
Ionization potential (IP) – theoretical considerations
To comprehend the experimentally obtained ionization potentials, ab initio calculations were performed, comparing the valence band position relative to the vacuum level using a state-of-the-art slab method (see Fig. 3a and Computational Details). In all studied systems magnetic ground state was assumed (an AFM-Neel arrangement for MnPS3 and an AFM-ZigZag spin ordering for the rest of the compounds), where the spin magnetic moments were aligned along the easy axis of magnetization, as reported in refs. 55,56. Partially filled d-orbitals exhibit strong Coulomb repulsion between electrons due to their localized nature, which is just partially accounted for in standard DFT calculations (LDA, GGA exchange functionals)57. To improve this description, the additional on-site Coulomb interaction U and Hund’s exchange JH of the half-filled 3d states are included.

a Calculation of the vacuum level for all employed systems based on the macroscopic planar average of the Hartree potential for 7-layer slab of MPX3 crystals in the direction normal to the surface. At the top, additional visualization of the geometric structure of the slab. Ionization potential (b–d). Position of the valence band peak relative to vacuum level for b different antiferromagnetic ordering of MnPS3, c as a function of the Hubbard parameter U for MnPS3, d for all employed bulk crystals and monolayers. VB denotes the energy of the valence band maximum.
The effect of electron correlations on IP
First, the effect of the U parameter on the results is examined. To this end, various U values (U = 3, 5, 7, 9 eV) were considered for a representative system (MnPS3, see Fig. 3c). The maximum difference between the IP calculations for U = 3 eV and U = 9 eV was 280 meV. Given that this family of materials is typically classified as relatively weakly correlated, the U = 9 eV value is unphysically high and was intentionally overestimated to assess the error of IP within the DFT + U framework. The resulting error, on the order of several hundred meV, is acceptable considering the complexity of the studied materials. Finally, the choice of a single U parameter for further calculations is justified. A recent self-consistent evaluation of the Hubbard parameter has reported U = 5 eV for MPX3 materials, regardless of the type of material in this family55. In our study, we focused on the accurate energetic positioning of electronic states near the band gap. For this purpose, we consider ARPES experiments particularly reliable40,58,59,60,61,62,63, as they provide direct insight into the dispersion and relative energies of occupied states. Based on this, fitting the calculated electronic band structures to experimental ARPES data yields U = 1.8 eV for MnPS358, U = 2.0 eV for NiPS359, and U = 1.2 eV for FePS360,61. These values suggest that correlation effects in this family of materials are moderate and that a mean-field DFT + U approach is sufficient to effectively reproduce the experimental photoemission spectra. Unlike strongly correlated materials such as cuprates64 or iridates65 where mean-field DFT + U is insufficient, the DFT + U method remains a valid approximation for MPX3 compounds. Based on these considerations, U = 3 eV and JH = 1 eV were set for all employed systems, following the Lichtenstein approach66 (which corresponds to Ueff = 2 eV in the Dudarev scheme)67.
The impact of magnetic state on IP
Next, the effect of various spin arrangements on ionization potentials is considered. Hence, various collinear AFM phases, using MnPS3 as an example (Fig. 3b) is considered. These energies for a fully relaxed system of each antiferromagnetic phase (Neel, ZigZag, Stripy) were determined. The difference between the Neel (magnetic ground state) and the ZigZag orders is around 25 meV - but for the Stripy ordering, which is energetically much less favorable, the difference is significant (316 meV).
IP – comparison theory vs. experiment
Let us now compare the theoretical results with experimental findings presented in the previous section. To account for possible variations in layer thickness, the performed theoretical analysis includes calculations for both monolayer and bulk systems. However, for direct comparison with the experimental data - obtained from bulk MPS3 crystals - the bulk parameters were considered. The theoretically obtained IP, defined here as IP = EVAC – EVBM (see Fig. 3d) correlates quite well with IP from experimental data (see Fig. 2a), fully reproducing experimental trends and differing in absolute value by a maximum of ~2.5%. In particular, the smallest and the largest absolute difference (ΔIP = |IPexp - IPth | ) were 65 meV and 159 meV for NiPS3 and for CoPS3, respectively. The IP of MPX3 materials falls in the range of 5.5-6.1 eV, which is generally similar to the ionization potential reported for transition-metal dichalcogenides (TMDs) with sulfur68, and higher than that of TMDs with selenium. The higher ionization potential reflects their deeper valence band levels, which can impact their optical properties and charge transfer behavior. Generally, our results demonstrated that IP energies obtained for the monolayers are larger than those obtained for bulk systems (Fig. 3d), which is in line with previous reports69. In bulk materials, electrons experience stronger dielectric screening from surrounding layers, which lowers the energy required to remove an electron. In other words, in monolayers, this screening is significantly reduced, leading to a higher ionization potential. The only material for which the difference seems significant is FePS3, in which prominent structural anisotropy exists, compared to other family members. Particularly, in FePS3, structural anisotropy occurs within the layer plane (lattice constant b ≠ √3a), so the iron sublattice is not precisely hexagonal. The magnitude of this anisotropy (defined as Δ = (b –a √3)/a) varies depending on whether the studied system is 3D or 2D (2.6% vs. 2.8%). It appears that due to this structural in-plane anisotropy, greater coupling with subsequent layers might be possible. However, investigations of the origins and consequences of FePS3 anisotropy are beyond the scope of this work. Nevertheless, this relationship is considered valuable to highlight when determining band alignment.
It is important to note that the presented band alignment diagrams are based on idealized, pristine surfaces. As such, this study should be viewed as exploratory and intended to establish a baseline for identifying promising material combinations. The actual performance of MPS3-based heterostructures will strongly depend on interfacial quality. Factors such as interface dipoles, defect-induced states, and potential band bending may significantly influence charge transfer dynamics19,32,33,34,35. Therefore, future experimental and theoretical efforts focusing on interfacial engineering and device-level fabrication will be crucial for validating and optimizing the proposed heterostructure concepts under realistic operational conditions.
Band alignments
The absorption spectra of MPX3 crystals are rich due to low-intensity optical transitions known as d–d transitions (see arrows in Fig. 2b), occurring below the absorption edge and the charge transfer excitations contributing to a strong absorption onset. As was mentioned before, ligand arrangement yields a nearly octahedral crystal field that splits the 3d states into even and odd levels, as presented schematically in Fig. 4a. Let us now take a closer look at the composition of the states that yield d–d transitions for all employed systems. The PDOS plots uncover hybridization between the occupied M(d) and S(p) orbitals below the Fermi level (see red-green shaded region in Fig. 4b) for all employed systems. On the other hand, the unoccupied energy bands close to the Fermi level are primarily composed of 3d states. Such states for the FePS3, CoPS3 and NiPS3 are well separated from the continuum of the bands and localized, whereas in the case of MnPS3, part of the empty d states hybridizes with ligand states (detailed explanation in ref. 70). These unoccupied 3d states, except in the case of MnPS3, can act as band gap traps. Additionally, the partially filled 3d states of transition metals determine the material’s magnetic properties, as their occupation affects the spin state and exchange interactions. On the other hand, the absorption edge most likely originates from optical transitions associated with charge transfer between the occupied ligand p states and unoccupied metal d states, which are denoted in violet in the excitation scheme shown in Fig. 4d (onset of a significant absorption in Fig. 2b). Qualitatively, the excitation and absorption edge energies align well with transitions arising from the electronic structure calculations.

a Visualization of the nearest environment of the transition metal ion (MS6 cluster) with a description of the splittings associated with the crystal field of this cluster. b Density of states with projection onto the transition metal (M), sulfur (S), and phosphorus (P) states. VBM is set to 0. c Calculated band alignments and band offsets for MPX3 bulk crystals, referenced to the vacuum level. The dotted line separates occupied (below) and unoccupied levels (above). Green color represents ligand region (1), while red denotes d states localized and unoccupied above the dotted line (2), and red-green shaded region – d states hybridized with p states below the dotted line (3). d Green and violet lines indicate the energy of d–d transitions (peak maxima, with green areas representing peak widths) and the absorption edge from absorption measurements (see Fig. 2b), respectively. Charge transfer excitation involves electron transfer from ligand states to ion d states, resulting in a ligand-to-metal transition. These transitions are typically more intense than d–d transitions and contribute to a strong absorption onset.
Heterojunctions
Understanding the band alignment in MPS3 compounds allows us to predict the types of heterojunctions that can form within this material system. This, in turn, facilitates a more informed evaluation of their potential applications in spintronics and photocatalysis. Localized, unoccupied 3d states of transition metals, well separated from ligand states, enable the control of the intrinsic magnetic order by choosing a material with suitable band alignment. Creating a heterostructure allows a charge transfer (electrons or holes) across the interface. This can selectively fill unoccupied localized 3d states of the MPX3 layer, modulating its magnetic properties. Beyond controlling the magnetic order in such heterostructures, optoelectronic devices can be more precisely designed by determining the nature and dispersion of the bands, which is crucial for optical transitions between the layers of the structure.
Additionally, when d states are localized away from ligands, they can participate in specific reactions without interference from ligand dynamics. This separation allows for more controlled reactivity, which is particularly beneficial in catalytic processes where selectivity is essential. Localized d states can facilitate electron transfer processes by providing distinct energy levels that are accessible for redox reactions.
The band alignment plays a crucial role in the efficiency of water-splitting reactions. The primary goal of the water-splitting reaction is to produce hydrogen (H2) and oxygen (O2) gases in hydrogen and oxygen evolution reactions (HER and OER), respectively, in a sustainable and efficient manner using sunlight as the energy source. HER occurs efficiently when the conduction band maximum (CBM) of the photocatalyst is above the hydrogen reduction potential \({\rm{E}_{{{\rm{H}}}^{+}/{H}_{2}}}\). Then, the photocatalyst can donate electrons to protons to form hydrogen. In the OER reaction, the VBM of the photocatalyst must be located below the oxygen oxidation potential \({\rm{E}_{{O}_{2}/{H}_{2}O}}\). Hence, it can provide holes efficiently to oxidize water molecules to oxygen. Both potentials depend on the pH value according to the formula:
respectively71,72. Only for MnPS3 case, CBM lies above the hydrogen reduction potential EH+/H2 in wide range of the pH of environment, whereas for FePS3 this condition is fulfilled for low pH, making them suitable as HER photocatalyst (see Fig. 5a). In addition, almost all employed MPX3 materials, except FePS3, have VBM located below the oxygen evolution potential, which allows them to function as photocatalysts with the holes driving OER. The previous report suggested that MPX3 materials are promising photocatalysts for both OER and HER applications24. However, these conclusions relied on overestimated theoretical values of the fundamental band gap; for example, HSE06 yields 3.01 eV for NiPS324, whereas experimental measurements report values between 1.4 and 1.5 eV. Moreover, the unoccupied d states were assumed to define the onset of the absorption edge. As a result, the band alignment in ref. 24 was misinterpreted. It is important to emphasize that the absorption edge was assumed to lie between the occupied states (VBM) and the unoccupied d states24. However, since these localized unoccupied d states typically fall within the fundamental band gap, the reported band gap would, in our interpretation, correspond to a d–d transition - further increasing the discrepancy between the theoretical prediction and experimental observations.

a The band alignments with the indicated hydrogen reduction potential \({\rm{E}_{{H}^{+}/{H}_{2}}}\) and oxidation potential \({\rm{E}_{{O}_{2}/{H}_{2}O}}\) in all pH range (blue areas). b Illustrative band diagram for MnPS3/NiPS3 heterostructure serving as a potential water-splitting system. The numbers (1–3) indicate the initial stages of the process that lead to the water-splitting reaction. (1) The semiconductors MnPS3 and NiPS3 absorb the light and create photogenerated carriers (electrons and holes). (2) Non-radiative interlayer recombination of the electron and hole at the interface (charge transfer – electron located at CBM in NiPS3 is transferred to VBM in MnPS3). (3) Photoexcited electrons in MnPS3 (photocathode) participate in the reduction of protons (H+) to hydrogen gas (H2) (HER), whereas photoholes at the NiPS3 (photoanode) oxidize water molecules, generating oxygen gas (O2).
Additionally, based on optimal band alignments, two heterostructures are theoretically proposed for water-splitting applications: MnPS3/NiPS3 and FePS3/NiPS3 (see Fig. 5a). However, MnPS3/NiPS3 could potentially work across a wider pH operating range and exhibits a favorable band alignment conducive to effective charge separation and water-splitting reactions. In contrast, the FePS3/NiPS3 system shows significantly smaller band offsets, which may lead to less efficient charge transfer and increased carrier recombination at the interface - factors that could limit its overall photocatalytic performance. Band alignment between MPS3 samples was evaluated using the vacuum-level alignment method applied to individual, pristine monolayers. This widely used first-principles approach allows for a qualitative prediction of band alignment types by comparing the relative positions of electronic states, without the need to explicitly model the full heterostructure or include interfacial effects. Consequently, charge transfer was not quantified explicitly; however, the resulting band offsets indicate which directions of electron transfer are energetically favorable. The proposal of MnPS3/NiPS3 and FePS3/NiPS3 as potential heterostructures for photocatalysis is exploratory and based on band alignment considerations derived from both experimental and theoretical analyses. This approach was taken due to the practical challenges associated with performing photoelectrochemical measurements on these materials, particularly their sensitivity to humidity, oxygen, and illumination, and anisotropic surface properties31,73,74,75,76. While these constraints limit experimental validation, they do not diminish the relevance or potential of our findings, which offer valuable guidance for future research in the field. In both configurations, NiPS3 could potentially serve as the photoanode77, efficiently transporting photogenerated holes to oxidize water molecules (OER reaction), while FePS3 (or MnPS3) acts as the photocathode, facilitating the transport of photogenerated electrons to participate in the reduction of protons (H⁺) to hydrogen gas (H2) (HER reaction) (see Fig. 5). Efficient charge separation can occur at the NiPS3/MnPS3 interface due to electron transfer from CBM of NiPS3 to the lower lying VBM of MnPS3 systems (see (2) in Fig. 5b), thus preventing radiative carrier recombination within each material. Note that both CBM and VBM are made from localized d states (as discussed above), and due to such localized states, an effective charge transfer occurs at the interface. Namely, the photoexcited electron is transferred from NiPS3 to recombine with the hole in MnPS3 at the interface. Hence, the photohole in NiPS3 and photoelectron in MnPS3 are more likely to take part in OER and HER reactions, respectively (see Fig. 5b).
Discussion
The electronic and optical properties of exfoliated magnetic MPS3 compounds - MnPS3, FePS3, CoPS3, and NiPS3 - were explored using X-ray and UV photoelectron spectroscopies, and optical absorption technique. Key electronic parameters, such as ionization potentials, were determined, ranging from 5.4 eV for FePS3 to 6.2 eV for NiPS3. The determined parameters allowed us to construct band diagrams for all studied MPS3 samples. Our analysis revealed that a heterostructure comprising MnPS3 and FePS3 forms a type I (straddling gap) heterojunction, whereas other configurations construct type II (staggered gap) heterojunctions. In addition, the ionization potential values reported here suggest promising potential for catalysis and energy conversion. In particular, the MnPS3 exhibits promising band alignment for photocatalytic applications in both HER and OER, whereas all employed MPX3 materials facilitate OER effectively.
The calculated ionization potentials closely match experimental data, reproducing trends with a maximum deviation of ~2.5% (65 meV for NiPS3 and 159 meV for CoPS3). The impact of magnetic ordering on ionization potentials was also investigated using MnPS3 as a case study. Our results confirm that electron correlations in these materials are moderate and that mean-field DFT + U methods can reliably reproduce experimental photoemission data. Moreover, the observed excitation and absorption edge energies correlate well with transitions derived from the electronic structure calculations (neglecting the excitonic effects). The resulting band alignment provides critical insights for tailoring the optoelectronic properties of heterostructures. The presence of d–d transitions and charge transfer states in MPX3 contributes to a rich and complex absorption spectrum, distinct from the simpler spectra of transition-metal dichalcogenides or black phosphorus. Namely, d–d transitions occur below the absorption edge, and the charge transfer excitations contribute to a strong absorption onset. The transitions in MPS3 are lower-energy than those in wide-band gap materials like h-BN, making them better suited for near-infrared and visible applications. Finally, MnPS3/NiPS3 and FePS3/NiPS3 heterostructures were proposed as potential candidates in water-splitting applications to effectively separate photogenerated electrons in MnPS3 and FePS3 (holes in NiPS3) and prevent their recombination in these materials, before they participate in HER (OER) reactions. Effective charge separation occurs at the interface through non-radiative interlayer recombination. Our findings offer a robust foundation for designing advanced 2D material-based heterostructures, enabling optimized performance in electronics, photovoltaics, and photocatalytic applications.
Methods
Sample preparation
MPS3 compounds were synthesized in quartz ampoules using the chemical vapor transport (CVT) method. The following high-purity starting materials were used as received: manganese (99.95%, -100 mesh, Abcr, Germany), iron (99.9%, -100 mesh, Abcr, Germany), cobalt (99.9%, -100 mesh, Alfa Aesar, Germany), nickel (99.99%, -100 mesh, Abcr, Germany), phosphorus (99.9999%, granules 2–6 mm, Wuhan Xinrong New Materials Co., China), sulfur (99.9999%, granules 2–6 mm, Wuhan Xinrong New Materials Co., China) and iodine (99.9%, granules, Fisher Scientific, Czechia). For the synthesis of MnPS3, FePS3, and NiPS3, the corresponding elements (sufficient for 30 g of the desired MPX3 compound) were placed in quartz ampoules along with 0.5 g of iodine and a 1 at.% excess of sulfur and phosphorus. The ampoules were melt-sealed under high vacuum (<1 × 10-3 Pa, oil diffusion pump with liquid nitrogen cold trap) using an oxygen-hydrogen welding torch. The ampoules were placed in a muffle furnace and heated at 450 °C for 25 h, at 500 °C for 50 h, and at 600 °C for 50 h, and finally at 70 °C for 25 h. The heating and cooling rates were 1 °C/min. The ampoule with pre-reacted MPX3 was placed in a two-zone furnace. First, the growth zone was heated at 750 °C, while the source zone was kept at 600 °C. After 2 days thermal gradient was reversed, and the source zone was heated at 750 °C and the growth zone at 650 °C for 10 days. The ampoule was cooled at room temperature and opened in an argon glovebox. For CoPS3, stoichiometric amounts of the elements corresponding to 15 g of the final compound were placed in a quartz ampoule with 0.5 g of iodine and a 1 at.% excess of phosphorus and sulfur. The ampoule was sealed under the same vacuum conditions as described above. The ampoule was placed in a muffle furnace and heated at 450 °C for 25 h, at 500 °C for 50 h, and finally at 560 °C for 3 weeks. The ampoule was cooled at room temperature and opened in an argon glovebox.
XPS/UPS measurements
Photoelectron experiments were performed using a hemispherical electron energy analyzer (Argus CU) with either UV or X-ray illumination. The used light sources included nonmonochromatic He I (21.21 eV) and monochromatic Al Kα (1486.6 eV). For UPS measurements, photoelectrons were collected with a 0.01 eV step size and a pass energy of 2 eV, while for XPS measurements, a 0.1 eV step size and a pass energy of 20 eV were applied. Binding energy values were referenced to the Fermi level of the electron analyzer, which was calibrated using a clean Ag reference sample. Samples were mounted on a plate using ultra-high vacuum-compatible, silver-filled conductive paste to ensure good electrical conductivity. For XPS and UPS measurements, we used exfoliated bulk crystals to ensure a clean, uncontaminated surface, as these are surface-sensitive techniques. Prior to measurement, each sample was exfoliated under ultra-high vacuum and then transferred to the analysis chamber.
Transmission/absorption measurements
Energy band gaps for the studied samples were determined through transmission (absorption) measurements. For these measurements, well-defined and uniform bulk MPS3 crystals were used. Exfoliation was not necessary, as the technique employed is bulk-sensitive and probes the entire thickness of the sample. The samples, each measuring 1 × 1 mm², had varying thicknesses: approximately 7 µm for CoPS₃, 13 µm for MnPS₃, 15 µm for FePS₃, and 18 µm for NiPS₃. Sample thicknesses were achieved through a polishing and lapping process using a Kemet 300 L machine with a Kemet Chem H polishing cloth. A suspension of Al₂O₃ powder with a grain size of 300 µm, mixed with deionized water, was used to control the polishing process. For optimal transmission measurements, both sides of each sample were dulled to prevent the formation of a Fabry-Perot interference effect. Each sample was secured to a brass holder with silver paste and aligned parallelly using 100 µm copper wire on either side, ensuring precise attachment. The transmission (absorption) spectra were collected using a “dark configuration” setup. In this setup, light from an OSRAM HLX 64642 150 W 24 V halogen lamp was modulated at 270 Hz by a STANFORD RESEARCH SYSTEMS SR540 CHOPPER and directed to a single-grating OMNI-λ 300 ZOLIX monochromator. Within the monochromator, the probe beam (white light) was dispersed before passing through the sample, which was positioned in a closed-cycle helium-cooled CTI-CRYOGENICS 22 JANIS cryostat, enabling measurements over a temperature range of 20–325 K. Transmission through the sample was detected by a HAMAMATSU S2386 Si diode, a PDA25K-EC GaP diode, and a PDA30G PbS diode. The resultant signal was measured using a lock-in technique with a STANFORD SR810 DSP phase-sensitive nanovoltmeter, capturing the spectra point by point along with the reference signal. The absorption α spectrum was calculated from the reflectance and transmission spectra using the formula:
where d - sample thickness, T - transmission signal, R - reflectance signal.
Theoretical calculations
The calculations were performed in the framework of DFT using the generalized gradient approximation within the PBE flavor, as implemented in the VASP software78. The ion-electron interactions were described by the projector augmented wave (PAW) method79,80. Plane-wave basis cutoff and Γ-centered Monkhorst-Pack81 k-point grid were set to 500 eV and 15 × 9 × 15, respectively. A Gaussian smearing of 0.03 eV was employed for the Brillouin zone (BZ) integration. The interlayer vdW forces were treated within the Grimme scheme using D3 correction82. Most of the results were obtained using the PBE + U method based on Lichtenstein’s approach66, with the effective on-site Hubbard U and Hund’s exchange J parameters assumed for the 3d orbitals. The spin-orbit coupling within the collinear treatment of magnetism was taken into account on top of the PBE + U scheme. The position of the atoms and unit cell were fully optimized within the PBE + U approach.
The work function and ionization potential were determined by adjusting the highest occupied eigenvalue of the bulk structure. This adjustment accounted for the energy difference between the vacuum level in slab calculations and the macroscopic planar average of the ionic and Hartree potential for the slab (excluding the exchange-correlation potential). For the slab calculations, 7 layers were taken and a vacuum corresponding to the length of the slab in the z direction (perpendicular to the plane of the layers). The work function (ϕ) and ionization potential (IP) in a solid are defined as ϕ = EVAC–EF and IP = EVAC–EVBM, where EVAC, EF, EVBM denote the energy of the vacuum level, Fermi level, and valence band maximum, respectively. The vacuum level (EVAC) was obtained using the macroscopic planar average of the ionic and Hartree potentials calculated for the slab. The slab represented a bulk surface and consisted of 7 layers and a vacuum thickness corresponding to the length of the slab in the direction normal to the surface. A vacuum of the thickness equal to 20 Å was added to mimic the isolated monolayer. Generally, for insulators and semiconductors, the ionization potential is larger than the work function due to the Fermi level position that is typically closer to mid-gap than the top of the valence band.
While this study focuses on ground state DFT-based band alignment and catalytic band edge positions, it does not include optical spectra or excitonic properties (e.g., via GW-BSE), as these effects are beyond the scope of the present work. However, dielectric screening and exciton binding energies are known to significantly influence charge carrier dynamics in MPX3 materials. A comprehensive discussion of these effects, including their dependence on dimensionality and composition, is provided in our earlier publications55,83.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request.
References
Kim, K. et al. Antiferromagnetic ordering in van der Waals 2D magnetic material MnPS3 probed by Raman spectroscopy. 2D Mater. 6, 041001 (2019).
Wang, Q. H. et al. The magnetic genome of two-dimensional van der Waals materials. ACS Nano 16, 6960–7079 (2022).
Kuo, C.-T. et al. Exfoliation and Raman spectroscopic fingerprint of few-layer NiPS3 Van der Waals crystals. Sci. Rep. 6, 20904 (2016).
Sivadas, N., Daniels, M. W., Swendsen, R. H., Okamoto, S. & Xiao, D. Magnetic ground state of semiconducting transition-metal trichalcogenide monolayers. Phys. Rev. B 91, 235425 (2015).
Chittari, B. L. et al. Electronic and magnetic properties of single-layer MPX3 metal phosphorous trichalcogenides. Phys. Rev. B 94, 184428 (2016).
Joy, P. A. & Vasudevan, S. Magnetism in the layered transition-metal thiophosphates MPS3 (M=Mn, Fe, and Ni). Phys. Rev. B 46, 5425–5433 (1992).
Lee, J.-U. et al. Ising-type magnetic ordering in atomically thin FePS3. Nano Lett. 16, 7433–7438 (2016).
Wang, X. et al. Raman spectroscopy of atomically thin two-dimensional magnetic iron phosphorus trisulfide (FePS3) crystals. 2D Mater. 3, 031009 (2016).
Liu, Q. et al. Magnetic order in XY-type antiferromagnetic monolayer CoPS3 revealed by Raman spectroscopy. Phys. Rev. B 103, 235411 (2021).
Sun, Y.-J., Tan, Q.-H., Liu, X.-L., Gao, Y.-F. & Zhang, J. Probing the magnetic ordering of antiferromagnetic MnPS3 by Raman spectroscopy. J. Phys. Chem. Lett. 10, 3087–3093 (2019).
Mai, T. T. et al. Magnon-phonon hybridization in 2D antiferromagnet MnPSe3. Sci. Adv. 7, eabj3106 (2021).
Kang, S. et al. Coherent many-body exciton in van der Waals antiferromagnet NiPS3. Nature 583, 785–789 (2020).
Zhang, Q. et al. Observation of giant optical linear dichroism in a zigzag antiferromagnet FePS3. Nano Lett. 21, 6938–6945 (2021).
Xing, W. et al. Magnon transport in quasi-two-dimensional van der Waals antiferromagnets. Phys. Rev. X 9, 011026 (2019).
McCreary, A. et al. Quasi-two-dimensional magnon identification in antiferromagnetic FePS3 via magneto-Raman spectroscopy. Phys. Rev. B 101, 064416 (2020).
Ghosh, A. et al. Spin-phonon coupling and magnon scattering in few-layer antiferromagnetic FePS3. Phys. Rev. B 103, 064431 (2021).
Zhang, X.-X. et al. Spin dynamics slowdown near the antiferromagnetic critical point in atomically thin FePS3. Nano Lett. 21, 5045–5052 (2021).
Wang, F. et al. New frontiers on van der Waals layered metal phosphorous trichalcogenides. Adv. Funct. Mater. 28, 1802151 (2018).
Ramos, M. et al. Photoluminescence enhancement by band alignment engineering in MoS2/FePS3 van der Waals heterostructures. ACS Appl. Mater. Interfaces 14, 33482–33490 (2022).
Brec, R. Review on structural and chemical properties of transition metal phosphorous trisulfides MPS3. Solid State Ion. 22, 3–30 (1986).
Le Flem, G., Brec, R., Ouvard, G., Louisy, A. & Segransan, P. Magnetic interactions in the layer compounds MPX3 (M = Mn, Fe, Ni; X = S, Se). J. Phys. Chem. Solids 43, 455–461 (1982).
Kurosawa, K., Saito, S. & Yamaguchi, Y. Neutron diffraction study on MnPS3 and FePS3. J. Phys. Soc. Jpn. 52, 3919–3926 (1983).
Du, K. et al. Weak Van der Waals stacking, wide-range band gap, and Raman study on ultrathin layers of metal phosphorus trichalcogenides. ACS Nano 10, 1738–1743 (2016).
Zhang, X., Zhao, X., Wu, D., Jing, Y. & Zhou, Z. MnPSe3 monolayer: a promising 2D visible-light photohydrolytic catalyst with high carrier mobility. Adv. Sci. 3, 1600062 (2016).
Kuzminskii, Y. V., Voronin, B. M. & Redin, N. N. Iron and nickel phosphorus trisulfides as electroactive materials for primary lithium batteries. J. Power Sources 55, 133–141 (1995).
Liang, Q. et al. General and scalable solid-state synthesis of 2D MPS3 (M = Fe, Co, Ni) nanosheets and tuning their Li/Na storage properties. Small Methods 1, 1700304 (2017).
Ismail, N., Madian, M. & El-Meligi, A. A. Synthesis of NiPS3 and CoPS and its hydrogen storage capacity. J. Alloy. Compd. 588, 573–577 (2014).
Chu, J. et al. High-performance ultraviolet photodetector based on a few-layered 2D NiPS3 nanosheet. Adv. Funct. Mater. 27, 1701342 (2017).
Byvik, C. E., Smith, B. T. & Reichman, B. Layered transition metal thiophosphates (MPX3) as photoelectrodes in photoelectrochemical cells. Sol. Energy Mater. 7, 213–223 (1982).
Aruchamy, A., Berger, H. & Lévy, F. Photoelectrochemical investigation of n‐ and p‐doped nickel phosphorous trisulfide (NiPS3). J. Electrochem. Soc. 136, 2261 (1989).
Oliveira, F. M., Paštika, J., Pires, L. S., Sofer, Z. & Gusmão, R. Photoelectrochemical activity of layered metal phosphorous trichalcogenides for water oxidation. Adv. Mater. Interfaces 8, 2100294 (2021).
Novoselov, K. S., Mishchenko, A., Carvalho, A. & Castro Neto, A. H. 2D materials and van der Waals heterostructures. Science 353, aac9439 (2016).
Pham, P. V. et al. 2D heterostructures for ubiquitous electronics and optoelectronics: principles, opportunities, and challenges. Chem. Rev. 122, 6514–6613 (2022).
Li, M.-Y., Chen, C.-H., Shi, Y. & Li, L.-J. Heterostructures based on two-dimensional layered materials and their potential applications. Mater. Today 19, 322–335 (2016).
Geim, A. K. & Grigorieva, I. V. Van der Waals heterostructures. Nature 499, 419–425 (2013).
Ryu, J., Kajale, S. N. & Sarkar, D. Van der Waals magnetic materials for current-induced control toward spintronic applications. MRS Commun. 14, 1113–1126 (2024).
Miyazaki, T. et al. UPS study of NiPS3 and FePS3 crystals using synchrotron radiation. Chem. Phys. 201, 539–546 (1995).
Budniak, A. K. et al. Spectroscopy and structural investigation of iron phosphorus trisulfide—FePS3. Adv. Optical Mater. 10, 2102489 (2022).
Tong, Y., Chen, P., Chen, L. & Cui, X. Dual vacancies confined in nickel phosphosulfide nanosheets enabling robust overall water splitting. ChemSusChem 14, 2576–2584 (2021).
Yan, M. et al. Correlations in the electronic structure of van der Waals NiPS3 crystals: an X-ray absorption and resonant photoelectron spectroscopy study. J. Phys. Chem. Lett. 12, 2400–2405 (2021).
Liu, J. et al. A robust 2D photo-electrochemical detector based on NiPS3 flakes. Adv. Electron. Mater. 5, 1900726 (2019).
Grodzicki, M. Properties of thin film-covered GaN(0001) surfaces. Mater. Proc. 2, 30 (2020).
Chandrasekharan, N. & Vasudevan, S. Magnetism and exchange in the layered antiferromagnet NiPS3. J. Phys. Condens. Matter 6, 4569 (1994).
Afanasiev, D. et al. Controlling the anisotropy of a van der Waals antiferromagnet with light. Sci. Adv. 7, eabf3096 (2021).
Mulliken, R. S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 23, 1833–1840 (1955).
Tinkham, M. Group Theory and Quantum Mechanics (Courier Corporation, 2003).
Kittel, C. & Luttinger, J. M. Crystalline stark splitting and microwave resonance absorption in paramagnetic salts. Phys. Rev. 73, 162–172 (1948).
Maekawa, S. et al. Physics of Transition Metal Oxides, Vol. 144 (Springer, Berlin, 2004).
Hubisz, J. L. Transition metal oxides: an introduction to their electronic structure and properties. Phys. Teach. 52, 574 (2014).
Tokura, Y. & Nagaosa, N. Orbital physics in transition-metal oxides. Science 288, 462–468 (2000).
Gnatchenko, S. L., Kachur, I. S., Piryatinskaya, V. G., Vysochanskii, Yu. M. & Gurzan, M. I. Exciton-magnon structure of the optical absorption spectrum of antiferromagnetic MnPS3. Low Temp. Phys. 37, 144–148 (2011).
Grasso, V., Neri, F., Perillo, P., Silipigni, L. & Piacentini, M. Optical-absorption spectra of crystal-field transitions in MnPS3 at low temperatures. Phys. Rev. B 44, 11060–11066 (1991).
Banda, E. J. K. B. Optical absorption of CoPS3 in the energy range 0.73 to 5.40 eV. Phys. status solidi (b) 135, K43–K47 (1986).
Banda, E. J. K. B. Optical absorption of NiPS3 in the near-infrared, visible and near-ultraviolet regions. J. Phys. C Solid State Phys. 19, 7329 (1986).
Rybak, M. et al. Magneto-optical anisotropies of two-dimensional antiferromagnetic MPX3 from first principles. Phys. Rev. B 109, 054426 (2024).
Kim, C. et al. Spin waves in the two-dimensional honeycomb lattice XXZ-type van der Waals antiferromagnet CoPS3. Phys. Rev. B 102, 184429 (2020).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Strasdas, J. et al. Electronic band structure changes across the antiferromagnetic phase transition of exfoliated MnPS3 flakes probed by μ-ARPES. Nano Lett. 23, 10342–10349 (2023).
Cao, Y. et al. Revealing the electronic structure of NiPS3 through synchrotron-based ARPES and alkali metal dosing. Preprint at https://doi.org/10.48550/arXiv.2407.02715 (2024).
Pestka, B. et al. Identifying band structure changes of FePS3 across the antiferromagnetic phase transition. ACS Nano 18, 32924–32931 (2024).
Nitschke, J. E. et al. Valence band electronic structure of the van der Waals antiferromagnet FePS3. Mater. Today Electron. 6, 100061 (2023).
Klaproth, T. et al. Origin of the magnetic exciton in the van der Waals antiferromagnet NiPS3. Phys. Rev. Lett. 131, 256504 (2023).
Nitschke, J. E. et al. Tracing the ultrafast buildup and decay of d-d transitions in FePS3. Newton 1, 100019 (2025).
Damascelli, A., Hussain, Z. & Shen, Z.-X. Angle-resolved photoemission studies of the cuprate superconductors. Rev. Mod. Phys. 75, 473–541 (2003).
Pärschke, E. M., Wohlfeld, K., Foyevtsova, K. & van den Brink, J. Correlation induced electron-hole asymmetry in quasi- two-dimensional iridates. Nat. Commun. 8, 686 (2017).
Liechtenstein, A. I., Anisimov, V. I. & Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467–R5470 (1995).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Retamal, J. R. D., Periyanagounder, D., Ke, J.-J., Tsai, M.-L. & He, J.-H. Charge carrier injection and transport engineering in two-dimensional transition metal dichalcogenides. Chem. Sci. 9, 7727–7745 (2018).
Komsa, H.-P. & Krasheninnikov, A. V. Native defects in bulk and monolayer MoS2 from first principles. Phys. Rev. B 91, 125304 (2015).
Harchol, A. et al. Tuning magnetic and optical properties in MnxZn1−xPS3 single crystals by the alloying composition. 2D Mater. 11, 035010 (2024).
Liu, J. et al. Diverse and tunable electronic structures of single-layer metal phosphorus trichalcogenides for photocatalytic water splitting. J. Chem. Phys. 140, 054707 (2014).
Chakrapani, V. et al. Charge transfer equilibria between diamond and an aqueous oxygen electrochemical redox couple. Science 318, 1424–1430 (2007).
Du, C.-F. et al. Layered trichalcogenidophosphate: a new catalyst family for water splitting. Nano-Micro Lett. 10, 67 (2018).
Ran, J. et al. NiPS3 ultrathin nanosheets as versatile platform advancing highly active photocatalytic H2 production. Nat. Commun. 13, 4600 (2022).
Sharma, S. et al. Stability of van der Waals FePX3 materials (X: S, Se) for water-splitting applications. 2D Mater. 10, 014008 (2022).
Mukherjee, D., Austeria, P. M. & Sampath, S. Two-dimensional, few-layer phosphochalcogenide, FePS3: a new catalyst for electrochemical hydrogen evolution over wide pH range. ACS Energy Lett. 1, 367–372 (2016).
Li, X. et al. High-yield electrochemical production of large-sized and thinly layered NiPS3 flakes for overall water splitting. Small 15, 1902427 (2019).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Holzwarth, N. A. W., Tackett, A. R. & Matthews, G. E. A Projector Augmented Wave (PAW) code for electronic structure calculations, part I: atompaw for generating atom-centered functions. Comput. Phys. Commun. 135, 329–347 (2001).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Birowska, M., Faria Junior, P. E., Fabian, J. & Kunstmann, J. Large exciton binding energies in MnPS3 as a case study of a van der Waals layered magnet. Phys. Rev. B 103, L121108 (2021).
Acknowledgements
R.K. acknowledges financial support from the National Science Centre (NCN), Poland [2022/45/B/ST7/02750]. All authors gratefully acknowledge the Polish high-performance computing infrastructure PLGrid (HPC Center: ACK Cyfronet AGH) for providing computer facilities and support within computational grant [PLG/2024/017490]. M.B. acknowledges financial support from the National Science Centre (NCN), Poland [2024/53/B/ST3/04258]. Calculations have been carried out using resources provided by the Wroclaw Centre for Networking and Supercomputing. Z.S. acknowledges financial support by project [LL2101] from the ERC-CZ program from the Ministry of Education, Youth and Sports (MEYS) and by the project Advanced Functional Nanorobots [CZ.02.1.01/0.0/0.0/15_003/0000444] financed by the EFRR.
Author information
Authors and Affiliations
Contributions
D.M.: Conceptualization, Formal analysis, Investigation (X-ray and UV Photoelectron Spectroscopies), Methodology, Visualization, Writing – original draft, Writing – review and editing. M.R.: Conceptualization, Formal analysis, Methodology, Theoretical calculations, Visualization, Writing – original draft, Writing – review and editing. R.B.: Investigation (Transmission/absorption). J.P.: Resources. U.A.: Resources. M.G.: Investigation (X-ray and UV Photoelectron Spectroscopies), Supervision, Writing – review and editing. D.H.: Supervision. M.B.: Conceptualization, Funding acquisition, Theoretical calculations, Supervision, Writing – review and editing. Z.S.: Funding acquisition, Resources, Supervision, Writing – review and editing. R.K.: Conceptualization, Funding acquisition, Supervision, Writing – review and editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no financial or non-financial competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Majchrzak, D., Rybak, M., Bartoszewicz, R. et al. Experimental and theoretical band alignment study of MPS3 (M = Mn, Fe, Co, Ni) for designing tailored 2D heterostructures. npj 2D Mater Appl 9, 53 (2025). https://doi.org/10.1038/s41699-025-00578-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41699-025-00578-w
