
Abstract
Recent advances in two-dimensional material nanopores have promoted significant progress in biosensing technology with single-molecule precision. The potential of detecting single amino acids using graphene and MoS2 nanopores is investigated here, using density functional theory combined with dielectric response calculations and quantum transport simulations. The electronic transport and the optical response of the nanopores are calculated, and their modifications in the presence of the selected amino acids are assessed. While the current-voltage characteristics of the nanopores show very modest modifications, the optical absorption spectra reveal quite evident modulations when an amino acid is present in the nanopore. The dielectric response of the MoS2 nanopores exhibits a broader spectrum of photon energy compared to the graphene ones. Specifically, the calculated optical sensitivity in distinguishing among amino acids in MoS2 nanopores reaches up to 90% compared to roughly 60% in graphene nanopores. For the latter, the calculated sensitivity in the transport mode, that is using the electronic current as a detection observable, is approximately 10 times lower. In the end, all observations are discussed in view of a potential broadband optical detection for protein detection platforms and biosensors.
Similar content being viewed by others


Introduction
Nanopores have been demonstrated as low-cost, label-free, and high-resolution platforms for advanced biotechnology applications and in particular for next-generation DNA sequencing1,2,3. The rapid development of nanopore science, including advances in sensing principles, materials engineering, and device integration, has been comprehensively reviewed4, providing a broader context for the evolution of nanopore technologies. The classic approach of nanopore sequencing relies on the measurement of the ionic current passing through a nanopore. When a molecule electrophoretically translocates through the nanopore, the ionic current can be modulated. The change in the ionic current can be used to detect and analyze the translocating molecule, for instance, to identify each nucleobase of a DNA molecule5. This was first demonstrated in the case of biological nanopores such as α-hemolysin6,7 and extended to Mycobacterium smegmatis porin A (MspA) and aerolysin8,9 and commercialization10,11,12. Representative commercial implementations include the MinION and PromethION platforms developed by Oxford Nanopore Technologies, which use biological nanopores for long-read DNA and RNA sequencing13.
In addition to biological nanopores, solid-state bulk nanopores have also drawn significant attention in parallel14,15,16,17,18. Compared with biological nanopores, solid-state bulk nanopores such as SiNx, SiO2, and Al2O3 provide better mechanical and chemical stability and higher flexibility in pore size and shape19,20. A common limitation of conventional solid-state nanopores based on materials such as SiN is their membrane thickness, typically on the order of 10 nm, though advanced fabrication techniques can locally thin these membranes down to a few nanometers21. In contrast, nanopores formed in two-dimensional materials such as graphene and MoS2 possess intrinsic atomic-scale thickness. The thickness of conventional solid-state nanopores results in an extended sensing region, making it difficult to accurately detect individual nucleobases for sequencing DNA molecules. To achieve single-base resolution in DNA sequencing, it is required that the membrane thickness be comparable to the distance between two DNA bases, i.e., approximately 0.34 nm. Driven by this pursuit, nanopores in two-dimensional (2D) materials have attracted significant attention22,23. Because of their sub-nanometer thickness, these atomically thin materials could, in principle, provide the highest spatial resolution in nanopore-based detection. To date, a variety of 2D material nanopores, including graphene, MoS2, hBN, and MXene, have been explored in the detection of individual DNA bases24,25,26,27,28,29,30,31,32,33,34,35. In addition to the DNA sequencing, hBN nanopores have also been investigated in the pore fabrication and characterization with controlled pore formation36.
However, the effective sensing length of a nanopore in a 2D membrane does not correspond to the physical thickness of the membrane, due to a dominating contribution of the access resistance31. This physical limitation is common to all ionic current measurements using solid-state nanopores and originates from the transition of ions between the bulk solution and the confined nanopore region. To overcome this limitation in 2D material nanopores, an alternative approach is to detect the transverse electronic current flowing within the sheet of 2D materials. This method leverages the direct interaction between molecule and nanopore, causing measurable changes in the transverse electronic current31. When compared to ionic current detection, a distinct advantage of transverse current detection is its higher-bandwidth measurement and thus higher temporal resolution in resolving fast translocation events. Recent advances in high-bandwidth electronics have significantly improved time resolution in solid-state nanopore experiments, enabling detection of ultrafast DNA dynamics37. Optical sensing approaches were originally proposed as complementary or parallelized alternatives38, particularly for large-scale array implementations, although they typically operate at lower intrinsic temporal bandwidth.
While electrical detection methods provide high sensitivity and fast response, optical detection techniques such as fluorescence39 and plasmon resonance40 provide unique advantages for nanopore-based biosensing. Experimentally, optical detection strategies used in solid-state nanopores provide high-sensitivity and multicolor readout capabilities that complement or surpass ionic-current measurements41, while recent developments in optical nanopore sensors demonstrate their ability to independently monitor many nanopores within high-density arrays for ultrasensitive, quantitative analysis of clinically relevant biomarkers42. These optical methods are insensitive to the specific buffer conditions, such as the ionic strength and pH value of the solution, and eliminate the need for electrodes, avoiding issues related to contact resistance, signal degradation, and complex device fabrication43. The direct interaction of light with the electronic structure of nanopores can be tuned to achieve high-contrast signal modulation upon binding single molecules. For instance, when combining nanopores with fluorescently tagged analytes, they create an optical readout system for accurate and sensitive detection of single molecules44.
The success of nanopore DNA detection has motivated the one-step-forward exploration of nanopore biotechnology for protein detection. The latter introduces additional complexity to nanopore devices compared to DNA. This complexity stems primarily from the existence of twenty amino acids, in contrast to four nucleic acids in DNA, resulting in a significant growth in the sensing data volume. At the same time, proteins are not as linearized molecules as DNA, neither have a uniform charge distribution that allows them to translocate smoothly through the pore. In view of detecting proteins and the post-translational modifications therein still utilizing the strengths of 2D materials, we investigate the detection potential of the latter and how the detection sensitivity can be improved. To this end, we probe electrical and optical signals of single amino acids confined within graphene or MoS2 nanopores. Our investigation explores the unique signatures produced by individual amino acids as they reside within these 2D materials nanopores, specifically the optical signatures. This optical approach overcomes the limitations of conventional electrical detection, providing a non-contact, chemical-robust, high-bandwidth method to precisely identify protein building blocks, which is crucial for advancing next-generation protein sensing platforms in view of potential biomedical applications. Moreover, optical readout modalities enable access to richer spectroscopic information that can be linked to the chemical composition and local environment of each amino acid, allowing subtle differences to be amplified and discriminated through distinct light-matter interactions.
Results
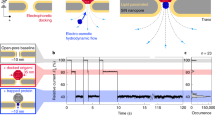
Using density-functional theory (DFT) combined with linear response and quantum transport formalisms, we investigate the electronic transport and optical response of graphene and MoS2 nanopores and their modifications of these in the presence of the selected amino acids. A sketch of the respective setup and sensing is provided in Fig. 1. Both nanopores were constructed with diameters of approximately 1.5 nanometers and functionalized by passivating the pore edges with hydrogen atoms to eliminate dangling bonds and mimic the edge reconstruction after drilling in the experiments. The graphene pore was opened from a single-layer graphene sheet, while the MoS2 pore was created within a monolayer sheet of the semiconducting MoS2 in the 2H phase, which is the most stable crystalline phase for MoS2. A set of representative amino acids, a basic aromatic (His), alkyl-chain non-polar (Ala, Gly, Val) of different sizes, and a non-polar aromatic (Phe) one. The theoretical details of modelling both nanopore setups and their electronic transport and optical response are presented in the Methods section.

a Left: Optical sensing setup using light at a frequency ω to detect an amino acid placed in the nanopore opened in the material membrane. Right: A top view of the membrane materials investigated here, graphene and MoS2, as denoted by the legends revealing an amino acid placed within the opening (nanopore) in these materials. b Electronic transport setup using the transverse electronic current I in response to the applied voltages V for the detection of amino acids residing in the graphene nanopore. c The amino acids (Ala, Gly, His, Phe, and Val) considered in both optical and transport simulations. d The modeling and calculation steps performed in order to calculate the conformational details (Step 1), the transport properties (Step 3), dielectric response (Step 4), and optical absorption (Step 5).
Conformational variability in the pore
We begin the analysis with the search for optimized conformations of the selected amino acids residing in the nanopores. To this end, we performed the single-point calculations for a wide range of rotations for each selected amino acid, as discussed in the previous section. The optimal conformation angles for the selected amino acids that are confined within graphene and MoS2 nanopores are summarized in Table 1. As can be seen, the optimal conformation (orientation) of the confined amino acid is highly dependent on the nanopore material and the amino acid type. As a representative example, Fig. 2 depicts the conformation energy landscapes for Ala confined within the two different nanopores, as a function of the two orientation angles, θ and ϕ. The panels (a, c) of this figure represent 2D heatmaps of the respective energy landscapes. The data in the figure clearly show that the energy landscape for Ala in the graphene nanopore is more rugged over a broader energy range [0, 2.7] eV, compared to the smoother landscape over a narrower energy range [0, 0.8] eV for Ala in the MoS2 nanopore. This difference in energy range directly reflects the strength and orientation dependence of the molecule-nanopore interaction obtained from our DFT analysis. The broader energy variation in graphene (up to ~ 2.7 eV) indicates stronger and more anisotropic coupling between the amino acid and the pore edge, whereas the significantly narrower variation in MoS2 (up to ~ 0.8 eV) suggests weaker and less orientation-sensitive interactions. This implies that the latter accommodates the molecule better, as the interactions are weaker. The weaker interaction in MoS2 could be understood from its surface chemistry and electronic structure. Graphene provides a continuous delocalized π-electron system, enabling stronger π-π stacking and dispersion interactions, particularly with aromatic residues. In contrast, MoS2 is a polar semiconductor composed of Mo-S bonds and lacks extended π conjugation at the pore edge, reducing aromatic orbital overlap and leading to a flatter conformational energy profile.

The conformation energy landscapes for Ala confined within graphene (a, b) and MoS2 (c, d). On the left, heatmaps of the conformational energy along the two rotational degrees of freedom represented by the angles θ and ϕ are shown. Cross sections of these along one of the angles (θ) are provided for the highest (Emax) and lowest (Emin) energy conformations are shown on the right. These insights on the conformational details are obtained by performing Step 1 in Fig. 1d.
Electronic transport across graphene nanopores
The lowest energy conformations revealed from the single point calculations were further geometrically optimized in order to further calculate the properties of the nanopores and their modifications due to the presence of the amino acids. We thus continue with the calculation of the electronic current-voltage (I-V) characteristics by performing quantum transport simulations of the graphene nanopore in the absence (open) and presence (blockade) of amino acids. In order to better visualize the amino acid-induced modulation in the transport characteristics, we calculate the transport sensitivity ηe from Eq. (8) with i = e across the nanopore material.
It should be noted that the transverse transport mechanism in graphene nanopores is dominated by the extended delocalized states of the graphene sheet. Because the nanopore edges are hydrogen-passivated in our model, the scattering potential introduced by a confined neutral amino acid remains highly localized. Consequently, the modification of the overall transmission function is modest, and the resulting current modulation remains small in magnitude (only 2–6%). This behavior differs from earlier theoretical and experimental studies on DNA nucleotides4,27, where larger molecular size, extended π-conjugation, and the charged phosphate backbone produce stronger electrostatic and orbital coupling to the graphene π-electron network, leading to more pronounced current variations. In contrast, most amino acids are comparatively small with strong similarities in their physical properties and less polarizable, resulting in weaker perturbations of the transverse conduction channels.
The electronic I-V characteristics of the graphene nanopore in the absence and presence of different amino acids are depicted in Fig. 3. Overall, the I-V characteristics reveal a nonlinear increase in the current with applied bias voltage, consistent with the electronic transport across graphene and other 2D nanopores for the electrical detection of single DNA nucleotides45,46. The nonlinear behavior originates from the bias-dependent modification of the transmission spectrum in the scattering region. As the applied bias increases, the bias window broadens, allowing a larger number of nonlinear energy-dependent transmission channels to contribute to the transport. In addition, higher bias can activate additional conduction pathways associated with the graphene electronic structure, thereby enhancing the total current in a nonlinear behavior. The underlying mechanism of the bias-induced modification of nonlinear energy-dependent transmission remains applicable to other nanoscale transport systems.

Importantly, the blockade currents for all amino acids follow the open current closely, revealing small modulations with respect to the open nanopore. This can be better visualized through the electrical sensitivity ηe (see Eq. (8)) at distinct values of the applied bias voltage. At a low bias (V = 0.2 V), ηe remains modest (< 3%), with Gly showing the weakest response (close to zero). As the bias increases, ηe is enhanced, with Phe showing the strongest response at V = 0.8 V, reaching a sensitivity of around 6%. This trend suggests that higher bias conditions increase the electronic transmission in the presence of blockade residues, thereby improving molecular discrimination. Moreover, amino acids with aromatic side chains, such as Phe, induce stronger blockade effects compared to smaller residues, like Gly, consistent with their expected stronger electronic interactions with the nanopore. These results point to differences in the nanopore modulation, especially for amino acids with distinct molecular structures and side-chains and with increasing bias. However, even for a high bias of 0.8 V, the sensitivities differ within 2–6% though. In a real nanopore environment, these will probably not be distinguishable, taken into account all the other sources of noise.
In experimental conditions, the presence of electrolyte and solvent can further influence transverse current signals. Variations in pH may modify the protonation state of amino acids, thereby altering their net charge and potentially enhancing electrostatic perturbations of the local potential landscape. On the other hand, increased salt concentration leads to ionic screening, which reduces long-range electrostatic interactions and is expected to decrease the modulation amplitude of the transverse current. Therefore, while the intrinsic modulation reported here remains modest, environmental variations of pH and ionic strengths can further modulate the electronic and optical responses. Note, though, that similar nanopore simulations including simple water have demonstrated that the characteristic transmission peaks are sustained and can be used for distinguishing the biomolecules47.
For completeness, we briefly comment on MoS2 nanopores in the transverse transport mode. Owing to its semiconducting nature and sizable bandgap, the transverse electronic current in pristine monolayer MoS2 at low bias is strongly suppressed in the absence of electrostatic gating48, and its carrier mobility is significantly lower than that of graphene under comparable conditions49. Consequently, though relative conductance modulation may occur upon molecular blockade, the absolute current magnitude is expected to remain small. Nevertheless, theoretical modeling of MoS2 nanopores for the transverse detection of DNA and protein molecules based on Molecular Dynamics and Boltzmann transport formalism32 has demonstrated that transverse current variations can arise from a self-consistent interaction among ions, charge carriers near the pore rim, and translocating biomolecules, with sensitivity depending on electrolyte concentration, pore size, nanoribbon geometry, and doping polarity. An experimental study on the transverse detection of DNA molecules using MoS2 nanopores31 has demonstrated electrically contacted nanoribbons integrated with nanopores and measured correlated ionic and transverse current signals during DNA translocation, suggesting a field-effect-type sensing mechanism based on direct charge or local potential modulation.
Amino acid dependent dielectric response
In order to search for other more amino acid specific observables beyond the electronic current, we turn to the dielectric response of the nanopores. The real and imaginary parts of the dielectric functions in the absence and presence of the selected amino acids for both the graphene and MoS2 nanopores are summarized in the left panels of Figs. 4, 5, respectively. The corresponding sensitivities, \({\eta }_{{\rm{Re}}[\varepsilon ]}\) and \({\eta }_{{\rm{Im}}[\varepsilon ]}\) obtained from Eq. (8) with \(i={\rm{Re}}[\varepsilon ]\) and \({\rm{im}}[\varepsilon ]\) are depicted in the right panels of these figures.

(Right) The respective sensitivities (\({\eta }_{{\rm{Im}}[\varepsilon ]}\) and \({\eta }_{{\rm{Re}}[\varepsilon ]}\)) of the selected amino acids obtained from Eq. (8) (see text) at two distinct photon energies ℏω, as described by the legends. The dielectric response is calculated by performing Step 4 in Fig. 1(d).
The dielectric response of the graphene nanopore system reveals a relatively narrow band behavior, characterized by two prominent resonance peaks in the imaginary component of the dielectric function, Im[ε], located near the photon energies 0.92 eV and 1.54 eV. Upon molecular blockade by one of the selected amino acids, the positions of these resonance peaks remain nearly unchanged. Their intensities, though, exhibit an amino acid-dependent reduction. The strongest modulation is induced by the aromatic residues in His and Phe, consistent with their larger polarizability and π-π interactions50, while Ala and Gly induce a comparatively weaker response. The corresponding sensitivities using either of the two components of the dielectric response, the real or imaginary one, indicate a good optical sensitivity reaching roughly 45–65% at the dominant resonances for His and Phe, but falling below 20% for weaker perturbations from the other amino acids. These results suggest that graphene nanopores can differentiate single amino acids through variations in dielectric resonance intensity, though limited to relatively narrow spectral windows. More specifically, using the dielectric mode for detecting the amino acids is one order of magnitude more efficient than the electronic transport mode, discussed earlier.
The enhanced response of aromatic residues in graphene can be directly correlated with their structural configuration and orientation relative to the graphene lattice. When aromatic side chains such as those of His and Phe adopt orientations that maximize π-π overlap with the delocalized graphene π-electron system, the perturbation of the electronic structure is amplified, leading to stronger modulation of the dielectric resonances. In contrast, non-aromatic residues lack extended conjugation and therefore couple more weakly to the graphene electronic states, resulting in reduced dielectric sensitivity.
The enhanced dielectric modulation of the graphene nanopores together with the observation of the richer conformational energy spectrum available in the MoS2 nanopores (see Fig. 2) provided the ground for repeating the dielectric response calculations for the latter material, as well. As evident from Fig. 5, the richer conformation spectrum in this nanopore is reflected in the markedly broadband dielectric response, with strong dispersive features in both the real and imaginary parts of the dielectric function spanning from the low energy regime (~0–0.4 eV) up to approximately 2 eV, especially in the imaginary component. These results indicate that MoS2 nanopores provide multiple energetic detection windows and robust dielectric signatures, a consequence of the strong dipole-field coupling and excitonic effects intrinsic to polar 2D semiconductors. A blockade due to the presence of an amino acid induces significantly stronger modulation than in graphene nanopores, particularly at low photon energies, where the dielectric response is highly sensitive to local electrostatic perturbations. The calculated sensitivities demonstrate large signal contrasts in the real and imaginary components. Specifically, \({\eta }_{{\rm{Im}}[\varepsilon ]}\) reaches values beyond 40% for all amino acids, including the weaker scatterers, while moving beyond 80% for the non-polar, non-aromatic Ala. On the other hand, \({\eta }_{{\rm{Re}}[\varepsilon ]}\) is substantially lower, in average falling within the 20–40% range. This trend is probably associated with the less varied spectrum of the real component of the dielectric function compared to the imaginary one (left of the same figure).

(Right) The respective sensitivities (\({\eta }_{{\rm{Im}}[\varepsilon ]}\) and \({\eta }_{{\rm{Re}}[\varepsilon ]}\)) of the selected amino acids obtained from Eq. (8) (see text) at three distinct photon energies ℏω, as described by the legends. The dielectric response is calculated by performing Step 4 in Fig. 1d.
The shift in sensitivity trends between graphene and MoS2 arises can be assigned to their different mechanisms of nanopore-molecule coupling. In graphene, the dielectric modulation is dominated by π-orbital interactions, enhanced by aromatic residues. In contrast, MoS2 is a polar semiconductor, and its dielectric response is strongly influenced by polarization effects. Consequently, even non-aromatic residues such as Ala can generate considerable modulation when their intrinsic dipole moments align favorably with the local polar environment of the MoS2 pore.
Although the sensitivity peaks are localized around specific resonance energies, the full spectral profile of Re[ε] and Im[ε] and the absorption across the entire photon-energy window constitutes a characteristic fingerprint for each amino acid. Therefore, practical and distinct optical identification should rely on multispectral analysis rather than a single scalar sensitivity value at one photon energy.
Amino acid-dependent optical absorption
In order to support and extend the findings on the dielectric response of the nanopores, we also present the calculated optical absorption coefficients α derived from frequency-dependent dielectric functions for both nanopores in the absence and presence of amino acids. The respective optical sensitivity ηα was calculated using Eq. (8) for i = α at certain photon energies reflecting the absorption peak intensities. The results for α and ηα are summarized in Fig. 6 for both nanopores.

For the optical absorption coefficients of the graphene nanopore (top left in the figure), two dominant absorption peaks are observed at 0.94 eV and 1.56 eV, corresponding to characteristic electronic transitions in the material. While the overall absorption spectra of all amino acids follow those of the open pore, small but distinct deviations emerge around the main peaks. At these resonance photon energies, we compute the optical sensitivity shown in the bottom panel. Phe and His exhibit the highest sensitivities, reaching up to 30–40% at the resonance photon energies, while Ala and Val display moderate values ( ~ 10–15%), and Gly shows a relatively weaker response. This indicates that aromatic amino acids induce stronger optical responses due to their delocalized π-electrons, which are more readily disturbed by an external field, leading to higher polarizability and more pronounced optical modulations compared to non-aromatic amino acids50.
In contrast to these trends, for the MoS2 nanopore, shown in the right of Fig. 6, a markedly different absorption landscape is observed, with a broad set of low-energy excitations and a dominant absorption feature at 1.8 eV. The corresponding sensitivities shown in the bottom panel are significantly enhanced compared to the graphene nanopores, particularly at lower photon energies. At ℏω = 0.33 eV, the sensitivity ηα is considerably enhanced reaching 70–90% for nearly all amino acids, suggesting a strong modulation of the optical absorption due to molecular confinement in the pore. At very low photon energies (ℏω = 0.07 eV), moderate sensitivities are observed (10–30%). Accordingly, even in the low-energy regime, the MoS2 nanopores remain optically responsive to molecular perturbations, though less than at ℏω = 0.33 eV. At higher energies (ℏω = 1.79 eV), the sensitivity is moderate (15–35%) directly correlating to the strong absorption characteristics. The fact that significant amino acid-dependent contrast persists at both the low-energy (0.07 eV) and high-energy (1.79 eV) regimes emphasizes the broadband optical biosensitivity of MoS2 nanopores.
It should be noted that though the absorption coefficient is mathematically derived from the real and imaginary parts of the dielectric function, it offers distinct experimental advantages. Optical absorption can be measured directly via transmission or reflection spectroscopy without requiring phase-sensitive techniques necessary for extracting the full complex dielectric function. Infrared and terahertz spectroscopies, in particular, provide experimentally accessible and robust platforms with comparatively lower systematic complexity. Therefore, even though α does not contain fundamentally new information beyond ε1 and ε2, it represents a practically convenient and experimentally reliable observable for nanopore-based optical sensing.
Discussion
The amino acid-dependent modifications in the dielectric response of the nanopores point toward the possibility of using relevant optical measurements to detect single amino acids and their sequences along peptides and proteins. In the optical readout scheme, the confined residue modulates the local dielectric environment, reshaping the nanopore’s optical absorption spectrum. As characteristic dielectric fingerprints are strongly connected to the intrinsic properties of single amino acids, such as the electronic polarizability, aromaticity, dipole moment, and side-chain size, they can serve as a robust identifier in biodetection. In contrast to conventional ionic blockade sensing, which relies on the ionic current modulation longitudinal to the nanopore, turning to optical/dielectric detection can significantly enhance the molecular specificity through the distinct spectral features and higher signal-to-noise ratio for high-sensitivity detection.
In realistic experiments, meaningful signal extraction would rely on differential measurements between blockade and open-pore states to suppress static background contributions. Techniques such as lock-in detection at controlled modulation frequencies, as well as plasmonic or cavity-enhanced field confinement, can further amplify local optical fields and increase signal-to-noise ratios. Although solvent environment, molecular labeling, or plasmonic enhancement may modify the absolute absorption spectrum, the relative contrast between blockade and open-pore configurations-the central quantity analyzed here-remains the key measurable parameter. Provided that this contrast exceeds the noise floor and is statistically averaged over repeated capture events, reliable identification remains feasible in realistic experimental conditions.
While the electrolyte environment, hydration structure, and protonation states are critical factors in experimental nanopore settings, our present theoretical work serves as a proof-of-principles baseline designed to isolate the nanopore–molecule interaction. In an aqueous solution, the surrounding medium (ϵwater ≈ 80) would screen electrostatic interactions and introduce spectral broadening. Since the key sensitivity metric, ηi = ∣Ablockade − Aopen∣/∣Aopen∣, relies on the relative contrast between configurations embedded in the same dielectric environment, first-order solvent screening effects are expected to partially cancel-out, leaving the observed broadband enhancement especially for MoS2 qualitatively robust. Regarding the protonation of the amino acids, while pH-dependent charge states would influence the local electrostatic perturbations, the increased net charge of zwitterionic amino acids would likely enhance the dielectric modulation, suggesting that the neutralized amino acids modelled here provide a conservative baseline. Hence, this work sets the ground for follow-up investigations including full dynamics and solvent effects through ab-initio Molecular Dynamics or other hybrid approaches.
An important advantage of using 2D materials for optical sensing lies in their atomically thin geometry, where enhanced light-matter interaction ensures that even subtle molecular perturbations can produce measurable spectral modulations. As shown in our results, graphene and MoS2 nanopores exhibit distinct optical response regimes: graphene produces resonance-localized sensitivity, while MoS2 provides a broadband response that remains pronounced even at low photon energies. Specifically, the direct comparison of the two materials, in our work, highlights the superior performance of MoS2 over graphene in the dielectric mode for optical nanopore sensing. While graphene exhibits a cleaner but narrower optical response with moderate amino acid selectivity, MoS2 shows broadband and high dielectric sensitivity across all selected residues, rendering this material more suitable for reliable optical identification. The enhanced response of MoS2 arises from its semiconducting nature and stronger interaction with molecular dipoles, whereas the weaker π-orbital perturbations in graphene limit its overall sensitivity. Therefore, MoS2 nanopores provide a more advantageous platform for optical single-molecule discrimination based on dielectric response.
In practice, biomolecules thread rapidly the pores assuming rich conformational changes. During translocation a real-time averaging over the large conformational space is mapped on the current signals. Note, though, that the optical dielectric response occurs on an electronic (femtosecond) timescale, meaning that the intrinsic signal can follow the atom-level conformational changes almost instantaneously, with resolution ultimately limited by photon detection rates rather than response speed. Furthermore, because MoS2 exhibits broadband sensitivity, identification can leverage multispectral pattern recognition, which is inherently more robust against orientational fluctuations than single-energy scalar observables. Our static configurations thus represent transiently trapped states, achievable through experimental methods like voltage modulation or viscosity control, and while this study focuses on establishing intrinsic material-dependent contrast.
In view of practical implementation, several optical modalities could be coupled to 2D nanopore devices. Broadband optical measurements, such as infrared optical absorption and terahertz time-domain spectroscopy, could be performed to track real-time changes in both the imaginary and real components of the dielectric function during molecular capture events. Resonant or cavity-enhanced approaches (e.g., plasmonic antennas or photonic crystals) can further increase the sensitivity through field amplification in the confined pore environments. By combining such optical enhancement schemes with controlled molecular translocation through the pore, a fully optical nanopore sequencing platform that is capable of distinguishing amino acids without using complex electrical current approaches could be realized.
In conclusion, we have investigated the sensing performance of 2D graphene and MoS2 nanopores in the detection of single amino acids based on their electronic transport, dielectric and optical response. Using quantum mechanical calculations combining density-functional theory, linear response theory, and non-equilibrium Green’s functions, we focused on assessing the modulations induced by the presence of single amino acids confined within the nanopores on these materials’ properties. Our simulations reveal a significant enhancement in the optical detection of single amino acids using 2D material nanopores and in particular MoS2 nanopores. Specifically, we have calculated the complex dielectric functions, optical absorption spectra, and transverse electronic currents of graphene and MoS2 nanopores in the presence of five selected amino acids (Ala, Gly, His, Phe, and Val). The simulations clearly revealed that graphene nanopores, exhibit a considerable enhancement in their biosensitivity in the optical mode (up to ~ 40% at one of the resonant peak photon energies) rather than in the electronic mode (less than ~ 6%). MoS2 nanopores demonstrated a considerably higher optical sensitivity (up to ~ 90% at one of the resonant peak photon energies), maintaining a strong amino acid-dependent contrast across a broad optical spectral window. Overall, while graphene nanopores show moderate detection sensitivities at resonant absorption peaks, MoS2 nanopores enable highly sensitive detection across a wider spectral window, highlighting the possibility of a broadband optical detection for protein detection platforms and biosensors.
Methods
As sketched in Fig. 1, we investigate the electronic transport and optical response of graphene and MoS2 nanopores and their modifications in the presence of the selected amino acids. Both nanopore setups were modeled and structurally optimized using the DFT approach implemented in the SIESTA code51,52.
Initially, the bare nanopores and isolated amino acid molecules were optimized separately. Subsequently, the nanopores were passivated with hydrogen atoms and re-optimized to account for structural relaxation upon edge functionalization. The optimized amino acids were then placed into the center of each functionalized nanopore, forming hybrid nanopore/biomolecule systems, which were subjected to further structural optimization. During this final step, the atomic positions of the nanopores were fixed while the amino acids were allowed to move inside and around the nanopores. All structural relaxations were performed using the conjugate gradient (CG) algorithm, with convergence criteria set to residual forces on atoms below 0.01 eV/Å. Exchange-correlation effects were treated using the generalized gradient approximation (GGA) of Perdew-Burke-Ernzerhof (PBE)53, along with norm-conserving Troullier-Martins pseudopotentials54 and a double-ζ polarized (DZP) basis set55. Long-range dispersion interactions were taken into account via the non-local van der Waals density functional (vdW-DF) correction56. A real-space mesh cutoff of 250 Ry and an energy shift of 0.01 Ry for the DZP basis set were used, with Brillouin zone sampling restricted to the Gamma point.
In order to address the conformational variability of the amino acids in the nanopores and ensure a good starting point for the geometry optimization of these hybrid systems, we have performed high-throughput single-point energy calculations of different molecular conformations in the nanopores. Specifically, we have sampled a wide range of orientations of the confined amino acid in the angular space, by scanning the orientation angles θ and ϕ around the axes x and y. These rotations allow the molecule to assume multiple distinct conformations along its free energy landscape. The optimal conformation of the amino acid confined within the nanopore is determined by the lowest energy conformation, after the sampling of all possible conformations of the amino acid in the (θ, ϕ) space.
The optical properties of the optimized nanopores were calculated using the SIESTA code, which computes the imaginary part of the frequency-dependent dielectric function using the linear response function57. Specifically, the optical response of materials can be described using the frequency-dependent complex dielectric function
where \({\varepsilon }_{1}(\omega )={\rm{Re}}[\varepsilon (\omega )]\) and \({\varepsilon }_{2}(\omega )={\rm{Im}}[\varepsilon (\omega )]\) are the real and imaginary parts of the dielectric function, respectively, at the optical frequency ω. The imaginary part of the frequency-dependent dielectric function is defined as57
where ε0 is the vacuum permittivity, e is the electron charge, m is the electron mass, the indices c and v represent the conduction and valence bands, respectively, ℏω the incident photon energy, \({E}_{c}\left({\bf{k}}\right.\) and \({E}_{v}\left({\bf{k}}\right.\) are the conduction and valence band energies at the wave vector k, respectively, pi,j is the momentum operator of the dipole transition between the conduction and valence bands, while f(Ec(k)) and f(Ev(k)) are the Fermi-Dirac distribution functions of the conduction and valence bands, respectively. The real part of the dielectric function \({\varepsilon }_{1}(\omega )={\rm{Re}}[\varepsilon (\omega )]\) is obtained from the imaginary part \({\varepsilon }_{2}(\omega )={\rm{Im}}[\varepsilon (\omega )]\) using the Kramers-Kronig transformation
where P denotes the Cauchy principal value and δ is the complex shift parameter. Finally, the optical absorption coefficient α(ω) is calculated from the real part ε1(ω) and the imaginary part ε2(ω) of the dielectric function58
with c the speed of light.
The electronic transport calculations were performed on the graphene nanopore device (see Fig. 1(b) for a schematic transport simulation setup). To this end, the non-equilibrium Green’s function (NEGF) approach within the DFT framework, as implemented in TranSIESTA and TBTrans59,60, was used to calculate the electronic transport coefficients. A 1 × 10 × 1 k-point grid was used in the TranSIESTA calculation, followed by a 1 × 100 × 1 k-point grid used in the TBTrans calculation. The most important quantity for the electronic transport calculations is Green’s function at a given energy (E) of the scattering region and electric bias (V) across the two electrodes:
where SS and HS are the overlap and Hamiltonian matrices of the scattering region, respectively, ΣL and ΣR are the self-energies taking into account the effects of the semi-infinite left (L) and right (R) electrodes, respectively. The electron charge density is calculated self-consistently using Green’s functions until convergence is achieved. The energy-dependent electron transmission coefficient, defining the electron transmission probability from the left electrode to the right one due to the scattering region as a function of the electron energy, can be calculated using the Landauer-Büttiker formula61,62:
where e is the electron charge, h is Plank’s constant, and ΓL and ΓR are the coupling matrices of the left and right electrodes, respectively, with Γα defined as \(i[{\Sigma }_{\alpha }-{\Sigma }_{\alpha }^{\dagger }]\) (α = L, R). The electric current can be calculated by the integration of the electron transmission coefficient weighted by the electron distribution functions of the left and right electrodes over the electron energy, which is given as
where f(E − μL) and f(E − μL) are the Fermi-Dirac distribution functions for the electrons at the chemical potentials μL = EF + V/2 and μR = EF − V/2 (with EF the Fermi energy of electrons at zero bias) in the left and right electrodes, respectively. It should be noted that the electronic transport calculations on the MoS2 nanopore device were not performed in this work. This is because the premise of TranSIESTA calculations is that the electrodes behave like bulk in the electrode regions of the scattering region. This means that the distance between the electrode and the scattering region should be the same as the screening length of the electrode. This is problematic for semiconducting systems such as the MoS2 nanopore device since they intrinsically have a very long screening length, which would make the convergence of the self-consistent NEGF calculations very difficult or even fail.
In order to better assess the efficiency of the nanopore in sensing and identifying the amino acid type, that is quantifying any amino acid induced modulation in the nanopore characteristics, we calculate the sensitivity ηi of the nanopore material as
where \({{\mathcal{A}}}_{open}\) and \({{\mathcal{A}}}_{blockade}\) are the observables (properties) of interest for the bare (open) and modulated (blockade) nanopores, corresponding to the absence and presence of an amino acid within the pore, respectively. The index i stands for the observable \({\mathcal{A}}\), that is sensing mode. Accordingly, for i = e the mode is the current (\({\mathcal{A}}=I\)), for \(i={\rm{Re}}[\varepsilon ]\) and \({\rm{Im}}[\varepsilon ]\) the mode is the real and imaginary parts of the dielectric response function, respectively, while for i = α the mode is the optical absorption coefficient. The sensitivities are calculated at different bias voltages for the electronic transport mode, and at distinct photon energies in the case of the other modes.
Data availability
The datasets generated and/or analyzed in the current study are available from the corresponding author on reasonable request.
References
Branton, D. et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 26, 1146–1153 (2008).
Niedringhaus, T. P., Milanova, D., Kerby, M. B., Snyder, M. P. & Barron, A. E. Landscape of next-generation sequencing technologies. Anal. Chem. 83, 4327–4341 (2011).
Fyta, M. Threading DNA through nanopores for biosensing applications. J. Phys.: Condens. Matter 27, 273101 (2015).
Traversi, F. et al. Detecting the translocation of DNA through a nanopore using graphene nanoribbons. Nat. Nanotechnol. 8, 939–945 (2013).
Manrao, E. A. et al. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotechnol. 30, 349–353 (2012).
Kasianowicz, J. J., Brandin, E., Branton, D. & Deamer, D. W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. 93, 13770–13773 (1996).
Stoddart, D., Heron, A. J., Mikhailova, E., Maglia, G. & Bayley, H. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proc. Natl. Acad. Sci. 106, 7702–7707 (2009).
Derrington, I. M. et al. Nanopore DNA sequencing with MspA. Proc. Natl. Acad. Sci. 107, 16060–16065 (2010).
Cao, C. et al. Single-molecule sensing of peptides and nucleic acids by engineered aerolysin nanopores. Nat. Commun. 10, 4918 (2019).
Ashton, P. M. et al. Minion nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat. Biotechnol. 33, 296–300 (2015).
Brown, C. G. & Clarke, J. Nanopore development at Oxford Nanopore. Nat. Biotechnol. 34, 810–811 (2016).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
Eisenstein, M. Oxford nanopore announcement sets sequencing sector abuzz. Nat. Biotechnol. 30, 295–296 (2012).
Dekker, C. Solid-state nanopores. Nat. Nanotechnol. 2, 209–215 (2007).
Lee, K. et al. Recent progress in solid-state nanopores. Adv. Mater. 30, 1704680 (2018).
Goto, Y., Akahori, R., Yanagi, I. & Takeda, K. -i Solid-state nanopores towards single-molecule DNA sequencing. J. Hum. Genet. 65, 69–77 (2020).
He, Y., Tsutsui, M., Zhou, Y. & Miao, X.-S. Solid-state nanopore systems: from materials to applications. NPG Asia Mater. 13, 48 (2021).
He, S. et al. Solid-state nanopore DNA sequencing: Advances, challenges and prospects. Coord. Chem. Rev. 510, 215816 (2024).
Haque, F., Li, J., Wu, H.-C., Liang, X.-J. & Guo, P. Solid-state and biological nanopore for real-time sensing of single chemical and sequencing of DNA. Nano Today 8, 56–74 (2013).
Fragasso, A., Schmid, S. & Dekker, C. Comparing current noise in biological and solid-state nanopores. ACS Nano 14, 1338–1349 (2020).
Yamazaki, H., Hu, R., Zhao, Q. & Wanunu, M. Photothermally assisted thinning of silicon nitride membranes for ultrathin asymmetric nanopores. ACS nano 12, 12472–12481 (2018).
Qiu, H., Sarathy, A., Schulten, K. & Leburton, J.-P. Detection and mapping of DNA methylation with 2d material nanopores. npj 2D Mater. Appl. 1, 3 (2017).
Qiu, H., Zhou, W. & Guo, W. Nanopores in graphene and other 2d materials: A decade’s journey toward sequencing. ACS Nano 15, 18848–18864 (2021).
Schneider, G. F. et al. DNA translocation through graphene nanopores. Nano Lett. 10, 3163–3167 (2010).
Merchant, C. A. et al. DNA translocation through graphene nanopores. Nano Lett. 10, 2915–2921 (2010).
Wells, D. B., Belkin, M., Comer, J. & Aksimentiev, A. Assessing graphene nanopores for sequencing DNA. Nano Lett. 12, 4117–4123 (2012).
Girdhar, A., Sathe, C., Schulten, K. & Leburton, J.-P. Graphene quantum point contact transistor for DNA sensing. Proc. Natl. Acad. Sci. 110, 16748–16753 (2013).
Farimani, A. B., Min, K. & Aluru, N. R. DNA base detection using a single-layer MoS2. ACS Nano 8, 7914–7922 (2014).
Feng, J. et al. Identification of single nucleotides in MoS2 nanopores. Nat. Nanotechnol. 10, 1070–1076 (2015).
Zhang, L. & Wang, X. DNA sequencing by hexagonal boron nitride nanopore: A computational study. Nanomaterials 6, 111 (2016).
Graf, M., Lihter, M., Altus, D., Marion, S. & Radenovic, A. Transverse detection of DNA using a MoS2 nanopore. Nano Lett. 19, 9075–9083 (2019).
Xiong, M., Graf, M., Athreya, N., Radenovic, A. & Leburton, J.-P. Microscopic detection analysis of single molecules in MoS2 membrane nanopores. ACS Nano 14, 16131–16139 (2020).
Yadav, P., Cao, Z. & Barati Farimani, A. DNA detection with single-layer Ti3C2 MXene nanopore. ACS Nano 15, 4861–4869 (2021).
Cao, Z., Yadav, P. & Barati Farimani, A. Which 2d material is better for DNA detection: Graphene, MoS2, or MXene? Nano Lett. 22, 7874–7881 (2022).
Prasongkit, J., Jungthawan, S., Amorim, R. G. & Scheicher, R. H. Single-molecule DNA sequencing using two-dimensional Ti2C (OH)2 MXene nanopores: A first-principles investigation. Nano Res. 15, 9843–9849 (2022).
Dai, C. et al. Evolution of nanopores in hexagonal boron nitride. Commun. Chem. 6, 108 (2023).
Lin, C.-Y. et al. Ultrafast polymer dynamics through a nanopore. Nano Lett. 22, 8719–8727 (2022).
Huang, S., Romero-Ruiz, M., Castell, O. K., Bayley, H. & Wallace, M. I. High-throughput optical sensing of nucleic acids in a nanopore array. Nat. Nanotechnol. 10, 986–991 (2015).
Cai, S., Sze, J. Y. Y., Ivanov, A. P. & Edel, J. B. Small molecule electro-optical binding assay using nanopores. Nat. Commun. 10, 1797 (2019).
Garoli, D., Yamazaki, H., Maccaferri, N. & Wanunu, M. Plasmonic nanopores for single-molecule detection and manipulation: toward sequencing applications. Nano Lett. 19, 7553–7562 (2019).
Gilboa, T. & Meller, A. Optical sensing and analyte manipulation in solid-state nanopores. Analyst 140, 4733–4747 (2015).
Fried, J. P., Wu, Y., Tilley, R. D. & Gooding, J. J. Optical nanopore sensors for quantitative analysis. Nano Lett. 22, 869–880 (2022).
Wang, Y. et al. Electrode-free nanopore sensing by DiffusiOptoPhysiology. Sci. Adv. 5, eaar3309 (2019).
Anderson, B. N. et al. Probing solid-state nanopores with light for the detection of unlabeled analytes. ACS Nano 8, 11836–11845 (2014).
Jena, M. K., Kumawat, R. L. & Pathak, B. First-principles density functional theory study on graphene and borophene nanopores for individual identification of DNA nucleotides. ACS Appl. Nano Mater. 4, 13573–13586 (2021).
Kumawat, R. L. & Pathak, B. Electronic conductance and current modulation through graphdiyne nanopores for DNA sequencing. ACS Appl. Electron. Mater. 3, 3835–3845 (2021).
Dou, M., Maier, F. C. & Fyta, M. The influence of a solvent on the electronic transport across diamondoid-functionalized biosensing electrodes. Nanoscale 11, 14216–14225 (2019).
Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. & Kis, A. Single-layer MoS2 transistors. Nat. Nanotech 6, 147–150 (2011).
Kim, T., Fan, S., Lee, S., Joo, M.-K. & Lee, Y. H. High-mobility junction field-effect transistor via graphene/MoS2 heterointerface. Sci. Rep. 10, 13101 (2020).
Guthmuller, J. & Simon, D. Linear and nonlinear optical response of aromatic amino acids: a time-dependent density functional investigation. J. Phys. Chem. A 110, 9967–9973 (2006).
Soler, J. M. et al. The siesta method for ab-initio order-N materials simulation. J. Phys.: Condens. Matter 14, 2745 (2002).
Garcia, A. et al. Siesta: Recent developments and applications. J. Chem. Phys. 152, 204108 (2020).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Troullier, N. & Martins, J. L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 43, 1993–2006 (1991).
Louwerse, M. J. & Rothenberg, G. Transferable basis sets of numerical atomic orbitals. Phys. Rev. B 85, 035108 (2012).
Klimes, J., Bowler, D. R. & Michaelides, A. Chemical accuracy for the van der waals density functional. J. Phys.: Condens. Matter 22, 022201 (2009).
Titantah, J. T. & Karttunen, M. Ab initio calculations of optical properties of silver clusters: cross-over from molecular to nanoscale behavior. Eur. Phys. J. B 89, 125 (2016).
Fox, M. & Bertsch, G. F. Optical properties of solids. Am. J. Phys. 70, 1269–1270 (2002).
Brandbyge, M., Mozos, J., Ordejon, P., Taylor, J. & Stokbro, K. Density-functional method for nonequilibrium electron transport. Phys. Rev. B 65, 165401 (2002).
Papior, N., Lorente, N., Frederiksen, T., Garcia, A. & Brandbyge, M. Improvements on non-equilibrium and transport green function techniques: The next-generation transiesta. Comput. Phys. Commun. 212, 8–24 (2017).
Landauer, R. Electrical resistance of disordered one-dimensional lattices. Philos. Mag. 21, 863–867 (1970).
Büttiker, M. Four-terminal phase-coherent conductance. Phys. Rev. Lett. 57, 1761 (1986).
Acknowledgements
The authors gratefully acknowledge the computing time provided to them on the high-performance computer Noctua2 at the NHR Center PC2. This is funded by the Federal Ministry of Education and Research and the state governments participating on the basis of the resolutions of the GWK for the national high-performance computing at universities (www.nhr-verein.de/unsere-partner). The computations for this research were performed using computing resources under project hpc-prf-meop. This work is part of the nanodiagBW consortium (project number 03ZU1208BI) funded by the German Federal Ministry of Education and Research (BMBF) within the Clusters4Future initiative. Funding from the German Funding Agency (DFG) under the project entitled “Novel complex nanopores for the detection of natural and mutated DNA” is greatly acknowledged.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L. Li conducted all simulations and prepared all figures/tables. M. Fyta conceived and supervised the research. L. Li and M. Fyta wrote and revised the manuscript. Both authors discussed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Fyta, M. Dielectric response of graphene and MoS2 nanopores in the detection of single amino acids. npj 2D Mater Appl 10, 47 (2026). https://doi.org/10.1038/s41699-026-00694-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41699-026-00694-1
