
Abstract
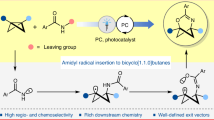
The increasing popularity of four-member rings in drug discovery has prompted the synthetic chemistry community to advance and reinvent old strategies to craft these structures. Recently, the strain-release concept has been used to build complex architectures. However, although there are many strategies for accessing small carbocyclic derivatives, the synthesis of azetidines remains underdeveloped. Here we report a photocatalytic radical strategy for accessing densely functionalized azetidines from azabicyclo[1.1.0]butanes. The protocol operates with an organic photosensitizer, which finely controls the key energy-transfer process with distinct types of sulfonyl imines. The radical intermediates are intercepted by the azabicyclo[1.1.0]butanes via a radical strain-release process, providing access to difunctionalized azetidines in a single step. This radical process is revealed by a combination of spectroscopic and optical techniques and density functional theory calculations. The power and generality of this method is illustrated with the synthesis of various azetidine targets, including derivatives of celecoxib and naproxen.

Similar content being viewed by others

Main
Nitrogen-containing heterocycles are one of the most abundant classes of molecules in drugs and agrochemicals1,2,3. In fact, more than 60% of currently marketed pharmaceuticals contain at least one N-heterocycle. Although planar systems, such as pyridine and its derivatives, still play a major role, the scientific community is moving towards alternative molecular architectures with elevated three-dimensionality and a high degree of sp3-hybridized carbon atoms (Fsp3)4,5. This trend is sustained by their greater metabolic stability and structural modularity relative to planar systems6,7. The azetidine core is considered a privileged scaffold because it can mimic pyridine, piperidine and pyrrolidine for bioisosteric replacement (Fig. 1a)8,9. Nevertheless, despite its promising pharmacokinetic properties relative to five- and six-membered nitrogen rings, azetidines have led to only nine approved drugs10,11,12,13. Among the four-membered heterocycles, sulfonamide derivatives are an important structural class (Fig. 1b). The discrepancy between the importance and prevalence of azetidine-bearing molecules is probably because they are challenging to synthesize.

a, Relevance of the azetidine scaffold and current prevalence in approved drugs. b, Selected examples of drugs featuring the azetidine core. c, Available synthetic strategies for constructing the azetidine core. d, Development of a RSR synthetic method, and the general mechanism of the synthesis.

Reaction conditions: 1 (0.1 mmol), 2 (0.05 mmol), PS (0.25 mol%, unless otherwise noted) in PhCF3 (otherwise noted), irradiated with a 50 W Kessil lamp under argon atmosphere at 40 °C. The yields in parentheses refer to the average of isolated products obtained from three independent reactions. 5TCzBN, 2,3,4,5,6-penta-(9H-carbazol-9-yl)benzonitrile; MeCzTrz, 9,9′,9″-(5-(4,6-diphenyl-1,3,5-triazin-2-yl)benzene-1,2,3-triyl)tris(3,6-di-methyl-9H-carbazole); TBCzTrz, 9,9′,9″-(5-(4,6-diphenyl-1,3,5-triazin-2-yl)benzene-1,2,3-triyl)tris(3,6-di-tert-butyl-9H-carbazole).
Classical approaches involve intramolecular displacement14,15 or lactam reduction16,17 (Fig. 1c). However, severe limitations still prevent the general application of these methods, including low functional group tolerance and the need for tailored precursors. More recently, the aza-Paternò–Büchi reaction has been popularized and used to access complex azetidines18,19,20. This is a milder synthetic approach than those mentioned above, requiring the use of visible light in the presence of commercially available photocatalysts. However, the reaction does not always display high levels of regio- and diastereoselectivity, and its stereochemical outcome is hard to predict. An alternative strategy uses strained 1-azabicyclo[1.1.0]butanes (ABBs)21,22,23,24,25,26,27. This strained molecule can undergo a double functionalization process in the presence of an electrophile (orange circle in Fig. 1c) and a nucleophile (blue circle in Fig. 1c)23,24,25, with the predictable formation of two new bonds in a single synthetic operation. Nonetheless, the generality of this process is hampered by the need to use a specific electrophilic partner and/or strong acidic conditions26,27.
Despite its latent relevance, there is still no radical strategy based on the strain release of ABBs. Such an approach, while unlocking uncharted radical reactivity, would provide access to difunctionalized azetidine variants, expanding the synthetic repertoire for its construction28. Advantageously, in recent decades, photocatalysis has been established as a pillar in synthetic catalysis, introducing new reactivities and non-obvious retrosynthetic disconnections29,30,31,32,33. In this context, radical strain-release (RSR) photocatalysis has emerged as a formidable tool for crafting small rings. Very recently, the state of the art of this field has been effectively compiled, highlighting the absence of N-heterocyclic scaffolds34. With only a handful of synthetic methods available, a general approach to access the azetidine core would have a great impact on the organic chemistry and medicinal chemistry communities. Seeking to address the latter, we defined as a synthetic blueprint the photocatalysed generation of two profitable radical intermediates which could react in predictable fashion with ABBs. Should such a reaction prove successful, the transient open-shell species would be incorporated, leading to the opening and double selective functionalization at N and C3 of the strained system (Fig. 1d).
Here we report the development of a radical light-driven method that transforms ABBs into densely functionalized C3–N sulfonyl azetidines. The strategy relies on the ability of a photosensitizer (PS) to promote the homolytic cleavage of a large variety of sulfonyl imine precursors. The two generated neutral radical intermediates react with the ABB, leading to its polarity-driven selective functionalization without the need for previous activation steps. This RSR protocol involves the generation of a key C3-centred radical, enabling the insertion of nitrogen, sulfur and hydrogen. The successful use of the ABBs under photocatalytic conditions reveals opportunities in RSR photocatalysis and more generally in free radical chemistry.
Results
Reaction development
To functionalize ABB 1, we needed to produce two suitable transient radical species. Because of the different electronic natures of the two atoms to be functionalized, namely nitrogen and carbon (C3), it was necessary to carefully consider the selection of the radical source. To prevent a possible unfruitful one-electron oxidation of the ABB through photoredox scenarios35, we elected to perform triplet sensitization, and used sulfonyl imine 2 as the reaction partner (Fig. 2). Preliminary optimization experiments led us to identify the optimal stoichiometry, between 1 and 2, and reaction time (Supplementary Tables 1 and 2). During the selection of the ideal PS, we found out that thioxanthen-9-one (TXO) 6 afforded full conversion of imine 2 with only 10% yield of product 3, along with major amounts of imine dimer 4 (Supplementary Table 3). This suggests that the concentration of the iminyl radical in the reaction mixture is critical. We thus reasoned that slowing down the iminyl radical formation might channel the reactivity towards the formation of the target azetidine 336,37,38. To this end, we investigated different classes of PSs at a reduced catalyst loading of 0.25 mol%, characterized by a sufficiently high T1 E0,0 (that is, the energy of the first triplet excited state) >2.55 eV, and a small ΔST (Supplementary Fig. 3 and Supplementary Table 3)39. This last parameter corresponds to the difference between the energies of the first singlet (S1) and triplet (T1) excited state and promotes a rapid S1 → T1 and T1 → S1 interconversion through intersystem crossing (ISC) and reverse intersystem crossing (RISC), respectively. We hypothesized that a small ΔST together with an efficient RISC—or in other words, an inefficient triplet sensitization event—would downregulate the concentration of the PS’s T1 state in solution, controlling the imine sensitization and preventing the formation of 4. Indeed, we observed a consistent trend in the relationship between the ΔST value and the selectivity of the process towards the azetidine 3 versus imine dimer 4 (Supplementary Table 3). Notably, other parameters can also influence the selectivity of the process, including the PS loading, which was kept as low as 0.25 mol%, λmax (maximum absorption wavelength), the PS’s absorption tail and the PS’s solubility (Supplementary Fig. 4). Considering all these aspects, we found the best performance for the PS 9, characterized by the lowest ΔST of 0.05 eV and a T1 E0,0 = 2.74 eV (calculated, 2.83 eV; see below). The design of the PS was inspired by recent progress in the field of thermally activated delayed fluorescence emitters and their use in photocatalysis40,41,42. The PS was thoroughly characterized in terms of its prompt and delayed lifetimes, ISC/RISC rates (Supplementary Equations 3 and 4 and Supplementary Table 4), T1 excited state energy, HOMO–LUMO difference and spin distribution at T1 (Fig. 3e and Supplementary Fig. 37). The molecular structure of the PS was unambiguously confirmed by single-crystal X-ray analysis (Supplementary Fig. 48). Quite surprisingly, across all the performed reactions, we never observed the formation of the reversed product isomer 5, in which the sulfonyl group is attached to the C3 position (see below).

a, Proposed reaction scenario suggesting the sensitization of 2, resulting in the generation of sulfonyl and iminyl radicals 10 and 11, respectively. Addition of 10 to 1 produces 12, which undergoes recombination with 11 leading to 3. b, Stern–Volmer plot shows 2 as the quencher. kSV is k of Stern–Volmer and kq is k of quenching. c, Left: the resulting transient absorption LFP spectrum of PS 9 in the presence of 2. The region between 420 nm and 520 nm (within the black circle) suggests that the sensitization of 2 by the triplet PS 9 leads to the radicals 10 + 11 because 10 is expected to absorb at this region. ΔOD, Δ optical density. Middle: flash photolysis of a solution of 2 in the presence of 1, leading to a prompt transient spectrum (time delay, 50 ns) with a maximum at 410 nm (orange trace) which can be assigned to the sulfonyl radical 10 (see left panel for comparison). The dark red trace with a maximum at 435 nm and a tail at longer wavelength (time delay, 750 ns) can be assigned to the radical intermediate 12 obtained by reaction of 1 with radical 10. mΔOD, milli Δ optical density. Right: the dependence of the transient signal at 450 nm on the concentration of 1. d, EPR studies using PBN as spin trap. The blue trace shows the experimental results conducted in the absence of 1; the red trace is the model reaction. e, Insights from DFT calculations. Unless otherwise stated, all values are reported in kcal mol−1.
Mechanistic investigations
We next investigated the mechanism of this RSR process (Fig. 3a). We initially assessed the feasibility of an energy transfer (EnT) step between the excited PS 9 and the sulfonyl imine 2. Time-correlated single-photon counting experiments demonstrated that the singlet excited state of PS 9 is reactive towards neither the sulfonyl imine 2 nor ABB 1. On the other hand, time-resolved luminescence experiments performed by laser flash photolysis (LFP) confirm that the triplet excited state of the PS is efficiently quenched by the sulfonyl imine 2 (bimolecular rate constant k = 2.6 × 108 M−1 s−1; Fig. 3b), whereas quenching by the ABB 1 is substantially less effective (k = 3.7 × 106 M−1 s−1; Supplementary Figs. 26–28). Transient absorption spectroscopic studies by LFP show that photoreaction of the PS 9 triplet with 2 leads to the formation of a subtle absorption signal in the 420–520 nm region (Fig. 3c, left panel), probably associated with the rapid generation of radicals 10 and 11 upon sensitization. However, its decay is faster than its formation, thus preventing full accumulation of the transient and its reliable attribution. We then focused on the homolytic cleavage of the nitrogen–sulfur bond and the reaction of the proposed radical intermediates with the ABB 1 to deliver the final N-tosyl azetidine 3. To gain insight into these steps, we performed LFP analysis upon direct excitation of 2 in the presence of the ABB 1. Under these conditions, we observed the prompt development of a transient absorption centred at 410 nm (Fig. 3c, middle panel; time delay, 50 ns), associated with the tosyl radical 10 (refs. 43,44). In the absence of other reagents, this species decays to the baseline within a few microseconds due to radical recombination (Fig. 3c, right panel). In the presence of 1, a new transient species subsequently forms, characterized by an absorption maximum at 435 nm and a pronounced tail at longer wavelengths. These transient changes can be reasonably assigned to the formation of the radical intermediate 12 (Fig. 3c, middle panel; time delay, 750 nm). Consistent with this attribution, the associated positive absorption, measured at 450 nm (Fig. 3c, right panel) increases with the concentration of the ABB 1, as expected based upon the bimolecular nature of the radical reaction between 10 and 1. The subsequent decay, occurring within a few microseconds, can be mainly assigned to the reaction of 12 with radical 11 and concurrent formation of the final product 3 (for further details, see Supplementary Figs. 33–35).
To further corroborate the mechanistic proposal, while gathering more information about the key intermediate 12, we performed electron paramagnetic resonance (EPR) experiments in the presence of the spin trapping reagent α-phenyl-tert-butylnitrone (PBN) and the PS 9. We first explored the homolytic cleavage of 2 in the absence of the ABB 1. Under these conditions, we detected mixed radicals, clearly identifying two main species (Fig. 3d, blue trace; for details, see Supplementary Fig. 29). The parameters of the EPR spectrum are consistent with the trapping of nitrogen- and sulfur-centred radicals in equal proportion, suggesting that these species are the sulfonyl and iminyl radicals 10 and 11. When performing the spin-trapping experiment under the reaction conditions (in the presence of 1), we obtained a more intense and completely different spectrum (Fig. 3d, red trace). This spectrum suggests that multiple radical species are trapped, but the major one (about 70%) is compatible with the carbon-centred radical 12. Notably, the reaction with PBN in the absence of 1 and 2 shows no radical formation, confirming that these reagents are the only source of the radical species in the reaction mixture.
Finally, we performed DFT calculations at the (U)M062x/def2TZVP level of theory, including a solvent model (IEFPCM, with toluene as the solvent) to evaluate the reaction’s energetic profile and rationalize the observed reaction outcome. To optimize computational resources, a sulfonyl imine featuring a mesyl group instead of the bulkier tosyl group was selected (2a; Fig. 3e). The S0 and T1 states of 2a and PS 9 have been computed. These calculations enabled us to elucidate the EnT process from the triplet state of PS 9 to the ground state of 2a, as outlined in the reaction scheme in Fig. 3e: 3(PS)∗ + 2a → PS + 3(2a)*. Remarkably, a Gibbs free-energy change (ΔG) of −9.2 kcal mol−1 (Fig. 3e, left) has been determined, underscoring the pronounced exergonic nature of the EnT process within the system. An examination of the spin-density distribution map of the relaxed triplet state of 9 (to optimize computational resources, the calculations of the ΔG of the energy transfer process were performed without taking into account the solvent model), combined with the observations acquired from the time-correlated single-photon counting experiments, which dictate that the triplet excited state is exclusively quenched, may diagnose that the EnT mechanism initiates via a collision complex involving the triazine moiety of PS 9 and the sulfonyl imine 2a (Supplementary Figs. 37 and 38). Next, the generation of the sulfonyl radical 10a and iminyl radical 11, resulting from the homolytic cleavage of the sulfonyl imine’s nitrogen–sulfur bond is determined to be exergonic, with a ΔG = −10.3 kcal mol−1. This step is accompanied by a relatively modest activation barrier (ΔG‡ = 8.1 kcal mol−1). Hence, we computed the insertion mechanism of the sulfonyl radical 10a at the C3 of the ABB 1, followed by radical–radical combination with the iminyl radical 11, ultimately yielding the final product 3a (Fig. 3e, right). Notably, the alternative structural isomer 5a, which remains unobserved experimentally, is theoretically computed to exhibit an energy of 21.9 kcal mol⁻1 higher than 3a. Thus, the absence of isomer 5a is more plausibly attributed to kinetic rather than thermodynamic factors. Indeed, our calculations support this scenario, revealing an energy barrier (ΔG‡) of 17.0 kcal mol−1 (TS2) between intermediate 12 (analogous to the benzyl radical) and the initial reactants 1 and 10a, while a substantially higher activation energy (ΔG‡ = 55.1 kcal mol−1) is determined between intermediates I2 and I3 (nitrogen-centred radical).
Generality of the method
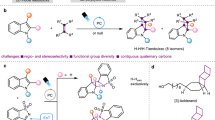
Having shed light on the reaction mechanism, we next evaluated the generality of this RSR photocatalytic process (Fig. 4). Various types of sulfonyl imines were competent substrates, including ketimine and aldimine precursors with electron-donating and electron-withdrawing groups, affording the azetidine products (3, 14–19) in up to 79% yield. Variations at the sulfonyl group were also tolerated well, and the corresponding products 18–27 were isolated in yields ranging from 47% to 80%. We next turned our attention to the ABB precursor. Here, diverse aryl and carbonyl groups were evaluated, delivering the corresponding azetidines (28–30) with yields of up to 84%. Additionally, 2-substituted ABBs furnished the products 31–34 in up to 82% yield, and 2.3:1 d.r. We obtained crystalline products for both diastereoisomers 33 and 34. By performing single-crystal X-ray analysis, we unambiguously determined their relative configurations. Finally, we applied the developed method to more systems with biologically relevant scaffolds. Remarkably, both the celecoxib-containing sulfonyl imine 35 and the naproxen-containing ABB 37 reacted smoothly, affording the corresponding azetidine products 36 and 38 in 71% and 66% yield, respectively. Notably, the mild reaction conditions allowed the preservation of the optical purity of the starting chiral naproxen derivative 37.

The reported yields refer to the average of isolated product obtained from three independent reactions. Unless otherwise noted, the reactions are performed at 0.05 mmol scale. CSD, Cambridge Structural Database (CSD 2305658, CSD 2305660, CSD 2305656, CSD 2305657, CSD 2305659); PMP, p-OMePh. aReaction performed in ethyl acetate.
Having proved the generality of the process for accessing C3–N-type azetidines via the activity of the key radical intermediate 12 (Fig. 3), we wondered if it would be possible to intercept this transient species with diverse single occupied molecular orbital (SOMO)philes. Considering the lower efficiency of aldimines in comparison to ketimines as a source of iminyl radicals45, we leveraged this feature by designing a three-component reaction manifold in which the ABB 1 and aldimines 39–41 react in the presence of the supersilane 42 (Fig. 5a, left). Remarkably, the reaction proceeded smoothly, affording 44–46 in up to 91% yield. Encouraged by these results, and given the importance of the SCF3 moiety for pharmaceuticals46,47, we tested phthalimide-SCF3 43. The reaction led to the corresponding thiotrifluoromethylated azetidines 47–50 in up to 71% yield, including two different celecoxib derivatives. This demonstrates that the designed methodology can easily unlock alternative types of reactivity while providing access to families of densely functionalized azetidines without additional optimization processes (for limitations of the protocol, see Supplementary Fig. 39).

a, Extension of the method to a three-component reaction with diverse SOMOphiles. b, Release and manipulations of the key amino functionalities of the products.
Synthetic utility
Finally, we performed various synthetic manipulations to reveal the key amino functionalities embedded in the azetidine products. The benzylated intermediate 51 can be accessed by reduction of 16 or reductive amination of 3. In both cases, there are very high yields (up to 95%) without the need for column chromatography. Hydrolysis of 3, 3a and 27 allowed the isolation of the hydrochloride salts 52, 53 and 54, respectively. Neutralization of 52 led to amino sulfonyl azetidine 55 without the need for purification steps. This is a key synthetic intermediate that can be easily transformed into tertiary amines, bearing a biologically relevant pyrrolidine such as 56. The same azetidine succeeded in tosyl group cleavage, giving rise to the two-vector 3-aminoazetidine 57, while providing access to a structural variant of the kinase modulators 58 (ref. 48). Lastly, by targeting the synthesis of amino acid 59 through a RuO4-mediated oxidative fission of the phenyl ring49,50, we obtained the unexpected yet attractive spiroazetidine scaffold 60 in 52% yield.
Conclusions
A key aspect of the devised synthetic platform is the selection and design of the PS, characterized by a small energy gap between the first singlet and triplet excited states. We speculate that this negligible difference favours an ideal balance between the inactive and active excited state species of the PS, resulting in the finely controlled sulfonyl imine activation. In addition, we proved that ABB is compatible with free-radical chemistry by means of an efficient, mild and selective radical strain release photocatalytic process. The developed synthetic platform allows easy access to a wide variety of densely functionalized azetidines with two exit vectors in high chemical yield. Mechanistic investigations revealed the pivotal activity of a transient carbon-centred radical located at the azetidine core, generated by the opening of the bicyclic compound, as this intermediate is suitable for engaging with nitrogen, sulfur and hydrogen atoms in new σ-bond formations. In conclusion, we foresee the use of ABBs in other types of radical strain release processes, expanding the synthetic repertoire for azetidine manufacturing.
Methods
General procedure for the difunctionalization of ABBs
In an oven-dried 10 ml Schlenk tube containing an oven-dried Teflon-coated stirring bar, the corresponding ABB (0.1 mmol, 2 equiv.) and sulfonyl imine (0.05 mmol, 1 equiv.) were dissolved in 0.6 ml of a solution of the PS 9 in PhCF3 (2.1 × 10−4 M; note that using this freshly distilled from phosphorus pentoxide is highly recommended). The Schlenk tubes were closed with suitable greased glass stoppers. The resulting solution was subjected to three cycles of free–pump–thaw to remove the oxygen, and in the last cycle, the vessel was filled with argon. The reaction was irradiated under stirring using a 50 W blue Kessil lamp (λmax = 456 nm) under argon atmosphere at 40 °C for 22 h. After completion, the resultant mixture was transferred to a 25 ml round-bottom flask and rinsing with ethyl acetate, and the volatiles were evaporated under reduced pressure. Purification of the resulting solid gave the desired difunctionalized azetidine.
General procedure for the difunctionalization of ABBs in the three-component variation
In an oven-dried 10 ml Schlenk tube containing an oven-dried Teflon-coated stirring bar, the corresponding ABB (0.1 mmol, 2 equiv.), sulfonyl aldimine (0.05 mmol, 1 equiv.) and SOMOphile (0.75 mmol, 1.5 equiv.) were dissolved in 0.6 ml of solution of the PS 9 in ethyl acetate (2.1 × 10−4 M; note that using this freshly distilled from phosphorus pentoxide is highly recommended). The Schlenk tubes were closed with suitable greased glass stoppers. The resulting solution was subjected to three cycles of free–pump–thaw to remove the oxygen, and in the last cycle, the vessel was filled with argon. The reaction was irradiated under stirring using a 50 W blue Kessil lamp (λmax = 456 nm) under argon atmosphere at 40 °C for 22 h. After completion, the resultant mixture was transferred to a 25 ml round-bottom flask and rinsing with ethyl acetate, and the volatiles were evaporated under reduced pressure. Purification of the resulting solid gave the desired difunctionalized azetidine.
Data availability
Details about materials and methods, experimental procedures, mechanistic studies, characterization data and NMR spectra are available in Supplementary Information. The atomic coordinates of the optimized models for all the computational studies are available as a separate public file. Crystallographic data are available from the Cambridge Structural Database with the following codes: PS 9 (CSD 2329152), 3 (CSD 2305658), 20 (CSD 2305660), 23 (CSD 2305656), 33 (CSD 2305657) and 34 (CSD 2305659). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/solutions/software/csd/. Additional data are available from the corresponding author upon request.
References
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Ricci, A. (ed.) Amino Group Chemistry: From Synthesis to the Life Sciences (Wiley-VCH, 2008).
Bhutani, P. et al. FDA approved drugs from 2015–June 2020: a perspective. J. Med. Chem. 64, 2339–2381 (2021).
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Lovering, F. Escape from flatland 2: complexity and promiscuity. MedChemComm 4, 515–519 (2013).
St. Jean, D. J. & Fotsch, C. Mitigating heterocycle metabolism in drug discovery. J. Med. Chem. 55, 6002–6020 (2012).
Shu, Y.-Z., Johnson, B. M. & Yang, T. J. Role of biotransformation studies in minimizing metabolism-related liabilities in drug discovery. AAPS J. 10, 178–192 (2008).
Andresini, M., Degennaro, L. & Luisi, R. The renaissance of strained 1-azabicyclo[1.1.0]butanes as useful reagents for the synthesis of functionalized azetidines. Org. Biomol. Chem. 18, 5798–5810 (2020).
Maetani, M. et al. Synthesis of a bicyclic azetidine with in vivo antimalarial activity enabled by stereospecific, directed C(sp3)–H arylation. J. Am. Chem. Soc. 139, 11300–11306 (2017).
Feskov, I. O. et al. 3-((Hetera)cyclobutyl)azetidines, “stretched” analogues of piperidine, piperazine, and morpholine: advanced building blocks for drug discovery. J. Org. Chem. 84, 1363–1371 (2019).
Rice, K. D. et al. Novel carboxamide-based allosteric MEK inhibitors: discovery and optimization efforts toward XL518 (GDC-0973). ACS Med. Chem. Lett. 3, 416–421 (2012).
Oizumi, K. et al. Antihypertensive effects of CS-905, a novel dihydropyridine Ca++ channel blocker. Jpn. J. Pharmacol. 51, 57–64 (1989).
Bauer, M. R. et al. Put a ring on it: application of small aliphatic rings in medicinal chemistry. RSC Med. Chem. 12, 448–471 (2021).
Kovács, E., Faigl, F. & Mucsi, Z. Regio- and diastereoselective synthesis of 2-arylazetidines: quantum chemical explanation of Baldwin’s rules for the ring-formation reactions of oxiranes. J. Org. Chem. 85, 11226–11239 (2020).
Betz, K. N., Chiappini, N. D. & Du Bois, J. Intermolecular sp3-C–H amination for the synthesis of saturated azacycles. Org. Lett. 22, 1687–1691 (2020).
Alcaide, B., Almendros, P. & Aragoncillo, C. β-Lactams: versatile building blocks for the stereoselective synthesis of non-β-lactam products. Chem. Rev. 107, 4437–4492 (2007).
Pitts, C. R. & Lectka, T. Chemical synthesis of β-lactams: asymmetric catalysis and other recent advances. Chem. Rev. 114, 7930–7953 (2014).
Franceschi, P., Cuadros, S., Goti, G. & Dell’Amico, L. Mechanisms and synthetic strategies in visible-light-driven [2 + 2]-heterocycloadditions. Angew. Chem. Int. Ed. 62, e202217210 (2023).
Rykaczewski, K. A. et al. Photochemical strategies enable the synthesis of tunable azetidine-based energetic materials. J. Am. Chem. Soc. 144, 19089–19096 (2022).
Becker, M. R., Wearing, E. R. & Schindler, C. S. Synthesis of azetidines via visible-light-mediated intermolecular [2 + 2] photocycloadditions. Nat. Chem. 12, 898–905 (2020).
Lopchuk, J. M. et al. Strain-release heteroatom functionalization: development, scope, and stereospecificity. J. Am. Chem. Soc. 139, 3209–3226 (2017).
Gianatassio, R. & Kadish, D. Direct alkylation of 1- azabicyclo[1.1.0]butanes. Org. Lett. 21, 2060–2063 (2019).
Fawcett, A., Murtaza, A., Gregson, C. H. U. & Aggarwal, V. K. Strain-release-driven homologation of boronic esters: application to the modular synthesis of azetidines. J. Am. Chem. Soc. 141, 4573–4578 (2019).
Funke, W. Synthesis and properties of 1-azabicyclo[1.1.0]butanes. Angew. Chem. Int. Ed. 8, 70–71 (1969).
Gianatassio, R. et al. Strain-release amination. Science 351, 241–246 (2016).
Musci, P. et al. Flow technology enabled preparation of C3-heterosubstituted 1-azabicyclo[1.1.0]butanes and azetidines: accessing unexplored chemical space in strained heterocyclic chemistry. Chem. Commun. 58, 6356–6359 (2022).
Hsu, C.-M. et al. Azetidines with all-carbon quaternary centers: merging relay catalysis with strain release functionalization. J. Am. Chem. Soc. 145, 19049–19059 (2023).
Marson, C. M. New and unusual scaffolds in medicinal chemistry. Chem. Soc. Rev. 40, 5514–5533 (2011).
Narayanam, J. M. R. & Stephenson, C. R. J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 40, 102–113 (2011).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013).
Skubi, K. L., Blum, T. R. & Yoon, T. P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 116, 10035–10074 (2016).
Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075–10166 (2016).
Dutta, S., Erchinger, J. E., Strieth-Kalthoff, F., Kleinmans, R. & Glorius, F. Energy transfer photocatalysis: exciting modes of reactivity. Chem. Soc. Rev. 53, 1068–1089 (2024).
Belloti, P. & Glorius, F. Strain-release photocatalysis. J. Am. Chem. Soc. 145, 20716–20732 (2023).
Beatty, J. W. & Stephenson, C. R. J. Amine functionalization via oxidative photoredox catalysis: methodology development and complex molecule synthesis. Acc. Chem. Res. 48, 1474–1484 (2015).
Lowe, J. T. et al. Synthesis and profiling of a diverse collection of azetidine-based scaffolds for the development of CNS-focused lead-like libraries. J. Org. Chem. 77, 7187–7211 (2012).
Mateos, J. et al. Unveiling the impact of the light source and steric factors on [2 + 2] heterocycloaddition reactions. Nat. Synth. 2, 26–36 (2023).
Mateos, J. et al. A visible-light Paternò–Büchi dearomatisation process towards the construction of oxeto-indolinic polycycles. Chem. Sci. 11, 6532–6538 (2020).
Strieth-Kalthoff, F., James, M. J., Teders, M., Pitzer, L. & Glorius, F. Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev. 47, 7190–7202 (2018).
Bryden, M. A. & Zysman-Colman, E. Organic thermally activated delayed fluorescence (TADF) compounds used in photocatalysis. Chem. Soc. Rev. 50, 7587–7680 (2021).
Lee, D. R. et al. Design strategy for 25% external quantum efficiency in green and blue thermally activated delayed fluorescent devices. Adv. Mater. 27, 5861–5867 (2015).
Zhang, D., Cai, M., Zhang, Y., Zhang, D. & Duan, L. Sterically shielded blue thermally activated delayed fluorescence emitters with improved efficiency and stability. Mater. Horiz. 3, 145–151 (2016).
Thoi, H. H., Ito, O., Iino, M. & Matsuda, M. Studies of sulfonyl radicals. 4. Flash photolysis of aromatic sulfones. J. Phys. Chem. 82, 314–319 (1978).
Ortica, F. et al. Mechanism of reaction and photoacid generation of N-oxysuccinimidoarylsulfonate PAGs: a laser flash photolytic study. Chem. Mater. 13, 2297–2304 (2001).
Tilby, M. J. et al. Photocatalytic late-stage functionalization of sulfonamides via sulfonyl radical intermediates. ACS Catal. 12, 6060–6067 (2022).
Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822–5880 (2018).
Zhou, Y. et al. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: new structural trends and therapeutic areas. Chem. Rev. 116, 422–518 (2016).
Substituted Cyclic Regulator of Protein Phosphatase 2A (PP2A) and How to Use It OY KR2023/145089 (RAPPTA Therapeutics, 2023).
Nunez, M. T. & Martin, V. S. Efficient oxidation of phenyl groups to carboxylic acids with ruthenium tetraoxide. A simple synthesis of (R)-γ-caprolactone, the pheromone of Trogoderma granarium. J. Org. Chem. 55, 1928–1932 (1990).
Burkhard, J. A., Guérot, C., Knust, H. & Carreira, E. M. Expanding the azaspiro[3.3]heptane family: synthesis of novel highly functionalized building blocks. Org. Lett. 14, 66–69 (2012).
Acknowledgements
This work was supported by MUR (Ministero dell’Università) PRIN 2020927WY3 (M.N., L.D.), Bando Prin 2022 PNRR – Missione 4: Istruzione e ricerca Componente C 2: Investimento 1.1 ‘Progetti di ricerca di Rilevante Interesse Nazionale’ – PRIN, finanziato dall’Unione Europea NextGenerationEU, Progetto: P2022WLAY7 - Chiral SUPRAmolecular Assemblies for Sustainable PHOTOCHEMical CO2 ValorisationSupraPhotoChem (https://www.mur.gov.it/it/pnrr/strumenti-di-attuazione/Linee-Guida-Soggetti-Attuatori/informazione-e-comunicazione), and (European Research Council) ERC-Starting Grant 2021 SYNPHOCAT 101040025 (L.D.). Chiesi Farmaceutici and D. Balestri are acknowledged for support with the D8 Venture X-ray equipment. P.C. thanks the University of Padova and the European Union—NextGenerationEU for financial support via the grant programme STARS@UNIPD, Frame-PSII and the MUR for a Young Researchers, Seal of Excellence fellowship (SOE_0000125). R.I.R. and V.C. acknowledge financial support from the European Union’s Horizon Europe research and innovation programme under Marie Sklodowska-Curie grant agreements ChirAzaL number 101105828 and PHOTO-STEREO number 101106125, respectively. A. Noble is gratefully acknowledged for insightful discussions and suggestions. We thank I. Fortunati and S. Mercanzin, Department of Chemical Sciences, University of Padova, for technical support.
Author information
Authors and Affiliations
Contributions
R.I.R. conceived the project and devised the experiments with L.D. and V.C. R.I.R., V.C., L.R. and S.V. carried out the reactions and isolated and characterized the products. L.D., R.I.R. and V.C. rationalized the experimental results. A.A., M.N. and M.B. performed the spectroscopic investigations using transient absorption spectroscopy and EPR. P.C. performed the DFT calculations. G.P. performed the X-ray analysis. L.D. and R.I.R. wrote the paper with contributions from all the authors. L.D. directed the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Catalysis thanks Adrian Whitwood and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–147, Advices 1–9, Equations 1–4 and Tables 1–10.
Supplementary Data (download TXT )
Computational data.
Supplementary Data 1
The .cif file of compound 3.
Supplementary Data 2
The .cif file of compound 20.
Supplementary Data 3
The .cif file of compound 23.
Supplementary Data 4
The .cif file of compound 33.
Supplementary Data 5
The .cif file of compound 34.
Supplementary Data 6
The .cif file of compound PS 9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rodríguez, R.I., Corti, V., Rizzo, L. et al. Radical strain-release photocatalysis for the synthesis of azetidines. Nat Catal 7, 1223–1231 (2024). https://doi.org/10.1038/s41929-024-01206-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41929-024-01206-4
This article is cited by
-
Homologative alkene difunctionalization
Nature Chemistry (2026)
-
Photochemical thiocarbonyl difluoride generation enables azetidine synthesis
Nature Communications (2026)
-
HFIP-assisted Brønsted acid-catalyzed ring opening of 1-azabicyclo[1.1.0]butane to access diverse C3-quaternary aza-azetidines and indole-azetidines
Nature Communications (2025)
-
A modular synthesis of azetidines from reactive triplet imine intermediates using an intermolecular aza Paternò–Büchi reaction
Nature Catalysis (2025)
-
Shining light to break planarity
Nature Catalysis (2024)
