
Abstract
Chrysosplenium sinicum, a traditional Tibetan medicinal plant, can successfully thrive in low-light environments for long periods of time. To investigate the adaptive evolution of shade plants in low-light environments, we generated a chromosome-scale genome assembly (~320 Mb) for C. sinicum by combining PacBio sequencing and Hi-C technologies. Based on our results, gene families related to photosynthesis and cell respiration greatly expanded and evolved in C. sinicum genome due to intracellular DNA transfer from organelle genome to nuclear genome. Under positive selective pressure, adaptive evolution of light-harvesting complex II (LHCII) component protein CsLhcb1s resulted in the expansion of threonine residues at the phosphorylation site of STN7 kinase, potentially establishing a crucial genomic foundation for enhancing C. sinicum’s adaptability in low-light environments. Through transcriptome and metabolome analysis, we identified chrysosplenol and chrysosplenoside as predominant flavonoid metabolites of C. sinicum and predicted their synthesis pathways. In addition, analysis of alternative splicing (AS) revealed that AS events help regulate state transition and flavonoid biosynthesis. The present study provides new insights into the genomes of shade plants exposed to low-light conditions and adaptive evolution of these genomes; in addition, the results improve our current knowledge on the biosynthetic and regulatory processes of chrysosplenol and chrysosplenoside.
Similar content being viewed by others


Introduction
Chrysosplenium sinicum Maxim (Chinese name “Zhonghuajinyao”, 2n = 24) is a perennial herb that belongs to the Chrysosplenium genus in the Saxifragaceae family1. The genus Chrysosplenium contains a large number of species (~83 species). These species generally prefer shade and dampness in growth environments and are widespread throughout Asia, America, Europe, and Africa, especially in temperate regions of Asia2,3. In particular, China contains diverse species from the genus Chrysosplenium. Chrysosplenium plants have been extensively used as a traditional Tibetan medicine in China and have been documented in various Tibetan pharmacopoeia monographs. The genus Chrysosplenium is described in “Tibetan Medicine Chronicles” to remove undesired heat and exert detoxification effects; in addition, the genus is used in medical recipes to treat fever, headache, acute icteric hepatitis, and acute liver necrosis. The flavonoids chrysosplenol and chrysosplenoside are the characteristic bioactive components within the genus Chrysosplenium4, which contains a large number of hydroxylated and methoxylated flavonoids. The demand for these traditional medicinal plants has increased in recent years, particularly for the species grown in China. However, despite the high medicinal value of the genus Chrysosplenium, molecular and genomic studies on this genus have rarely been carried out5.
In nature, plants are classified into sun or shade plants based on their ability to adapt to sunlight. In general, sun plants requires high levels of light exposure during their life cycle to grow, whereas shade plants often require considerable shade to grow6. In agriculture, environmental light intensity is among the most critical factors that influence crop development. As an abiotic stress, low-light exposure results in compromised photosynthesis, thus reducing the potential crop yields7,8. For instance, persistent cloudy weather or rainfall can reduce rice yields in India9,10. For long-term survival under low-light conditions, the photosynthesis system in shade plants has evolved to function optimally and use sunlight more efficiently. Since the biomass accumulation of crops is highly correlated with the photosynthetic rate under low-light conditions11, it would be beneficial to study the mechanism by which shade plants tolerate low-light environments.
Photosynthesis in plants utilizes captured light energy to produce ATP and NADPH through two major pigment-protein complexes, namely photosystem I (PSI) and photosystem II (PSII). The nuclear-encoded light-harvesting complex (LHC) (also known as the antenna complex), which increases the optical absorption cross-section of PSI and PSII12, captures solar energy and delivers it to the reaction center P700 in PSI and P680 in PSII13. Light-harvesting complex I (LHCI), which is composed of four subunits (Lhca1-4), connects to the PSI core via the PsaF or PsaJ subunits14. The antenna complex of PSII includes the trimeric light-harvesting complex II (LHCII), which consists of Lhcb1, Lhcb2, and Lhcb3 and three monomeric antennae (Lhcb4, Lhcb5 and Lhcb6)15. In addition, the complex contains a set of trimeric LHCIIs that can move between PSI and PSII in a process called state transitions, which allows photosynthetic organisms to adapt to changing light conditions16. In state 1, LHCII is almost exclusively attached to PSII. In state 2, the phosphorylation of LHCII by STN7 dissociates LHCII from PSII and attaches it to PSI17,18. PSI contains more chlorophyll a and stable red-shifted pigments and therefore can effectively utilize enriched far-red light under low-light conditions19. Studies on long-term dark-adapted plants have demonstrated that LHCII in state 2 increases the size of the PSI antenna and transfers energy very efficiently to PSI20, potentially improving the efficiency by which plants utilize light under low-light conditions. Therefore, to achieve better photosynthetic efficiency under low-light conditions, it is worthwhile to investigate whether the LHCII within shade plants tends to occur in state 2.
The intracellular transfer of DNA from the organelle genome to the nuclear genome has been a driving force in the evolution of eukaryotic genomes. Eukaryotic plants have evolved in a highly variable growth environment in which abiotic stresses increase the frequency of DNA transfer from organelles to the nucleus21; therefore, nucleus-localized organelle DNA may be essential for plant adaptive evolution under environmental stress, such as low-light stress. The constant transfer of organelle DNA has contributed to the great diversity of new genetic material for plant adaptive evolution22,23, and it provides the raw material for the generation of new functional genes24,25. In most cases, the nuclear genes that originate from organelle DNA transfer are nonfunctional, and most of these genes are eliminated rapidly from the nuclear genome26,27. However, in some rare cases, the transferred genes can acquire a suitable nuclear promoter for transcriptional activation and thus subsequently become functional. Many proteins encoded by these genes can translocate back into their original organelle to perform specific physiological functions if they can also capture a suitable transit peptide28. The remaining proteins perform other new functions that are not related to their previous functions29. However, the contribution of nucleus-localized organelle DNA to the adaptability of shaded plants to low-light environments remains unclear.
Flavonoids, which are a group of secondary metabolites that are abundant in plants, exhibit remarkable medicinal bioactivities, such as antioxidant, anti-inflammatory, and anti-infection effects. As a traditional Tibetan medicine, C. sinicum contains a high abundance of flavonoids. Chrysosplenol and chrysosplenoside are the characteristic bioactive components within C. sinicum, and belong to a special class of flavonoids. However, limited research has been conducted on the biosynthesis of flavonoids in Chrysosplenium plants, impeding the further use of this genus. On the other hand, flavonoids are important metabolites for overcoming physiological stress when plants are challenged by various biological and non-biological environmental factors. Therefore, the high accumulation of chrysosplenol and chrysosplenoside in C. sinicum may be attributed to its special characteristics in low-light environments.
In this study, we report a chromosomal-level genome assembly of C. sinicum, a traditional Tibetan medicine plant that predominantly thrives in low-light and humid forests as well as alpine shrubs at elevations ranging from 500 to 3550 m. In addition, the whole-genome sequence of C. sinicum, including its nuclear, chloroplast, and mitochondrial sequences, was included to extend our knowledge on the adaptation basis of shade plants in low-light environments; in addition, these results revealed potential gene-editing sites for enhancing crop tolerance to low-light stress. Genes related to the synthesis of chrysosplenol and chrysosplenoside were identified by analyzing metabolomic and transcriptomic data generated from three different tissues (roots, stems, and leaves). Additionally, alternative splicing (AS) analysis was performed to investigate the regulatory role of AS events in tissue-specific gene expression. In conclusion, the present study investigated the adaptive evolution of shade plants in low-light environments based on the C. sinicum genome and investigated the metabolite composition, flavonoid biosynthesis, and tissue-specific expression regulation within the Chrysosplenium plant.
Results
Genome assembly and annotation
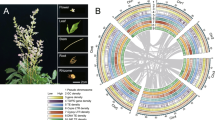
C. sinicum (2n = 24)30, a traditional Tibetan medicine plant, always grows in cold, moist shade beneath trees (Fig. 1a; Supplementary Fig. 1). Several technologies have been integrated to sequence and assemble its genome (Supplementary Table 1). K-mer analysis (k = 17) revealed that the genome size was ~333.50 Mb, with a repeat sequence content of 47.09%, and the estimated heterozygosity proportion was 0.4% (Supplementary Fig. 2; Supplementary Tables 2 and 3). Flow cytometry estimations also supported the estimated genome size of the K-mers (Supplementary Fig. 3). Using the PacBio Sequel platform, we produced a total of 39.80 Gb (119× in depth) of high-quality long PacBio reads with an N50 length of 16,478 bp (Supplementary Table 4). In addition, 105.26 Gb (316× in depth) of short paired-end reads were obtained (Supplementary Table 5). A total of 145.06 Gb (435× in depth) of C. sinicum DNA sequence was de novo assembled into 633 contigs, and the contig N50 was 2.13 Mb (Supplementary Table 6). The Hi-C data were then used to construct pseudo-chromosomes, and a total of 41.2 Gb (124× in depth) of Hi-C clean data were produced (Fig. 1b; Supplementary Table 7). The final assembly of C. sinicum was 321.79 Mb, with a scaffold N50 of 25.0 Mb (Table 1; Supplementary Table 8).

a Photographs of C. sinicum grow in low-light environments. b Genome-wide Hi-C map of C. sinicum. Post-clustering heatmap showing the density of Hi-C interactions between contigs. c Circos plot of the C. sinicum genome assembly. The density of genes in the track (A), GC content (B), simple sequence repeats (C), and long terminal repeat retrotransposons (D) were calculated using a 250 kb non-overlap window. The innermost layer shows syntenic blocks. The bandwidth is proportional to the syntenic block size.
The functional completeness of the assembly was evaluated by performing Benchmarking Universal Single-Copy Orthologues (BUSCO) analysis using the compleasm tool31. In total, 93.42% (2173 of 2326) of the core eudicot genes were present in the C. sinicum genome, including 2117 (91.01%) completely aligned genes (Supplementary Table 9). The consensus quality value (QV) and genome completeness were evaluated using the Merqury tool32, which yielded a QV of 43.1 and genome completeness of 95.7%. Additionally, the alignment results showed that 98.25% of the short reads could be aligned to the genome (Supplementary Table 10). The results suggested that the C. sinicum assembly is a high-quality reference genome. Repetitive elements accounted for 48.22% (155.16 Mb) of the C. sinicum genome, with long terminal repeats (LTRs) comprising 36.38% (56.45 Mb) (Supplementary Fig. 4; Supplementary Table 11). An inverse correlation between transposable elements (TEs) and gene density was observed on the chromosomes (Fig. 1c).
Three strategies, ab initio prediction, homologous prediction, and transcription-based prediction, were combined to construct gene models for the C. sinicum genome. A total of 27,441 protein-coding genes were predicted, with an average gene length of 3931 bp and an average coding sequence length of 1188 bp (Supplementary Table 12). Overall, 27,064 (98.63%) of the predicted genes had functional descriptions, according to Gene Ontology (GO) terms (17,124 genes; ~62.40%), KEGG (11,195 genes; ~40.80%)33, and the NR database (24,425 genes; ~89.01%) (Supplementary Table 13). GO term analysis revealed that 35.56%, 14.25%, and 50.19% of the predicted genes were involved in the biological process, cellular component, and molecular function categories, respectively (Supplementary Fig. 5). In addition, 1422 transcription factor (TF) encoding genes were identified, covering 70 TF gene families. Among them, the AP2/ERF, bHLH, MYB, and NAC families included the most genes, with 120 (8.44%), 105 (7.38), 97(6.82), and 83 (5.84%) genes, respectively (Fig. S6; Supplementary Data 1). Notably, the number of genes in the FAR-RED-IMPAIRED RESPONSE1 (FAR1; 60, 4.22%) family in C. sinicum genome was significantly greater than that in closely related plants (Supplementary Fig. 6).
Genome evolutionary analysis
Evolutionary analyses of plastid DNA sequences and the Kalanchoe fedtschenkoi nuclear genome placed Saxifragales as a sister group to rosids, and together, they constitute a large clade of superrosids1,34,35. However, there are inconsistencies in the nuclear gene tree based on a single Saxifragales species. To further validate the phylogenetic placement of Saxifragales and elucidate its divergence time from rosids, a phylogenomic tree of four Saxifragales plants, eight rosid plants, and eight other monocot and dicot plants was constructed using single-copy genes (Fig. 2a and Supplementary Fig. 7; Supplementary Data 2). Consistent with previous studies, the phylogenomic tree revealed that Saxifragales is a sister to the rosids (Fig. 2a). The divergence of Saxifragaceae (exemplified by C. sinicum) from Crassulaceae (exemplified by K. fedtschenkoi) occurred approximately 8–105 million years ago (MYA), while the separation between Saxifragales and rosids (exemplified by Rosales) took place approximately 104-123 MYA (Fig. 2a).

a Inferred phylogenetic tree reconstructed from single-copy orthologous genes shared by 20 species. Divergence timings were estimated using MCMCTree and are indicated by blue bars at the internodes with the 95% highest posterior density. Ea. Early, M. Middle, La. Late, Ju. Jurassic, Pa. Paleogene, P. Paleocene, Eo. Eocene, O. Oligocene, Ne. Neogene, Mi. Miocene. Green block: Superasterids; orange block: Saxifragales; blue block: Rosids. b Dot plot illustrating the comparative analysis of C. sinicum-grape plants; the black dots represent the syntenic gene blocks. The red boxes highlight several major duplication events between C. sinicum and grape. c Distribution of the synonymous substitution rates (Ks) for pairs of syntenic paralogues in C. sinicum and three other plant species. d Karyotype evolution of C. sinicum and related species.
Ancient whole-genome duplication (WGD) events are important driving forces of the evolution of plant lineages36,37. We examined grapes as a reference for studying genome duplication events in C. sinicum, because its genome did not undergo additional duplication after ancestral gamma hexaploidization38,39. The analysis of collinearity and synteny depth indicated that the grape genome contained one to two syntenic blocks in C. sinicum (Fig. 2b; Supplementary Fig. 8). Moreover, the distribution of synonymous substitution rates (Ks) in both reciprocal best hit (RBH) paralogous gene pairs and syntenic block gene pairs showed a single peak (Fig. 2c; Supplementary Fig. 9). These results provide strong evidence that an independent WGD event occurred in C. sinicum after the shared gamma-WGT event.
Intragenomic collinearity analysis of C. sinicum revealed that the predominant types of gene duplication were dispersed duplication (40.29%, 9627) and WGD/segmental duplication (30.56%, 7303), in contrast to tandem duplication (5.78, 1380) and proximal duplication (5.42, 1294) (Supplementary Table 14; Supplementary Data 3). Interestingly, the significantly enriched GO terms of genes originating from WGD or segmental duplication were closely associated with plant photosynthesis (Supplementary Fig. 10; Supplementary Data 4). Orthologous genes exhibiting signs of positive selection usually undergo adaptive divergence40. To explore the biological function of positively selected genes (PSGs) in the C. sinicum genome, we conducted a positive selection analysis on single-copy genes from ten Superrosids plants (Supplementary Fig. 11). Our findings revealed 132 PSGs in the C. sinicum genome (Supplementary Data 5). Moreover, functional classification analysis indicated that these PSGs are actively involved in crucial biological processes such as DNA replication, DNA recombination, and protein phosphatases (Supplementary Fig. 12; Supplementary Data 6).
The association between chromosomal reorganization and karyotype evolution has been substantiated in previous studies41. The genome of Superrosids plants evolved from a pre-γ ancestral eudicot karyotype (AEK) with 7 protochromosomes to a post-γ AEK karyotype with 21 protochromosomes42. To investigate chromosomal reorganization in the C. sinicum genome, we mapped the AEK genes onto the chromosomes of seven Superrosids species (C. sinicum, V. vinifera, E. grandis, M. truncatula, F. vesca, P. trichocarpa, and A. thaliana) (Fig. 2d). The V. vinifera genome has the closest resemblance to the ancestral chromosomal composition, while other Superrosids species underwent varying degrees of chromosome fusion. The C. sinicum genome experienced complex chromosome fusions, with each chromosome originating from a minimum of four ancestral chromosomes; an exception was chromosome 4, which originated from only two ancestral chromosomes (AEK1 and AEK5) (Fig. 2d; Supplementary Table 15).
Comparative genomics of gene families
C. sinicum is a shade plant that can adapt to well to low-light conditions. To elucidate the genomic basis underlying the distinctive phenotypic traits of C. sinicum, we characterized its gene families as unique or shared by comparing them with those of three other Saxifragales plants, namely, Rhodiola crenulate, Kalanchoe laxiflora, and Kalanchoe fedtschenkoi. The comparative analysis revealed a total of 9908 gene families distributed across all four genomes, with an additional 807 gene families exclusively found in C. sinicum (Fig. 3a). GO enrichment analysis of the 807 unique gene families, comprising 1649 genes, revealed eleven significant GO terms. Notably, the following terms were directly associated with photosynthesis: ‘photosynthesis, light reaction’, ‘photosynthetic electron transport chain’, and ‘photosynthesis’ (Fig. 3b; Supplementary Data 7). This suggests that the evolution of photosynthesis-related gene families may play a pivotal role in facilitating C. sinicum’s adaptability to low-light conditions.

a Venn diagram of shared orthologous gene families among the genomes of four Saxifragales species. b GO enrichment results of 807 C. sinicum-specific gene families. c Statistics of expanded and contracted gene families in C. sinicum. d Venn diagram of C. sinicum-specific gene families and expanded gene families. e GO enrichment results of 609 rapidly evolving (expanded) gene families in C. sinicum. The relevant terms associated with photosynthesis are denoted in green, while those pertaining to respiration are indicated in blue. f Functional annotation of the expanded gene families in C. sinicum. The left panel shows the gene family size among 11 species and the right panel shows the functional annotation and classification of gene families. Cs C. sinicum, Rc Rhodiola crenulata, Kf Kalanchoe fedtschenkoi, Vv Vitis vinifera, Eg Eucalyptus grandis, Mt Medicago truncatula, Fv Fragaria vesca, Pt Populus trichocarpa, Csi Citrus sinensis, At Arabidopsis thaliana, Tc Theobroma cacao. RE rapidly expanded. NE normal expanded. U C. sinicum specific.
The analysis of gene family expansion and contraction in C. sinicum revealed that 1580 and 6204 gene families were classified as expanded and contracted, respectively. Among these gene families, 1021 were recognized as rapidly evolving (609 expanded and 412 contracted) (Fig. 3c), while a subset of 104 gene families exhibited rapid expansion and were unique families (Fig. 3d). The rapidly evolving gene families may provide potential insight into the adaptability of C. sinicum to low-light environments. Functional enrichment analysis of these 609 rapidly expanded gene families revealed that their functions were closely associated with the process of plant photosynthesis and respiration (Fig. 3e and Supplementary Fig. 13; Supplementary Data 8). We also performed functional enrichment analysis on two sets of families: 104 rapidly expanding and unique families, and 505 rapidly expanding but non-unique families. The results revealed that terms related to photosynthesis were enriched in both sets, while terms associated with protein phosphorylation were exclusively enriched in the unique families. Conversely, respiratory-related terms were solely enriched in the non-unique families (Supplementary Fig. 14; Supplementary Data 8). The evolution of the electron transport chain (such as electron transfer flavoprotein) used for cellular respiration in mitochondria allows plants to adapt to sucrose starvation induced by darkness43. Multiple GO terms associated with cellular respiration, such as ‘aerobic respiration’, ‘respiratory electron transport chain’, and ‘ATP synthesis coupled electron transport’, were significantly enriched in rapidly expanding gene families; thus, the rapidly evolving gene families involved in cellular respiration may also play a crucial role in enhancing C. sinicum’s adaptability to low-light conditions (Fig. 3e; Supplementary Data 8).
To further investigate the evolution of gene families related to photosynthesis and cellular respiration in C. sinicum, a functional classification was performed on expanded gene families. The initial steps of photosynthesis, also known as light reactions, involve two large membrane protein complexes referred to as photosystem I and photosystem II. Among the expanded gene families, two gene families comprising 14 genes were implicated in the function of photosystem I, while ten gene families consisting of 56 genes were related to photosystem II (Fig. 3f). Notably, the gene family known as the LHCII component protein Lhcb1, which was previously found at significantly greater levels in shade-tolerant plants than in light-resistant plants44, was expanded in C. sinicum. Three of the expanded gene families were involved in the electron transport chain of respiration, and among them, the electron transfer flavoprotein: ubiquinone oxidoreductase (ETFQO) family is crucial for the ability of plants to withstand extended darkness43,45 (Fig. 3f). Moreover, functional classification analysis revealed that other gene families associated with photosynthesis and respiration, such as ‘phytochrome’, ‘photosynthetic electron transport chain’, and ‘mitochondrial respiratory chain complex I’, were expanded (Fig. 3f).
Intracellular DNA transfer in Chrysosplenium sinicum
Among the genes associated with photosynthesis, we observed a significantly lower number of repetitive elements in the proximal region (upstream and downstream 50 kb) of rapidly expanded genes than in other genes (Fig. 4a). To determine the distribution of genes with fewer repetitive elements in the proximal region, we counted the number of repetitive elements near each gene at the whole-genome scale and arranged them based on their chromosomal order (Fig. 4b). The analysis revealed the presence of multiple large gene clusters, which exhibited a significantly lower number of repetitive elements and were distributed across multiple chromosomes (Fig. 4b; Supplementary Fig. 15). Among these gene clusters, we identified five gene clusters associated with photosynthesis and cellular respiration through functional annotation (Fig. 4b; Supplementary Data 9). The five gene clusters included 47.62% rapidly expanded photosynthesis-related genes and 20% rapidly expanded cell-respiration-related genes. Cluster 1 comprised 19 genes related to photosynthesis and was located on Chr03. Clusters 2, 3, and 4 were located on Chr04. Specifically, Cluster 2 consisted of 24 genes related to cellular respiration, while Clusters 3 and 4 comprised a total of 44 and 24 genes associated with photosynthesis, respectively. Cluster 5, located on Chr11, exhibited the greatest number of photosynthesis-related genes (49 genes) (Fig. 4b; Supplementary Data 9). The intracellular transfer of DNA from the organelle genome to the nuclear genome is widespread and ongoing in eukaryotes46, constituting a driving force in the evolution of eukaryotic genomes. Given the relatively lower number of repetitive sequences in organelle genomes, these gene clusters may have originated from the transfer of organelle DNA.

a Statistics of the number of repetitive elements in the proximal region of rapidly expanded genes and not rapidly expanded genes. Student’s t-test (two-sided, pvalue = 1.41E-14): *P < 0.05; **P < 0.01; ***P < 0.001. b Overview of the abundance of repetitive elements in the proximal region (upstream and downstream 50 kb) of genes on Chr03, Chr04, and Chr11. The abscissa shows the arrangement of genes along a chromosome. c Gene map of the C. sinicum chloroplast genomes. Genes are color-coded to indicate functional groups. d Distribution of NUPT and NUMT sequences on the 12 chromosomes of C. sinicum. e Collinearity between the chloroplast fragments and Cluster 5. The asterisk-marked genes represent novel recombination genes. The ordinate of coverage of long reads represents log10(number of long-reads coverage). f Schematic structure and homology between accD in chloroplasts and the genomic region on Chr03 containing Cs3G0076700 and Cs3G0076800. g Schematic structure and homology between Cs4G0091400 on Chr04 and the chloroplast region containing psaC and ndhD. The gray blocks represent regions that are homologous.
To verify the origin of these gene clusters, we assembled the complete genomes of the chloroplasts and mitochondria of C. sinicum. The length of the chloroplast genome was 153,427 bp (Fig. 4c). The mitogenome was assembled as three circular molecules (chromosome 1: 233,531 bp, chromosome 2: 116,502 bp, chromosome 3: 45,818 bp) (Supplementary Fig. 16). The distribution of nucleus-localized plastid or mitochondrial DNAs (NUPTs or NUMTs) suggested that Clusters 1, 3, 4, and 5 were composed of NUPT fragments derived from the chloroplast genome, while Cluster 2 consisted of NUMT fragments originating from mitochondrial genome transfers (Fig. 4d). These findings indicate that intracellular DNA transfer served as a driving force behind the rapid expansion of gene families associated with photosynthesis and cell respiration in C. sinicum, potentially facilitating its adaptive evolution to low-light environments.
Homology analysis between chloroplast genes and nuclear genes revealed that genes in four photosynthesis-related clusters originated from two distinct fragments of the chloroplast genome, namely fragment 1 and fragment 2. (Fig. 4c). In the nuclear genome of plants, NUPTs and NUMTs frequently exhibit nonrandom arrangements, forming loose or tight clusters based on their physical linkage degrees47. Collinearity analysis revealed significant synteny between Clusters 1 and 5 within the chloroplast genome, as the gene arrangement in these two clusters closely resembled that observed in chloroplast genes (Fig. 4e; Supplementary Fig. 17). Moreover, collinearity analysis of Cluster 3 and 4 with the chloroplast genome was conducted; however, no significant synteny was observed between them (Supplementary Fig. 17). Interestingly, Clusters 3 and 4 exhibited significant synteny with Cluster 5 and most genes within Clusters 3 and 4 were identified as WGD or segmental duplication types; thus, WGD or segmental replication potentially contributed to the generation of Clusters 3 and 4 (Supplementary Fig. 18; Supplementary Data 3). These results indicated that the rapidly expanded gene families associated with photosynthesis in the C. sinicum may be attributed to the combined effects of intracellular DNA transfer and segmental or WGD replication.
During the early phase of eukaryote evolution, the nuclear transfer of organellar DNA resulted in the massive relocation of entire genes to nuclear chromosomes24. Researchers believe that the recent transfer of organelle DNA to the nucleus mainly yielded nonfunctional nuclear sequences. However, new studies have suggested that the exons of novel genes in the nuclear genome can be generated by the insertion of organellar DNA48. In C. sinicum, the recruitment of preexisting organellar protein domains to novel nucleus-encoded proteins is a prevalent phenomenon. The novel recombination genes, which integrate one or more organelle gene domains, accounted for 24.06% (32) of the genes in the four photosynthesis-related clusters (Supplementary Fig. 19). For example, Cs4G0094400 integrates the domains of four organelle genes, namely rpl14, rpl16, rps3, and rpl22, as exons, while the recombination of the organelle gene accD results in the generation of two novel genes, Cs3G0076700 and Cs3G0076800 (Fig. 4f; Supplementary Fig. 20). To confirm that the novel genes found in the nuclear genome were inserted fragments from organelle DNA rather than from assembly mistakes, we examined the coverage of long reads for these clusters. The results revealed that the long reads for these clusters were highly covered, and the novel genes were completely covered within the long reads (Fig. 4e; Supplementary Fig. 17). Furthermore, we amplified the novel genes from nuclear DNA using PCR, and the sequencing data demonstrated that these genes were identical to the genome assembly results (Supplementary Fig. 17; Supplementary Data 10).
Approximately 26.11% of the genes in the five clusters were expressed in different tissues of C. sinicum (Supplementary Data 11). Notably, a chloroplast transfer gene (Cs11G0121300) called the large subunit of RUBISCO (RBCL) exhibited a high level of expression (Supplementary Fig. 21; Supplementary Data 11). Cluster 2, which originated from the mitochondrial genome, exhibited the greatest proportion of expressed genes, as 14 out of 24 (58.33%) genes were expressed (Supplementary Fig. 21; Supplementary Data 11). Furthermore, most of the novel recombination genes exhibited negligible expression levels, except for Cs4G0091400, which was produced from an inversion of a fragment between the organelle genes psaC and ndhD, and displayed a relatively high expression level (Fig. 4g and Supplementary Fig. 21; Supplementary Data 11). Based on the extensive expression of these genes derived from organellar DNA transfer, the genes may provide the foundation for the adaptation of C. sinicum to low-light environments.
Adaptive evolution of the CsLhcb1s subfamily in Chrysosplenium sinicum
The LHC super-gene family encodes light-harvesting chlorophyll a/b-binding proteins, which constitute the antenna systems of the photosynthetic apparatus. These antenna systems efficiently capture solar energy and transfer it to PSI and PSII, thereby providing electrons for subsequent dark reactions. (Fig. 5a)49. The structure of the antenna complexes in different types of photosynthetic organisms is remarkably diverse, suggesting that early organisms adapt to specific light environments through the evolution of antenna complexes50. A total of 25 genes of the LHC family were identified in C. sinicum, including five Lhca genes and 20 Lhcb genes (Supplementary Data 12). A comparative genomic analysis between Saxifragales and Rosids revealed that the Lhcb1 subfamily expanded in three Saxifragales species, namely C. sinicum, R. crenulate, and K. laxiflora (Fig. 5b). Moreover, through gene structure analysis, we observed a significant increase in intron sequences within the CsLhcb1s of C. sinicum compared to those of closely related species (Supplementary Fig. 22). The gains and losses of introns are key factors in the evolution of genomes, and major evolutionary transitions may be accompanied by massive gains and losses of introns51,52. These results indicate that the Lhcb1 subfamily of C. sinicum possibly underwent adaptive evolution in response to low-light conditions.

a Photosynthetic light response pathway map in C. sinicum. b Statistics of gene numbers within each subfamily of the LHC superfamily were analyzed for four Saxifragales species and eight Rosids species. c Multiple sequence alignment of amino acid sequences of Lhcb1 genes from C. sinicum and three other Saxifragales species. The amino acid highlighted in red serves as the recognition site of STN7 kinase. d Expression and phylogenetic profiles of genes in the LHC superfamily in roots, stems, and leaves from C. sinicum. e Prediction of the phosphorylation potential of threonine residues surrounding the STN7 kinase recognition site in CsLhcb1s. PKC protein kinase C, unsp non-specific protein kinase.
Positive selection analysis using the program Codeml of PAML53 enables us to better understand the evolutionary process. A total of seven positively selected sites were detected, indicating that the CsLhcb1s of C. sinicum were subjected to positive selective pressure (Supplementary Fig. 23; Supplementary Data 13). The positive selection of specific amino acid residues in the CsLhcb1s may influence the adaptability of C. sinicum to low-light environments.
Photosynthetic organisms adapt to a variety of light conditions by redistributing light excitation energy between PSI and PSII through state transitions54. The reversible association of LHCII between PSI and PSII facilitates the adaptation of plants to diverse light conditions. The relocation of LHCII from PSII to PSI (state transition from state 1 to state 2) is initiated by the phosphorylation of specific threonine residues at the N-terminus of the Lhcb1 and Lhcb2 proteins by STN7 kinase18,55. The CsLhcb2 protein was strictly conserved at the phosphorylation site of STN7 kinase (Supplementary Fig. 24). Among the five CsLhcb1s proteins, a noticeable increase in the number of threonine residues adjacent to the phosphorylation site of STN7 kinase was observed compared to that in closely related species (Fig. 5c and Supplementary Fig. 25). Among them, CsLhcb1.5, CsLhcb1.8, CsLhcb1.9, and CsLhcb1.12 contain two threonine residues adjacent to the phosphorylation site of STN7 kinase, while CsLhcb1.10 contains three threonine residues near the phosphorylation site of STN7 kinase (Fig. 5c and Supplementary Fig. 25). Furthermore, a positively selected site is located near the phosphorylation site of STN7 kinase, implying that natural selection influences the expansion of threonine residues at the phosphorylation site of STN7 kinase in C. sinicum’s CsLhcb1s proteins (Fig. 5c, Supplementary Figs. 23 and 25; Supplementary Data 13).
Among the LHC family members of C. sinicum, CsLhca1, CsLhca4, CsLhca2.1, CsLhcb1.13, CsLhcb2, and CsLhcb5 exhibited high expression levels in leaf tissue (Fig. 5d). The CsLhcb1s, which contain multiple threonine residues at the phosphorylation site of STN7 kinase, are expressed in leaf tissue (Fig. 5d; Supplementary Data 12). Thereafter, we performed a prediction analysis on the threonine residues adjacent to the phosphorylation site of STN7 kinase in the Lhcb1 family using NetPhos 3.1 server56. Through this tool, threonine residues were identified as potential phosphorylation sites that may be susceptible to kinase-mediated phosphorylation (Fig. 5e). These results suggest that the adaptive evolution of CsLhcb1s potentially plays a crucial role in enhancing the ability of C. sinicum to adapt to low-light environments.
In addition to the LHC family of C. sinicum, we further identified the gene families involved in photosynthesis and analyzed their replication and expansion. With respect to photosystem I, tandem and WGD/segmental duplications increased the gene numbers of the PsaA (photosystem I P700 chlorophyll a apoprotein A1) and PsaB (photosystem I P700 chlorophyll a apoprotein A2). Within photosystem II, the genes of multiple protein subunits, including cp43, cp47, PsbE, PsbF, PsbO, and PsbQ, were expanded through WGD/segmental replication. Furthermore, WGD/segmental duplication also expanded the gene families of cytochrome f (PetA), cytochrome b6 (PetB), and subunit IV (PetD) within the cytochrome b6f complex (Supplementary Data 14).
Synthetic pathway of chrysosplenol and chrysosplenoside
Flavonoids, which are widely distributed in nature and exhibit diverse pharmacological activities, are important active constituents for clinical applications of species in the genus Chrysosplenium5. A total of 594 metabolites were identified in the three tissues (roots, stems, and leaves) of C. sinicum, including 128 (21.6%) flavonoids, 108 (18.2%) lipids, 104 (17.5%) phenolic acids, 63 (10.6%) organic acids, and 59 (9.9%) amino acids and derivatives (Fig. 6a). Chrysosplenol and chrysosplenoside represent the predominant flavonoid metabolites in C. sinicum, constituting 54.07% of the total flavonoid content (Supplementary Data 15 and 16). Distinct differences were observed among samples from different tissues in the PCA score plots and metabolite heatmaps of the samples, indicating that metabolite accumulation was tissue-specific (Supplementary Fig. 26). Through differentially abundant metabolite analysis, we observed that chrysosplenol and chrysosplenoside levels were significantly increased in the leaf tissue (Fig. 6b; Supplementary Fig. 27).

a Pie graph of metabolite classification in C. sinicum. b Top ten metabolites of flavonoids in C. sinicum. Heatmap showing the differential content of the top ten flavonoids in three tissues (root, stem, and leaf). c Presumptive biosynthesis pathway of chrysosplenol and chrysosplenoside in C. sinicum. Heatmap showing the expression levels of genes involved in the biosynthesis of chrysosplenol and chrysosplenoside in three tissues (roots, stems, and leaves). Dashed arrows represent the presumed biosynthesis pathway. PAL phenylalaninammo-nialyase, C4H trans-cinnamate 4-monooxygenase, 4CL 4-coumarate-CoA ligase, CHI chalcone isomerase, F3H naringenin 3-dioxygenase, FLS flavonol synthase, F3′H flavonoid 3′-monooxygenase, F3′5′H flavonoid 3′,5′-hydroxylase. d Correlation analysis between genes and metabolites. Pale green circles denote metabolites chrysosplenol and chrysosplenoside. The orange circles denote synthetase.
Combined with the chemical structure and the flavonoid biosynthesis pathway in KEGG, we predicted the synthetic pathway of chrysosplenol and chrysosplenoside in C. sinicum (Fig. 6c). In the presumptive synthetic pathway, chrysosplenol B, chrysosplenol C, and chrysosplenol D are formed through hydroxylation and methylation of kaempferol, while chrysosplenol E, chrysosplenol F, and chrysosplenol G are generated via hydroxylation and methylation of quercetin. Finally, O-glycosyltransferases (UGTs) serve as the glycosylation enzymes responsible for synthesizing chrysosplenoside from chrysosplenol (Fig. 6c). Chalcone synthase (CHS) is the first committed enzyme in the conserved flavonoid biosynthesis pathway among plants57. A total of nine CsCHS genes were identified in C. sinicum, and the CsCHS gene family expanded. A heatmap revealed that three CsCHSs (CsCHS.3, CsCHS.4, and CsCHS.7) were differentially expressed in leaf tissue, which was consistent with the observed accumulation pattern of chrysosplenol and chrysosplenoside (Fig. 6c; Supplementary Data 17). Additionally, a total of 57 CsUGTs were identified in C. sinicum, among which 14 CsUGTs exhibited differential upregulation in leaf tissue (Fig. 6c; Supplementary Data 17).
A correlation analysis was then performed using Pearson’s correlation coefficient (>0.95) to further explore the genes involved in chrysosplenol and chrysosplenoside synthesis and examine the correlation between gene expression and metabolite content. The results revealed significant correlations between the expression of CsCHS.3, Cs4CL.5, one flavonoid o-methyltransferase (AOMT), two isoflavone 2′-hydroxylases (CYP81Es), and seven CsUGTs and the content of chrysosplenol and chrysosplenoside, suggesting that these genes play roles in the biosynthesis of chrysosplenol and chrysosplenoside (Fig. 6d, Supplementary Data 18). The AOMT enzyme methylates hydroxyl groups at the 3′ and 5′ carbon positions in the B ring of flavonoids, while the CYP81E-encoded enzyme catalyzes the hydroxylation of isoflavones to produce 2′-hydroxyisoflavones (Supplementary Fig. 28). These two enzymes potentially participate in chrysosplenol synthesis through hydroxylation and methylation processes. The expression of flavonoid biosynthetic genes is regulated by the transcription factors MYB and bHLH58,59. Significant associations were detected between the contents of chrysosplenol and chrysosplenoside and multiple genes encoding bHLH and MYB transcription factors (Fig. 6d; Supplementary Data 18).
Gene regulation among different tissues in Chrysosplenium sinicum
Alternative splicing (AS) enriches the diversity of proteins and phenotypic traits by generating multiple transcripts from a single gene. To determine the function of AS in gene regulation in C. sinicum, we analyzed the different splice isoforms in our dataset and identified a total of 14,173 AS events, which were categorized into the following types across three tissues (root, stem, and leaf): retained intron (IR), skipping exon (SE), alternative 5ʹ/3ʹ splice sites (A5SS/A3SS), mutually exclusive exons (MX), and alternative first or last exons (AF/AL). A high frequency of RIs (36.94%) was identified, followed by A3SS, A5SS, SE, AF, AL, and MX (Fig. 7a).

a Overview of the seven different types of alternative splicing (AS) events and their frequencies in the three tissues of C. sinicum. b Pie graph for the percentages of differentially alternatively spliced genes. c KEGG enrichment results of 5239 alternatively spliced genes. d Comparison of differentially expressed genes (DEGs) and differentially alternatively spliced genes (DASGs) among different tissues. e AS variants for genes involved in photosynthesis and flavonoid biosynthesis. For each gene, AS-covering and total long-read counts are shown for AS variants in different tissues, and differential expression of AS variants is indicated by ΔPSI. ‘Up’ and ‘down’ represent up-regulated and down-regulated, respectively, between tissues.
The 14,173 alternative splicing events corresponded to 5239 alternatively spliced genes (ASGs). KEGG enrichment analysis of the 5239 alternatively spliced genes revealed “protein kinases” as the most significant category (Fig. 7b). Among these alternatively spliced genes, 25.6% were classified as differentially alternatively spliced genes (DASGs) (Fig. 7c; Supplementary Data 19). Specifically, when comparing roots to stems, 447 differential AS events were identified for 339 DASGs; when comparing leaves to stems, 1394 differential AS events were detected for 1043 DASGs; and when comparing leaves to roots, 1155 differential AS events were observed for 893 DASGs (Fig. 7d; Supplementary Data 19).
We also identified genes with differential expression between different tissues. A total of 4097, 5199, and 6379 differentially expressed genes (DEGs) were identified in the roots vs. stems, leaves vs. stems, and leaves vs. roots, respectively. The number of DEGs was nearly twelve times greater than the number of DASGs (4097 versus 339) between the roots and stems, approximately five times greater than the number of DASGs (5199 versus 1043) between the leaves and stems, and approximately seven times greater than the number of DASGs (6379 versus 893) between the leaves and roots (Fig. 7d). The intersection of DEGs and DASGs accounted for 2.0%, 4.3% and 4.1% in the roots vs. stems, leaves vs. stems and leaves vs. roots, respectively (Fig. 7d; Supplementary Data 19-20).
To elucidate the functions of genes within the DEGs and DASGs identified, a functional enrichment analysis was performed. The consistently enriched terms ‘flavonoid biosynthesis’ and ‘photosynthesis-antenna proteins’ were detected across all comparable groups of DEGs (Supplementary Fig. 29–31). The enrichment analysis revealed that the DASGs were associated with several GO terms, including ‘protein kinases’, ‘kinase activity’, ‘calmodulin binding’, ‘protein dephosphorylation’, and ‘protein serine/threonine phosphatase activity’ (Supplementary Figs. 29–31). Protein kinases play significant roles in regulating plant responses to various abiotic stresses60, indicating that AS potentially regulates protein phosphorylation and responds to abiotic stress. As a protein kinase, the A3SS-type AS variant of CsSTN7 exhibited decreased express levels in stems vs. leaves, suggesting that AS helps regulate the LHCII state transition. Two CsFAR1s also exhibited alternative splicing events (Fig. 7e). In addition to DEGs, AS events for genes in the flavonoid biosynthesis pathway, such as CsUGT.35 and CsUGT.41, may represent another mechanism by which flavonoid biosynthesis is regulated (Fig. 7e).
Discussion
Low-light intensity is widely recognized as a prevalent abiotic stress that negatively impacts photosynthesis and leads to a reduction in crop yield potential. The growth of cucumber is impeded under low-light stress, resulting in compromised cucumber quality and reduced yields61,62. In the wet season in India and Southeast Asian countries, low-intensity weather often leads to a significant yield loss in rice cultivation63. Therefore, breeders and researchers have focused on developing or generating new crop varieties with enhanced tolerance to low-light conditions64,65,66. In the present study, we selected a classic shade plant, C. sinicum, as a model and investigated the mechanism underlying the low-light tolerance of its chromosome-scale genome. Our results demonstrated that the rapid expansion of photosynthesis-related gene families through intracellular DNA transfer and the adaptive evolution of CsLhcb1s could establish a crucial genomic foundation for the increased adaptability of the shade plant C. sinicum to low-light environments.
Our comparative genomic analysis of gene families revealed that numerous photosynthesis-related gene families with low repetitive element density in C. sinicum rapidly expanded, which was attributed to intracellular DNA transfer and segmental/WGD replication (Fig. 4e and Supplementary Fig. 17). By analyzing the density of repetitive elements surrounding nuclear genes at the chromosome level, we successfully detected large-scale intracellular DNA transfer within chromosomes (Fig. 4b). In the C. sinicum genome, a total of five large gene clusters originating from the organelle genome were identified (Fig. 4b, d). Interestingly, four gene clusters derived from the chloroplast genome are associated with the photosynthesis process, while a single cluster originating from the mitochondrial genome is involved in the cellular respiration process. The photosynthetic system is directly correlated with the efficiency by which plants utilize light energy, and cellular respiration is intricately connected to a plant’s capacity to survive in dark environments43,45. Moreover, these genes, which are located within the five clusters associated with photosynthesis and cellular respiration processes, are extensively expressed in leaf tissue (Supplementary Fig. 21). These results strongly support our speculation that intracellular DNA transfer was a driving force in the adaptive evolution of C. sinicum towards long-term survival in low-light environments.
In this study, we observed a significant increase in the number of threonine residues adjacent to the STN7 kinase phosphorylation site in the CsLhcb1s from C. sinicum, compared to those from closely related species (Fig. 5c). The adaptive evolution of CsLhcb1s may contribute to the adaptability of C. sinicum to low-light environments. In land plants, 15–20% of LHCII exhibits transferability during state transitions. A previous investigation revealed that more LHCII is associated with PSI (in state 2) after long-term of dark adaptation in plants20. Therefore, we speculated that the number of threonine residues is positively related to the susceptibility of LHCII to phosphorylation by STN7 kinase, resulting in an increased proportion of transferable LHCII associated with PSI (in state 2). As daylight passes through the vegetative canopy, the red-to-far-red light (R/Fr) ratio of daylight decreases due to the absorption of red light by photosynthetic pigments, resulting in a higher proportion of far-red light in low-light environments67,68. The enrichment of far-red light results in the preferential excitation of PSI19,69. An increased proportion of LHCII associated with PSI (in state 2) can significantly increase the size of the PSI antenna and transfer energy efficiently to PSI, thereby improving a plant’s ability to efficiently utilize far-red light. This may be an adaptive strategy that enables shade plants to persist in long-term low-light environments. However, the additional threonine near the STN7 phosphorylation site within CsLhcb1s may act as a competitor for the original threonine, therefore diminishing the susceptibility of this active site to STN7 kinase-mediated phosphorylation. Alternatively, the FAR1 gene family, known for its ability to inhibit the shade avoidance response by repressing hypocotyl elongation70, also underwent rapid expansion in the C. sinicum genome, suggesting that shade tolerance over shade avoidance is a strategy employed by shade plants to cope with competition for light.
C. sinicum, which belongs to the Chrysosplenium genus, is an ideal plant for studying adaptive evolution in low-light environments and has significant medicinal value as a traditional Chinese Tibetan medicine. However, the metabolic characteristics of Chrysosplenium in relation to the synthesis and accumulation of flavonoid compounds remain unclear. In this study, we performed metabolomic analysis of three tissues (roots, stems, and leaves) of C. sinicum, and our analysis revealed that the predominant flavonoids in this species are chrysosplenol and chrysosplenoside (Fig. 6b; Supplementary Data 15 and 16). Chrysosplenol and chrysosplenoside, which are rarely found in other plant sources, are widely distributed in species of the genus Chrysosplenium; thus, these flavonoids may be associated with the unique characteristics of this genus. Modern research has demonstrated that chrysosplenol and chrysosplenoside possess a broad range of pharmacological properties. For example, chrysosplenol B and chrysosplenol C exhibit a notable inhibitory effect against rhinoviruses71, while chrysosplenol D exhibits anticancer activity against oral squamous cell carcinoma72. In this study, we propose a putative pathway for chrysosplenol and chrysosplenoside synthetic based on their chemical structure and the KEGG flavonoid biosynthesis pathway (Fig. 6c). Additionally, based on the transcriptome and metabolome data, we identified key genes involved in synthesis via correlation analysis (Fig. 6c, d). Although the synthetic routes presented are incomplete, the preliminary results for the synthesis of characteristic flavonoid metabolites in the Chrysosplenium genus will contribute to the utilization of these biological resources within the genus.
AS events play a crucial regulatory role in plants by enhancing the diversity of the proteome while simultaneously governing the abundance of the transcriptome. In the C. sinicum, we found that STN7 kinase and multiple UGT genes underwent AS events in three tissues (roots, stems, and leaves). The STN7 kinase phosphorylates LHCII, which then migrates from PSII to PSI (state 2) and thus rebalances the light excitation energy between the photosystems. Several studies have provided evidence for the pivotal involvement of UGTs in the biosynthesis of flavonoids within plant organisms73,74. The differential alternative splicing of the STN7 and UGT genes indicated that AS events may play a regulatory role in photosynthesis and flavonoid accumulation across the three tissues.
In summary, the genome of C. sinicum, a classic shade plant, was assembled in this study via comparative genomics analysis to investigate the adaptive evolution mechanism that allows shade plants to thrive in long-term low-light environments. Our results demonstrated that the expansion of photosynthesis-related gene families, caused by intracellular DNA transfer, and the adaptive evolution of the Lhcb1 family potentially established a crucial genomic foundation for the increased adaptability of C. sinicum to low-light environments. Additionally, we explored the biosynthesis and regulatory mechanisms of the flavonoid compounds present in C. sinicum, chrysosplenol and chrysosplenoside in different tissues using metabolome and transcriptome analysis and predicted their biosynthetic pathways and key enzymes.
Methods
Plant material
The C. sinicum plant used in this study was obtained from Badagongshan National Natural Reserve in Hubei province, China. The root apex was utilized for karyotype and flow cytometry examination to determine the plant’s genome size and diploidy. A single C. sinicum plant was selected for a de novo genome assembly. Three different tissues (roots, stems, and leaves) were collected during the reproductive growth stage for transcriptome and metabolome analysis. The freshly collected samples were promptly frozen in liquid nitrogen and subsequently stored at −80 °C.
Whole-genome sequencing and Hi-C sequencing
Whole-genome DNA sequencing was conducted using the MGIseq and PacBio platforms. Genomic DNA was extracted from young leaves using a modified CTAB extraction method. Short-read sequencing libraries were constructed following the manufacturer’s standard protocol and performed sequencing on the MGIseq platform. The PacBio long-reads library was prepared using the SMRTbell Express Template Prep kit 2.0 (Pacific Biosciences) according to the manufacturer’s instructions and then sequenced on the PacBio Sequel II platform.
The plant used for PacBio sequencing was also utilized in the construction of the Hi-C sequencing library. Hi-C data was used to assist scaffolding. The purified cross-linked DNAs were first digested, followed by blunt-end-repair and tagging with biotin. Subsequently, a Hi-C library was constructed by capturing and PCR-enriching DNA fragments containing biotin. Finally, the library sequencing was conducted utilizing the PE150 strategy on the Illumina HiSeq platform.
Genome assembly and quality evaluation
The genome size and heterozygosity of C. sinicum were evaluated using k-mer frequency analysis75 and DNA flow cytometry76. For PacBio assemblies, the Falcon software (v0.2.2)77 was used to assemble the long reads into contigs. To improve the accuracy of single-base, the contigs were initially polished using SMRTLink software (v5.1) with PacBio data, followed by a secondary polished using Pilon software (v1.22)78 with short-reads data. We used Juicer software (v1.5.6)79 to align the Hi-C sequencing reads to the contigs, followed by JucieBox software (v1.5.3)80 for completing visual correction of the interaction map, ultimately obtaining 12 pseudo-chromosomes of C. sinicum.
The BWA-MEM software (v0.7.12)81 was used to evaluate the quality of the genome assembly by mapping short-read DNA sequencing data to the assembly and calculating the mapping rates. The functional completeness of our assembly was assessed using compleasm (v0.2.6) tool31 for BUSCO analysis, with eudicots_odb1082 serving as the reference database. Merqury (v1.3) tool32 was employed to evaluate QV and genome completeness.
Repeat analysis
A repeat library was constructed from the assembled C. sinicum genome using RepeatModeler (open-1.0.11) (http://repeatmasker.org/RepeatModeler). The repeat sequence library obtained from RepeatModeler software was combined with RepBase (v20181026) (http://www.girinst.org/repbase) to create a comprehensive repeats library. The DeepTE83 and TEsorter (v1.4.6)84 tools were employed for the classification of repeats belonging to the “Unknown” category in the repeat library. Subsequently, this repeat library was utilized in RepeatMasker (OPEN-4.0.7) (http://www.repeatmasker.org) to identify repetitive elements within the C. sinicum genome.
Genome annotation
After masking the repetitive sequences using the RepeatMasker software, we used the MAKER pipeline (v2.31.10)85 to predict the gene set by incorporating three different approaches: Ab initio prediction, homology-based prediction, and transcript-based prediction. The Augustus software (version 3.3.1)86 was used for Ab initio prediction, with the Solanum lycopersicum selected as the prediction model. Homology-based prediction utilized protein sequences from four sequenced plants (including Arabidopsis thaliana, Vitis vinifera, Kalanchoe fedtschenkoi, and Rhodiola crenulata), as well as proteins from the uniport database (from https://www.uniprot.org/uniprotkb). Protein sequences were mapped to the genome using tBlastn (v2.7.1+). For transcript-based prediction, RNA-seq reads were trimmed using Trimmomatic (v0.36)87 and then assembled using Trinity (v2.8.4)88.
The function annotation of predicted protein sequences using InterProScan (version 5.39-77.0)89 and eggNOG-mapper (v2.0.1)90,91. InterProScan was run with the options ‘-iprlookup -goterms -pa -f tsv’ to generate the GO annotation for proteins. The iTAK (v1.7)92 software was used to predict transcription factor92.
Genome evolution and whole-genome duplication
The evolution of the C. sinicum genome was investigated by selecting 20 representative plant genomes, including species from Poales (Oryza sativa93), Ranales (Aquilegia coerulea94, Nelumbo nucifera), Caryophyllales (Beta vulgaris95), Asterales (Helianthus annuus96), Lamiales (Mimulus guttatus97), Solanales (Solanum lycopersicum98), Gentianales (Coffea canephora99), Saxifragales (C. sinicum, Kalanchoe fedtschenkoi35, Kalanchoe laxiflora, Rhodiola crenulata100), Vitales (Vitis vinifera39), Myrtales (Eucalyptus grandis101), Fabales (Medicago truncatula102), Rosales (Fragaria vesca103), Malpighiales (Populus trichocarpa104), Sapindales (Citrus sinensis105), Brassicales (Arabidopsis thaliana106), and Malvales (Theobroma cacao107). Orthofinder (v2.3.3)108 was used to identify orthologous groups with default parameters. The single-copy orthologous genes across the analyzed genomes were extracted from the output of Orthofinder. We construct the species tree based on these single-copy orthologous genes using Raxml (v8.2.12)109 package. MCMCTREE software (from PAML v4.9i)53 was used to calculate the divergence times. Subsequently, we corrected these divergence times based on the calibration of Monocot and Dicot (>1.15 Mya and <3.08 Mya) as well as Rosids and Asterids (>1.11 Mya and <1.31 Mya) divergence times from the TIMETREE database (http://www.timetree.org/)110. The visualization of the species tree used R package MCMCtreeR (v1.1)111.
JCVI112 was used to search for pairwise synteny regions between C. sinicum and grape genomes. The command ‘python -m jcvi.graphics.dotplot’ was utilized to generate dotplots for visualizing the pairwise synteny of genomes. The command ‘python -m jcvi.compara.synteny depth -histogram’ was utilized to analyze the synteny pattern between the genomes. ParaAT (https://github.com/wonaya/ParaAT) and KaKs_Calculator (version 2.0)113 were used to calculate Ks values for each gene pair. Based on the pairwise synteny region of C. sinicum versus AEK42, we generated the karyotype of C. sinicum genome.
Comparative genomics and visualizations
The C. sinicum-specific gene families were extracted from the Orthofinder (v2.3.3) results of four Saxifragales species. We conducted KEGG and GO enrichment analyses on gene sets using R package clusterProfiler (v4.6.2)114. The R package ggplot2 (v3.4.3) was utilized to visualize the results generated from the enrichment analysis. The duplicate_gene_classifier module from MCScanX115 was used to classify genes into distinct categories, including segmental/WGD duplicates, tandem duplicates, proximal duplicates, dispersed duplicates, and singletons. The expansion and contraction of gene families in each lineage were identified using CAFE (V4.2)116 software. Based on the repetitive elements identified in C. sinicum genome, we used bedtools (v2.27.1)117 program to count the number of repetitive elements in the proximal region (upstream and downstream 50 kb) of genes.
Mitochondria and chloroplast genome assembly
The chloroplast genome of C. sinicum was obtained from our previously submitted dataset (NC_051987.1)118. After removing fragments of chloroplast genome using Blastn (v2.7.1+) program, we used GetOrganelle (v.1.7.5)119 software to perform de novo assembly of a draft mitogenome with parameters ‘-R50 -k 21,45,65,85,105,127 -F embplant_mt’. Subsequently, we aligned the PacBio long reads to the draft mitogenome using minimap2 (v2.17)120 software and extracted these aligned long reads using samtools (v1.7)121. Based on the aligned PacBio long reads, we separately conducted mitogenome assembly using Canu (v2.2)122 and Flye (v2.9)123. The two results were combined to generate a complete mitogenome with three rings.
We annotated the mitogenome of C. sinicum by referencing the mitogenome of Heuchera parviflora (KR559021) using three software programs: GeSeq (v2017)124, Geneious Prime (2021.2.2), and IPMGA (v2022) (http://www.1kmpg.cn/mgavas). The mitogenome map of C. sinicum was generated utilizing OGDRAW (v1.3.1)125 software. The NUPTs and NUMTs were identified by conducting Blastn (v2.7.1+) searches against nuclear genomic sequences, using the chloroplast and mitochondrial genome as query sequences.
Positive selection analysis
The positive selection analysis of Lhcb1s was conducted for the C. sinicum and seven other Superrosids species. The amino acid sequences of Lhcb1s were aligned using T-COFFEE (v13.45.33)126, and then transformed into nucleic acid sequences using ParaAT. The codeml program, an integral part of the PAML (v4.9) software, was employed to conduct positive selection analysis through the branch-site model, with the CsLhcb1s from C. sinicum designated as the foreground branch127.
Transcriptome and metabolome analysis
The samples for transcriptome sequencing were collected from multiple plants at the reproductive growth stage within the same population. After the collection and processing of root, stem, and leaf tissues, each containing three biological replicates, we follow the manufacturer’s instructions to extract total RNA using Trizol (Invitrogen, CA, USA). The purified mRNA was fragmented into small pieces using a fragmentation buffer. First-strand cDNA was synthesized using random hexamer-primed reverse transcription, followed by the synthesis and purification of second-strand cDNA. The repaired (using A-Tailing Mix and RNA Index Adapters) cDNA fragments were amplified by PCR and the products were purified by Ampure XP Bead to obtain the final RNA-seq library. Subsequently, all these libraries for RNA-seq were sequenced utilizing the PE150 strategy on the Illumina HiSeq platform.
To quantify the gene expression level, the RNA-seq clean reads from three tissues were mapped to the C. sinicum genome using Hisat2 (v2.1.0)128 with default parameters. Then we used StringTie (v1.3.4d)129 to quantify the expression level of genes, employing default parameters. Differentially expressed genes were identified using R package DESeq2 (v1.38.3)130. Gene exhibiting a difference of at least twofold change and false discovery rate (q-value) ≤ 0.05 were considered to be significantly differentially expressed.
The plants used for metabolome analysis were obtained from the same batch as the transcriptome. Samples (roots, stems, and leaves) for the metabolome analysis were collected during the reproductive growth stage, with each tissue containing three biological replicates pooled from multiple plants. Metabolome analysis was conducted using a widely targeted metabolomics method by Wuhan Metware Biotechnology Co., Ltd (Wuhan, China) (http://www.metware.cn/). In brief, the freeze-dried C. sinicum tissues were crushed using a mixer mill (MM 400, Retsch) with a zirconia bead for 1.5 min at 30 Hz. The 100 mg tissue powder was weighted and extracted overnight at 4 °C with 1.2 ml 70% aqueous methanol. Following centrifugation at 12,000 rpm for 10 min, the extracts were filtrated (SCAA-104, 0.22μm pore size) before UPLC-MS/MS analysis (UPLC, SHIMADZU Nexera X2, www.shimadzu.com.cn/; MS, Applied Biosystems 4500 Q TRAP, www.appliedbiosystems.com.cn/). Metabolite characterization was performed using the self-built database MWDB (metware database) of Metware Biotechnology, and metabolite quantification was conducted using a scheduled multiple reaction monitoring (MRM) method131. The Python package SciPy (v1.11.2) was used to calculate Pearson’s correlation coefficient between gene expression and metabolite content. We screened the candidate gene set potentially involved in the synthesis of chrysosplenol and chrysosplenoside using a Pearson’s correlation coefficient threshold greater than 0.95. Subsequently, we performed screening of enzymes involved in flavonoid hydroxylation, methylation, and glycosylation within the candidate gene set using KEGG function annotation. Finally, we identified the candidate genes by analyzing their specific enzyme catalytic sites within the flavonoid ring.
Full-length transcript analysis
The high-quality full-length transcript (RQ > 0.9) was generated using IsoSeq (v3.3.0) (https://github.com/PacificBiosciences/IsoSeq) based on PacBio long reads. These full-length transcripts were then corrected with short-reads data via LoRDEC (v0.9)132 software, and subsequently mapped to the assembled C. sinicum genome through minimap2 (v2.17). Finally, the redundant transcripts were collapsed using the cDNA_Cupcake program (v8.7) (https://github.com/Magdoll/cDNA_Cupcake).
The SUPPA2 (v.2.3)133 software was utilized to identify alternative splicing (AS) events from non-redundant full-length transcripts, and the percentage spliced in index (PSI) value was computed to quantify the inclusion levels of AS events based on the transcripts' TPM values. If the difference in PSI of an AS event between the two tissues surpasses a stringent threshold (false discovery rate ≤ 0.05, |ΔPSI | > 0.1), it is classified as differentially AS events.
Statistics and reproducibility
The statistical significance of the difference in the number of repetitive elements in the proximal region of genes was determined using the Student’s t-test (*P < 0.05; **P < 0.01; ***P < 0.001). The statistical significance of GO and KEGG terms was evaluated using Fisher’s exact test in combination with FDR correction for multiple testing (P < 0.05). Three biological replicates were prepared for each tissue under the same condition. The mean FPKM value computed from the three replicates represents the final FPKM for the genes.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The whole-genome sequencing data (including short reads, Pacbio long reads, Hi-C interaction reads, and genome assembly), and RNA-seq data for the three tissues have been deposited in the National Genomics Data Center under the accession PRJCA005831. Source data underlying Fig. 3 are presented in Supplementary Data 7 and 8; source data underlying Fig. 5 are presented in Supplementary Data 12–14; source data underlying Fig. 6 are presented in Supplementary Data 15–18; source data underlying Fig. 7a and Fig. 7c–e are presented in Supplementary Data 19 and 20. Uncropped and unedited gel image (PCR) of inserted organelle genes is included in Supplementary Fig. 32. The annotation information for the mitochondrial genome and nuclear genome, as well as source data for Figs. 4 and 7b, are deposited in a Figshare repository (https://doi.org/10.6084/m9.figshare.24558607.v2). All other data is available from the corresponding author upon reasonable request.
References
Group TAP. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20 (2016).
POWO. Plants of the World Online. Facilitated by the Royal Botanic Gardens, Kew., Published on the Internet http://www.plantsoftheworldonline.org/ (2024).
Soltis, D. E. et al. Phylogenetic relationships and evolution in Chrysosplenium (Saxifragaceae) based on matK sequence data. Am. J. Bot. 88, 883–893 (2001).
Yang, Y., Zhang, Y., Shi, G. & Lu, R. Advances in studies on chemical constituents and structure-activity of Chrysosplenium. Nat. Prod. Res. Dev. 17, 253–257 (2005).
Zhao, J. et al. A review of the genus Chrysosplenium as a traditional Tibetan medicine and its preparations. J. Ethnopharmacol. 290, 115042 (2022).
Cuzzuol G. R. F. & Milanez C. R. D. Morphological and physiological adjustments in juvenile tropical trees under contrasting sunlight irradiance (ed. Najafpour, M. M.) 501–518 (IntechOpen, 2012).
Tian, Y., Sacharz, J., Ware, M. A., Zhang, H. & Ruban, A. V. Effects of periodic photoinhibitory light exposure on physiology and productivity of Arabidopsis plants grown under low light. J. Exp. Bot. 68, 4249–4262 (2017).
Kaiser, E., Morales, A. & Harbinson, J. Fluctuating light takes crop photosynthesis on a rollercoaster ride. Plant Physiol. 176, 977–989 (2018).
Venkateswarlu, B. Influence of low light intensity on growth and productivity of rice, Oryza sativa, L. Plant Soil 46, 713–719 (1977).
Liu, Q. H., Zhou, X. B., Yang, L. Q., Li, T. & Zhang, J. J. Effects of early growth stage shading on rice flag leaf physiological characters and grain growth at grain-filling stage. Ying Yong Sheng Tai Xue Bao 20, 2135–2141 (2009).
Qu, M. et al. Leaf photosynthetic parameters related to biomass accumulation in a global rice diversity survey. Plant Physiol. 175, 248–258 (2017).
Dall’Osto, L., Bressan, M. & Bassi, R. Biogenesis of light harvesting proteins. Biochim. Biophys. Acta 1847, 861–871 (2015).
Scholes, G. D., Fleming, G. R., Olaya-Castro, A. & Van Grondelle, R. Lessons from nature about solar light harvesting. Nat. Chem. 3, 763–774 (2011).
Mazor, Y., Borovikova, A. & Nelson, N. The structure of plant photosystem I super-complex at 2.8 Å resolution. eLife 4, e07433 (2015).
Jansson, S. A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci. 4, 236–240 (1999).
Allen, J. F. State transitions-a question of balance. Science 299, 1530–1532 (2003).
Tikkanen, M. et al. Phosphorylation-dependent regulation of excitation energy distribution between the two photosystems in higher plants. Biochim. Biophys. Acta 1777, 425–432 (2008).
Bellafiore, S., Barneche, F., Peltier, G. & Rochaix, J.-D. State transitions and light adaptation require chloroplast thylakoid protein kinase STN7. Nature 433, 892–895 (2005).
Wientjes, E., Roest, G. & Croce, R. From red to blue to far-red in Lhca4: How does the protein modulate the spectral properties of the pigments? Biochim. Biophys. Acta 1817, 711–717 (2012).
Chukhutsina, V. U., Liu, X., Xu, P. & Croce, R. Light-harvesting complex II is an antenna of photosystem I in dark-adapted plants. Nat. Plants 6, 860–868 (2020).
Wang, D., Lloyd, A. H. & Timmis, J. N. Environmental stress increases the entry of cytoplasmic organellar DNA into the nucleus in plants. Proc. Natl Acad. Sci. USA 109, 2444–2448 (2012).
Sheppard, A. E. et al. Transfer of plastid DNA to the nucleus is elevated during male gametogenesis in tobacco. Plant Physiol. 148, 328–336 (2008).
Stegemann, S., Hartmann, S., Ruf, S. & Bock, R. High-frequency gene transfer from the chloroplast genome to the nucleus. Proc. Natl Acad. Sci. USA 100, 8828–8833 (2003).
Timmis, J. N., Ayliffe, M. A., Huang, C. Y. & Martin, W. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–135 (2004).
Lloyd, A. H. & Timmis, J. N. The origin and characterization of new nuclear genes originating from a cytoplasmic organellar genome. Mol. Biol. Evol. 28, 2019–2028 (2011).
Matsuo, M., Ito, Y., Yamauchi, R. & Obokata, J. The rice nuclear genome continuously integrates, shuffles, and eliminates the chloroplast genome to cause chloroplast–nuclear DNA flux. Plant Cell 17, 665–675 (2005).
Sheppard, A. E. & Timmis, J. N. Instability of plastid DNA in the nuclear genome. PLoS Genet. 5, e1000323 (2009).
Stegemann, S. & Bock, R. Experimental reconstruction of functional gene transfer from the tobacco plastid genome to the nucleus. Plant Cell 18, 2869–2878 (2006).
Martin, W. et al. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl Acad. Sci. USA 99, 12246–12251 (2002).
Funamoto, T. & Zhou S-l Chromosome studies of three species of Chrysosplenium (Saxifragaceae) in Heilongjiang Province, People’s Republic of China. Chromosome Bot. 5, 81–85 (2010).
Huang, N. & Li, H. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39, btad595 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Zeng, L. et al. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 214, 1338–1354 (2017).
Yang, X. et al. The Kalanchoë genome provides insights into convergent evolution and building blocks of crassulacean acid metabolism. Nat. Commun. 8, 1899 (2017).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Adams, K. Genomic clues to the ancestral flowering plant. Science 342, 1456–1457 (2013).
Murat, F. et al. Karyotype and gene order evolution from reconstructed extinct ancestors highlight contrasts in genome plasticity of modern rosid crops. Genome Biol. Evol. 7, 735–749 (2015).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Fitch, W. M. Distinguishing homologous from analogous proteins. Syst. Zool. 19, 99–113 (1970).
Ren, G. et al. The genome sequence provides insights into salt tolerance of Achnatherum splendens (Gramineae), a constructive species of alkaline grassland. Plant Biotechnol. J. 20, 116–128 (2022).
Murat, F., Armero, A., Pont, C., Klopp, C. & Salse, J. Reconstructing the genome of the most recent common ancestor of flowering plants. Nat. Genet. 49, 490–496 (2017).
Ishizaki, K. et al. The critical role of Arabidopsis electron-transfer flavoprotein:ubiquinone oxidoreductase during dark-induced starvation. Plant Cell 17, 2587–2600 (2005).
Mishanin, V. I., Trubitsin, B. V., Benkov, M. A., Minin, A. A. & Tikhonov, A. N. Light acclimation of shade-tolerant and light-resistant Tradescantia species: induction of chlorophyll a fluorescence and P700 photooxidation, expression of PsbS and Lhcb1 proteins. Photosynth. Res. 130, 275–291 (2016).
Ishizaki, K. et al. The mitochondrial electron transfer flavoprotein complex is essential for survival of Arabidopsis in extended darkness. Plant J. 47, 751–760 (2006).
Hazkani-Covo, E., Zeller, R. M. & Martin, W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6, e1000834 (2010).
Richly, E. & Leister, D. NUPTs in sequenced eukaryotes and their genomic organization in relation to NUMTs. Mol. Biol. Evol. 21, 1972–1980 (2004).
Noutsos, C., Kleine, T., Armbruster, U., DalCorso, G. & Leister, D. Nuclear insertions of organellar DNA can create novel patches of functional exon sequences. Trends Genet. 23, 597–601 (2007).
Levin, G. & Schuster, G. LHC-like proteins: the guardians of photosynthesis. Int. J. Mol. Sci. 24, 2503 (2023).
Blankenship, R. E. Early evolution of photosynthesis. Plant Physiol. 154, 434–438 (2010).
Babenko, V. N., Rogozin, I. B., Mekhedov, S. L. & Koonin, E. V. Prevalence of intron gain over intron loss in the evolution of paralogous gene families. Nucleic Acids Res. 32, 3724–3733 (2004).
Sharpton, T. J., Neafsey, D. E., Galagan, J. E. & Taylor, J. W. Mechanisms of intron gain and loss in Cryptococcus. Genome Biol. 9, R24 (2008).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Depège, N., Bellafiore, S. & Rochaix, J.-D. Role of chloroplast protein kinase Stt7 in LHCII phosphorylation and state transition in chlamydomonas. Science 299, 1572–1575 (2003).
Leoni, C. et al. Very rapid phosphorylation kinetics suggest a unique role for Lhcb2 during state transitions in Arabidopsis. Plant J. 76, 236–246 (2013).
Blom, N., Sicheritz-Pontén, T., Gupta, R., Gammeltoft, S. & Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 (2004).
Ferrer, J. L., Austin, M. B., Stewart, C. & Noel, J. P. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiol. Biochem. 46, 356–370 (2008).
Dubos, C. et al. MYB transcription factors in Arabidopsis. Trends Plant Sci. 15, 573–581 (2010).
Goossens, J., Mertens, J. & Goossens, A. Role and functioning of bHLH transcription factors in jasmonate signalling. J. Exp. Bot. 68, 1333–1347 (2017).
Zhu, J. K. Abiotic stress signaling and responses in plants. Cell 167, 313–324 (2016).
Ai, X., Wang, X., Guo, Y. & Xing, Y. Effects of suboptimal temperature and low temperature under low light intensity on stomatal characteristics and chloroplast ultrastructure of cucumber seedlings. Zhongguo Non. Ye Ke Xue 39, 2063–2068 (2006).
Gommers, C. M., Visser, E. J., St Onge, K. R., Voesenek, L. A. & Pierik, R. Shade tolerance: when growing tall is not an option. Trends plant Sci. 18, 65–71 (2013).
Viji, M., Thangaraj, M. & Jayapragasam MJBp. Low irradiance stress tolerance in rice (Oryza sativa L.). Biol. Plant. 39, 251–256 (1997).
Rao, C. & Murty, K. Swarnaprabha, a low light tolerant high yielding variety. IRRN 12, 7 (1987).
Dutta, S. S. et al. Marker–trait association for low-light intensity tolerance in rice genotypes from Eastern India. Mol. Genet. Genom. 293, 1493–1506 (2018).
Kumar, A. et al. Role of sedoheptulose-1,7 bisphosphatase in low light tolerance of rice (Oryza sativa L.). Physiol. Mol. Biol. Plants 26, 2465–2485 (2020).
Ruberti, I. et al. Plant adaptation to dynamically changing environment: the shade avoidance response. Biotechnol. Adv. 30, 1047–1058 (2012).
Yang, F. et al. Effect of interactions between light intensity and red-to- far-red ratio on the photosynthesis of soybean leaves under shade condition. Environ. Exp. Bot. 150, 79–87 (2018).
Morosinotto, T., Breton, J., Bassi, R. & Croce, R. The nature of a chlorophyll ligand in Lhca proteins determines the far red fluorescence emission typical of photosystem I. J. Biol. Chem. 278, 49223–49229 (2003).
Liu, Y. et al. Arabidopsis FHY3 and FAR1 regulate the balance between growth and defense responses under shade conditions. Plant Cell 31, 2089–2106 (2019).
Tsuchiya, Y. et al. Antiviral activity of natural occurring flavonoids in vitro. Chem. Pharm. Bull. 33, 3881–3886 (1985).
Hsieh, M. J. et al. Chrysosplenol D triggers apoptosis through heme oxygenase-1 and mitogen-activated protein kinase signaling in oral squamous cell carcinoma. Cancers 13, 4327 (2021).
Peng, M. et al. Differentially evolved glucosyltransferases determine natural variation of rice flavone accumulation and UV-tolerance. Nat. Commun. 8, 1975 (2017).
Pei, T. et al. Characterization of UDP-glycosyltransferase family members reveals how major flavonoid glycoside accumulates in the roots of Scutellaria baicalensis. BMC Genom. 23, 169 (2022).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Preprint at https://doi.org/10.48550/arXiv.1308.2012 (2013).
Dolezel, J. & Bartos, J. Plant DNA flow cytometry and estimation of nuclear genome size. Ann. Bot. 95, 99–110 (2005).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1, e323 (2021).
Yan, H., Bombarely, A. & Li, S. DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 36, 4269–4275 (2020).
Zhang, R. G. et al. TEsorter: an accurate and fast method to classify LTR-retrotransposons in plant genomes. Hortic. Res. https://doi.org/10.1093/hr/uhac1017 (2022).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinform. 48, 4.11.11–14.11.39 (2014).
Keller, O., Kollmar, M., Stanke, M. & Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–d314 (2019).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Zheng, Y. et al. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 9, 1667–1670 (2016).
Ouyang, S. et al. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 35, D883–D887 (2007).
Filiault, D. L. et al. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. eLife 7, e36426 (2018).
McGrath, J. M. et al. A contiguous de novo genome assembly of sugar beet EL10 (Beta vulgaris L.). DNA Res. https://doi.org/10.1093/dnares/dsac1033 (2023).
Badouin, H. et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546, 148–152 (2017).
Hellsten, U. et al. Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc. Natl Acad. Sci. USA 110, 19478–19482 (2013).
Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Denoeud, F. et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345, 1181–1184 (2014).
Fu, Y. et al. Draft genome sequence of the Tibetan medicinal herb Rhodiola crenulata. GigaScience 6, 1–5 (2017).
Bartholomé, J. et al. High-resolution genetic maps of Eucalyptus improve Eucalyptus grandis genome assembly. N. Phytol. 206, 1283–1296 (2015).
Tang, H. et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genom. 15, 312 (2014).
Shulaev, V. et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116 (2011).
Zhang, S. et al. Chromosome-scale assemblies of the male and female Populus euphratica genomes reveal the molecular basis of sex determination and sexual dimorphism. Commun. Biol. 5, 1186 (2022).
Wu, G. A. et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 32, 656–662 (2014).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210 (2012).
Motamayor, J. C. et al. The genome sequence of the most widely cultivated cacao type and its use to identify candidate genes regulating pod color. Genome Biol. 14, r53 (2013).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819 (2017).
Puttick, M. N. MCMCtreeR: functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics 35, 5321–5322 (2019).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Wang, D., Zhang, Y., Zhang, Z., Zhu, J. & Yu, J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 8, 77–80 (2010).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Wu, Z. et al. Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genom. 21, 621 (2020).
Jin, J. J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatic 25, 2078–2079 (2009).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Tillich, M. et al. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic acids Res. 45, W6–W11 (2017).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64 (2019).
Notredame, C., Higgins, D. G. & Heringa, J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 (2000).
Yang, Z., Wong, W. S. & Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118 (2005).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Chen, W. et al. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: application in the study of rice metabolomics. Mol. plant 6, 1769–1780 (2013).
Salmela, L. & Rivals, E. LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514 (2014).
Trincado, J. L. et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 19, 40 (2018).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 32170207), the Fund for Scientific Research Platforms of South-Central Minzu University (No. PTZ24019), Fund for Academic Innovation Teams of South-Central Minzu University (No. XTZ24020 and CZD24002). The authors appreciate Wen Huang for collecting plant samples and thank Yongmei Luo and Xiaoyan Wang for their experimental support.
Author information
Authors and Affiliations
Contributions
H.L. and J.T. coordinated the project, and conceived and designed the experiments. S.L. and Z.W. assembled and performed bioinformatics analyses of the genomes. T.Y. and J.W. performed the mitochondria genome assembly. J.X. performed the full-length transcript analysis. S.A. and E.Q. prepared samples for sequencing. S.L. wrote the manuscript. K.M. performed the experiments. H.L., S.L., J.L., and R.Q. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Vincent Courdavault and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: David Favero and Luke Grinham. A peer review file is available
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, S., Wu, Z., Yang, T. et al. The Chrysosplenium sinicum genome provides insights into adaptive evolution of shade plants. Commun Biol 7, 1004 (2024). https://doi.org/10.1038/s42003-024-06701-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06701-9
This article is cited by
-
Assembly and analysis of the first complete mitochondrial genome sequencing of main Tea-oil Camellia cultivars Camellia drupifera (Theaceae): revealed a multi-branch mitochondrial conformation for Camellia
BMC Plant Biology (2025)
-
A chromosome-level genome of Astilbe chinensis unveils the evolution of a terpene biosynthetic gene cluster
Nature Communications (2025)
