
Abstract
Fungus-growing termites, like Odontotermes obesus, cultivate Termitomyces as their sole food source on fungus combs which are continuously maintained with foraged plant materials. This necessary augmentation also increases the threat of introducing non-specific fungi capable of displacing Termitomyces. The magnitude of this threat and how termites prevent the invasion of such fungi remain largely unknown. This study identifies these non-specific fungi by establishing the pan-mycobiota of O. obesus from the fungus comb and termite castes. Furthermore, to maximize the identification of such fungi, the mycobiota of the decaying stages of the unattended fungus comb were also assessed. The simultaneous assessment of the microbiota and the mycobiota of these stages identified possible interactions between the fungal and bacterial members of this community. Based on these findings, we propose possible interactions among the crop fungus Termitomyces, the weedy fungus Pseudoxylaria and some bacterial symbiotes. These possibilities were then tested with in vitro interaction assays which suggest that Termitomyces, Pseudoxylaria and certain potential bacterial symbiotes possess anti-fungal capabilities. We propose a multifactorial interaction model of these microbes, under the care of the termites, to explain how their interactions can maintain a predominantly Termitomyces monoculture.
Similar content being viewed by others

Introduction
Termites primarily utilize plant-derived lignocellulosic materials as sources of nutrition. Lignocellulose is a recalcitrant material that only some basidiomycete fungi can digest completely1. Termites have evolved symbiotic associations with different groups of organisms (bacteria, protists, and fungi) to achieve lignocellulose digestion2. In all non-Termitidae termites and some Termitidae, it is achieved directly through nutritional symbiosis with gut-dwelling microbial symbionts3. However, the termites of the subfamily Macrotermitinae (Family: Termitidae) have evolved an indirect way of utilizing the lignocellulose-degrading capabilities of the basidiomycete fungus, Termitomyces. This fungus subsists on plant materials, brought into the termite mound by workers, and thrives on a spongy lignocellulosic structure called the fungus comb4,5. Termitomyces utilizes the lignocellulose to grow6, whereas termites use the growing fungal nodules as food7. This association, which first evolved in tropical Africa around 35 MYA5,8,9, has become so successful that fungus cultivation in termites is considered a canonical example of the evolution of agriculture in animal societies10. As this crop fungus has become the sole source of nutrition for these termites, it has led to the evolution of many unique behavioral phenotypes associated with their successful cultivation11. These phenotypes include, but are not limited to, (1) the construction of the specialized fungus-growing beds (combs) and the earthen mounds10,12 to protect them; (2) the use of semi-digested plant material13, feces and soil14,15 to build the combs and in the process seeding the combs with the inoculum of Termitomyces13,16 regulation of temperature16, relative humidity16,17and CO2 levels17 and (4) continuous maintenance of fungus gardens to ensure ideal growing conditions for Termitomyces.
However, these ideal growing conditions for Termitomyces can also be conducive to the growth of other, non-desirable fungi, similar to the invasion of weeds in human agricultural fields. In addition, the continuous and unidirectional flow of dead and decaying plant materials into these mounds increases the threat of introducing potential non-specific fungi from the environment. These decaying plant materials can harbor a diverse array of saprotrophic communities of fungi and bacteria18,19. Indeed, several such non-specific fungi have been isolated from different fungus-growing termites15,20,21,22. Many of these have the potential to outcompete the fungal cultivar Termitomyces and can result in a severe reduction of available nutrients for the termites. The termite-fungus symbiosis, therefore, should be under strong selection pressure to evolve mechanisms that can prevent the growth of such non-specific fungi. This selection pressure can act on the termites directly, enabling them to constantly seek out non-specific fungi for elimination through digestion23. However, recent reports20 indicate that such a strategy of selective de-weeding does not reduce non-specific fungal load as these spores survive the passage through the termite gut. The near absence of any other active fungi in healthy fungus combs, which are dominated by Termitomyces24, points to mechanisms that actually prevent these fungi from proliferating within the combs. This could be due to the direct or indirect use of the resident microbial community of the fungus comb, by the termites, in preventing the growth of any non-specific fungi25,26,27,28. Some indirect evidence for this hypothesis can be derived from the compositional similarity of the microbiota of many different fungus-growing insects29. This is a remarkable example of convergent evolution as most of these symbiotic microbial communities are dominated by the bacteria Pseudomonas30,31,32 which has also been shown to inhibit weedy fungus33. The ubiquitous presence of Pseudomonas can be evidence for selection33 to include bacterial symbiotes34 within the resident microbiota, which can hinder the growth of weedy fungi. Accordingly, some in vitro studies have found many resident bacteria acting as secondary symbiotes capable of preventing Pseudoxylaria35,36,37, the most prevalent non-Termitomyces fungal growth observed within decaying termite fungal gardens. Several such bacterial secondary symbiotes have since been identified, including Bacillus26,37, Streptomyces38,39, Burkholderia33 and Pseudomonas33 which can act as anti-fungal agents. Another line of evidence for this hypothesis comes from the ability of Termitomyces themselves to hamper the growth of some non-specific fungi when cultured on plates that previously grew Termitomyces24,40. However, what remains unknown is how and to what extent Termitomyces can act against them.
The current research on how fungus-growing termites raise a weed-free crop has primarily been restricted to mechanisms by which Pseudoxylaria is controlled. However, these studies lack a comprehensive assessment of the threat from other non-specific fungi. Many sequencing approaches have revealed variations in the bacterial communities from these fungus combs33,41,42, but since there are no reports of the corresponding core-mycobiota from any fungus-growing termites, it is difficult to know the roles of other fungi in this symbiosis and the relationship between the microbiota and the mycobiota. In this study, we try to answer these questions by using the widespread fungus-growing termite from India, Odontotermes obesus. First, to assess the threat from the non-specific fungi, we identify the pan-mycobiota of this symbiosis by amplifying the internal transcribed spacer (ITS) gene fragment and generating amplicons on the Nanopore platform of the fungal comb and termite castes. We augment this estimation by including samples from the decaying stages of the fungus comb, after removal of the termites, to identify which non-specific fungi remain suppressed in an active comb but become visible with the progressive decay. Second, to identify any interactions between the myco- and microbiota of these combs, we characterize the community-wide changes in the abundance of microbes from these decaying combs by using nanopore platform and confirm the results using qPCR for three specific microbes proven to be important in this association33. Third, as these temporal changes of the decaying community of microbes highlight the possibility of a few important interactions, we empirically validate these by isolating different fungi from the colonies and testing their growth in the presence of specific microbes. Fourth, we test whether putative bacterial symbiotes, identified in a previous study using these same termite mounds33, also show anti-fungal properties against these non-specific fungi. Finally, we propose a model to explain the role of these interactions in the disease-free growth of Termitomyces in the fungus gardens of O. obesus.
Results
O. obesus colonies harbor a vast array of non-specific fungi
The pan-mycobiota of O. obesus established from the combs (fresh and decaying), alates, workers and nymphs (Fig. 1 and Supplementary Fig. S1) yielded over 3.9 × 105 high-quality Nanopore reads (Table 1), with the highest number of reads from female alates (~1.06 × 105 reads) and the lowest from the 120 h-old comb (~4.0 × 103 reads). The rarefaction curves (Supplementary Fig. S4a) showed an exhaustive sampling of major workers and female alates, indicating sufficient coverage. The number of identified fungal OTU’s ranged from 197 (in major workers) to 51 (in minor workers), with 418 unique fungal genera across O. obesus. Figure 2 indicates the presence of the fungal OTU’s having a relative abundance of greater than 0.05% in any one of the eleven samples. The termite-free incubation of the comb yielded 236 different non-specific fungi other than Pseudoxylaria24,43,44. These include 81 fungi alone from the 24 h-old comb, which reduced to 69 after 120 h of incubation. The highest fungal diversity was found from 72 h-old comb which showed 110 different fungi. This indicates that many of these fungi are actively suppressed in the combs for Termitomyces to flourish. The detailed taxonomic classification and abundances of these OTU’s are tabulated in supplementary data 1. Community similarities, using UniFrac distances in a weighted PCoA plot, indicated greater similarity among the mycobiota of the decaying combs and between both alates and major workers (Supplementary Fig. S4c). Though the combs were collected from just two mounds, the number of reads and the fungal diversity that this study has uncovered represent a far more detailed estimate of the microbial data for any other fungus-growing termite.

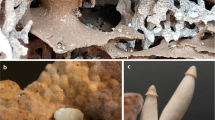
a, b Show the two mounds of O. obesus used in the study, c closeup view of fungus comb of O. obesus, d different worker castes of termites present over the fungus comb, e male and female alates emerging out from the gaps in the mound during the monsoon season from one of the mounds in this study.

Fungal OTU’s that accounted for >0.05% abundance in any of the six samples are shown in the heatmap. The numbers below each pie chart indicates the percentage of Termitomyces OTU’s obtained. The genera mentioned in bold were also obtained in culture.
The dynamics of the micro- and mycobiome in the decaying fungus comb
To determine how the micro- and mycobiota of fungus combs change after the removal of termites, we incubated fresh fungus combs for 120 h and enumerated their temporal changes every 24 h (Supplementary Data 1 and 2). Over 1.59 × 105 fungal OTU’s were identified across 120 h of incubation. The highest number of reads were obtained from the 24 h-old comb (0.58 × 105) and the lowest from 120 h-old comb (0.04 × 105). The mycobiota survey confirmed (Fig. 2, Supplementary Tables S5, and Supplementary Data 1) previous observations of a fresh comb being dominated by Termitomyces24,45 as 89% of the OTU’s were identified as Termitomyces. However, with each passing day, these combs revealed the proliferation of various other fungi. The relative abundance of Termitomyces was highest in the fresh fungus combs (90.7%), which drastically decreased after 24 h (2.1%) and was almost undetectable by 72 h (Fig. 3 and Supplementary Fig. S8). This was accompanied by a corresponding increase in Pseudoxylaria OTU’s (Fig. 3 and Supplementary Fig. S8) which increased from 2.6% by 24 h and reached the maximum in 48 h (26.1%) but then also experienced a reduction by 120 h (12.2%). The temporal changes in the abundances of these two fungi were also accompanied by a steady increase of the non-specific fungal OTU’s which jumped from ~10% in fresh comb (0 h) to 81% by 96 h of incubation. Thus, the increase in abundance of these non-specific fungi seems to be negatively correlated with Termitomyces and Pseudoxylaria (Supplementary Fig. S8).

a Changes in the physical appearance of the decaying fungus comb, b the relative abundances, and c copy numbers of Termitomyces and Pseudoxylaria in decaying fungus comb as determined by Nanopore Sequencing (bubble plot) and qPCR (box plot). Fungal OTU’s that had relative abundance of at least >0.3% in any of the six samples are shown in the bubble plot and the size of the bubble indicates the abundance of the fungus. (Uncult. = Uncultured fungus).
qPCR estimation to confirm the OTU abundance from the nanopore platform
Since qPCR is a more sensitive estimator of actual abundance46, we confirmed the OTU data with qPCR estimation of copy number variations of Termitomyces and Pseudoxylaria using the same DNA sources used to generate the OTU’s. The 95% confidence intervals of these copy numbers were used to assess the changes (Supplementary Table S6) and indicate a similar pattern of dynamics between the OTU and qPCR data for both these fungi (Fig. 3 and Supplementary Fig. S6). As expected, the OTU data revealed a lower threshold of the number of reads in comparison to qPCR estimates.
The corresponding microbiota of this incubation was estimated from over 1.16 × 105 high-quality Nanopore reads (Supplementary Table S7) across these same six samples, with the highest number of identified reads from 48 h-old comb (~0.36 × 105 reads) and the lowest from the fresh comb (~0.12 × 105 reads). The rarefaction curves (Supplementary Fig. S5a) indicated relatively exhaustive coverage of bacterial species diversity except for 48 h-old comb. A weighted PCoA analysis of the Unifrac distances among these samples showed a limited clustering (Supplementary Fig. S5c), indicating differential temporal abundances of some of these bacterial genera (Supplementary Fig. S6). Significantly, the magnitude of the changes in the bacterial community was far less severe than in the mycobiome. This indicates relatively more resilient bacterial communities than fungal ones in these combs. Subsequently, to determine the correlation between the potential bacterial symbiotes that have inhibitory effects against Pseudoxylaria33, the abundances of Pseudoxylaria and the bacterial symbiotes were analyzed by comparing their number of OTU’s. This comparison also revealed a negative association (Supplementary Fig. S7a, b). This shows the possibility that symbiotic bacteria may play a defensive role against the weedy fungus which results in healthy combs having low incidence of Pseudoxylaria.
To test any interaction between the bacteria and the fungi, we selected Pseudomonas, a secondary symbiote identified from these same mounds33. The number of copies of Pseudomonas remained largely unchanged till 48 h, then increased after 72 h and came down after 120 h (Supplementary Fig. S7b). One-way ANOVA conducted with Termitomyces, Pseudoxylaria and Pseudomonas showed significant variation between the groups (P = 0.00081, F = 7.512, df = 2). Post hoc Tukey tests revealed insignificant pairwise site differences in the copy numbers of Termitomyces and Pseudoxylaria (P = 0.102) and Pseudoxylaria and Pseudomonas (P = 0.364).
Thus, these complex patterns of the abundances of the three major microbes, Termitomyces, Pseudoxylaria and the bacterial symbiotes, raises the possibility of a synergistic role in preventing non-specific fungi.
Members of both the micro- and mycobiota can prevent non-specific fungi
The temporal dynamics of the micro- and mycobiota, detailed above, suggested a crucial role for both Termitomyces and Pseudoxylaria. It appears that the initial high abundance of Termitomyces, and from 48 h onwards, the abundance of Pseudoxylaria is negatively correlated with the presence of other non-specific fungi (Supplementary Fig. S8). Therefore, we tested whether these two fungi can inhibit the growth of some of these non-specific fungal strains in culture. We began by looking at the capability of Termitomyces against Pseudoxylaria and found that Pseudoxylaria easily grew over Termitomyces (Supplementary Fig. S9), indicating that the crop fungus cannot prevent the most prominent weedy fungus. However, as Fig. 4 indicates, Termitomyces can inhibit fourteen other fungi in direct interaction assays (Supplementary Fig. S12). Termitomyces showed maximum inhibition (more than 50%) against Alternaria-MN913751, Curvularia-MN913753, Rhizomucor-MN913756, Syncephalastrum-MN913763 and Ustilago-MN913772 (Fig. 4, Supplementary Fig. S12 and Supplementary Table S8) and limited inhibition (less than 50%) against nine other fungi, Aspergillus-MN913749, Aspergillus-MN913750, Diaporthe-MN913762, Fusarium-MN913754, Lasiodiplodia-MN913758, Paraconiothyrium-MN913759, Penicillium-MN913771, Phialotubus- MN913760 and Phoma-MN913761 (Fig. 4, Supplementary Fig. S12, and Supplementary Table S8). However, the interaction with Rhizomucor-MN913756 and Diaporthe-MN913762, was not conclusive as the patterns of growth were too ambiguous to clearly differentiate these two fungi from Termitomyces. The two fungi, Mucor-MN913757 and Trichoderma-MN913767 overgrew Termitomyces, showing no sign of inhibition. However, these results indicate that Termitomyces possess the capability of preventing many ecologically relevant non-specific fungi to a variable degree.

The middle panel shows representative pictures of Termitomyces and Pseudoxylaria interactions with the non-specific fungi with the top panel showing the growths in control plates. The phylogenetic analysis of ITS sequences (as shown in the right panel) was run on MEGAX with the K2+g substitution model using Talaromyces as the outgroup. (represents >75 bootstrap values).
Pseudoxylaria showed variable degrees of inhibition against all of the non-specific fungi tested (Fig. 4 and Supplementary Fig. S13). The maximum degree of inhibition (more than 50%) was shown against Alternaria-MN913751, Diaporthe-MN913762, Penicillium-MN913771, Rhizomucor-MN913756, Syncephalastrum-MN913763, Trichoderma-MN913767, and Ustilago-MN913772 (Fig. 4 and Supplementary Fig. S13) and limited inhibition (less than 50%) was shown against Aspergillus-MN91375049, Aspergillus-MN913750, Curvularia-MN913753, Fusarium-MN913754, Lasiodiplodia-MN913758, Mucor-MN913757, Paraconiothyrium-MN913759, Phialotubus-MN913760 and Phoma-MN913761 (Fig. 4 and Supplementary Fig. S13). Interaction of Pseudoxylaria against Alternaria-MN913751 and Syncephalastrum-MN913763 showed vertical growth of both these interacting fungi. However, the two fungi could still be identified, and their zones of growth were easily evaluated (Fig. 4 and Supplementary Fig. S13).
These interaction assays reveal that both Termitomyces and Pseudoxylaria mirror each other in their inhibitory effects (Table S8). As Fig. 4 indicates, certain non-specific fungi are inhibited by both. The exceptions are Mucor-MN913757 and Trichoderma-MN913767 where the major inhibition is shown only by Pseudoxylaria while Phialotubus-MN913760 is mostly inhibited by Termitomyces. However, Aspergillus-MN913749, Aspergillus-MN913750, Fusarium-MN913754, Lasiodiplodia-MN913758, Mucor-MN913757, and Phoma-MN913761 were only marginally inhibited by Termitomyces and Pseudoxylaria and hence these fungi were then assayed with the bacterial symbiotes (Bacillus, Burkholderia, Pseudomonas, and Streptomyces).
Seven of the eight non-specific fungi were inhibited by at least one of the bacterial symbiotes. Fusarium-MN913755 was not inhibited by any of the bacterial strains (Fig. 5 and Supplementary Figs. S15–S22). As Fig. 5 indicates, out of the eight bacterial strains, six showed a significant capability to prevent the growth of some of these fungi (Supplementary Table S8). These in vitro interaction assays suggest that termites possess the multifaceted capability of preventing the growth of non-specific fungi through Termitomyces, Pseudoxylaria and bacterial symbiotes.

The right panel shows the pictures of each assay where bacteria were streaked at the center and fungal plaques were placed equidistant to the bacterial growth. Whisker over horizontal bars represent standard deviation, and number indicates the type of inhibition shown by the bacterial strains (1= clear zone of inhibition, 2= reduced growth near bacteria, 3= contact inhibition, 4= negligible inhibition).
Discussion
The fungus gardens of O. obesus appear to be a monoculture of Termitomyces, but the pan-mycobiota indicates that the threat from many potential non-specific fungi remain suppressed in a fresh comb. A total of 418 different fungal genera were successfully annotated from the nearly 2.69 × 105 OTU’s generated from the Nanopore platform (Table 1), with 236 unique genera from the fungus comb. This remains a conservative estimate as rarefaction curves of the fungal OTU’s from many samples (Supplementary Fig. S4a) did not reach saturation. These results are contrary to the studies in Macrotermes bellicosus by Bos et al.20 and Macrotermes natalensis and different species of Odontotermes by Otani et al.24, which reported significantly fewer non-specific fungi with relatively low abundances of non-Termitomyces genera from fungus combs which ranged from <0.001% (454 sequencing datasets) to 0.07% relative abundances (MiSeq sequencing datasets) of non-specific fungi20,24. Only one of the four samples from ref. 20 identified 75% of the reads as Termitomyces and 24% as Xylaria. These discrepancies could be the result of methodological differences or could be characteristic of O. obesus colonies. Our initial assumption of workers bringing in potentially non-specific fungi was also supported as the major workers revealed the presence of 197 different fungal OTU’s. The abundance of such fungi in the alates, with only 0.6–12% relative abundance of Termitomyces (Fig. 2), also suggests vertical transmission of these non-specific fungi. How these fungi are suppressed by alates during nest founding and establishing Termitomyces monocultures merits future studies47. Thus, our results suggest that the ideal growing conditions for Termitomyces can also harbor a vast array of non-specific and weedy fungi and identify the magnitude of the ‘fungal threat’ that this symbiosis needs to overcome. Consequently, these results indicate the efficiency of termites suppressing the growth of non-cultivar fungi, together with the efficiency of Termitomyces and bacterial strains. These combined actions of these microbial symbiotes ensure successful proliferation of healthy termite colonies.
Termites are reported to select the best candidates among the different strains of Termitomyces available for cultivation and propagation8,48. Natural selection should favor both choosing (termites) and the chosen (cultivar strain) partners if the cultivar is adept at preventing the proliferation of non-specific fungi. As Fig. 4 indicates, Termitomyces can indeed inhibit several non-specific fungi obtained from these same mounds, but strikingly, not the most prominent weed, Pseudoxylaria (Supplementary Fig. S9). However, as Supplementary Fig. S8 illustrates, the presence of both Termitomyces and Pseudoxylaria is correlated with the non-specific fungi being in check for at least the first 24–48 h. This can be crucial evidence for the synergistic effects of both Termitomyces and Pseudoxylaria in preventing other non-specific fungi and maintaining a healthy comb. Therefore, we tested Pseudoxylaria for anti-fungal properties and, as Fig. 4 indicates, it shows some inhibitory effects against almost all the fungi tested. Moreover, the eight bacterial symbiotes previously identified33 also show similar growth inhibitory capabilities. Thus, our results point to the presence of microbes, both bacterial and fungal, with fungicidal capabilities present in this symbiosis. Given the magnitude of the “fungal threat” to this monoculture, this seems unsurprising. However, what remains unknown is what role, if any, selection has played in bringing about the composition of this community and how precisely these microbes bring about a successful monoculture.
These results further confirm the presence of termites is indispensable for a functioning monoculture, as their removal initiates the proliferation of non-specific fungi. This indicates that the termites need constant weeding to keep any such non-specific fungi at bay. Previous studies23 reported that O. obesus workers bury any visible Pseudoxylaria when given portions of cultured hyphae, indicating that they can recognize this as a contaminant to be removed, possibly through olfactory cues49. However, the qPCR data indicated that Pseudoxylaria is present in appreciable numbers even in fresh combs (Supplementary Fig. S8) and is obviously prevented from proliferating. This can be indicative of a dynamic equilibrium where the growth of Pseudoxylaria is tolerated by the termites to some extent, perhaps to prevent other non-specific fungi, but is also simultaneously inhibited from taking over the comb. This can be achieved by constant weeding out of Pseudoxylaria outbreaks within a functioning comb. However, how termites perform such a selective de-weeding remains unknown. As Fig. 3 indicates, Pseudoxylaria starts proliferating after 24 h and the termites probably have less than 48 h to remove it as by that time Termitomyces experiences a drastic reduction (Supplementary Fig. S8). This decline progresses steadily with the corresponding increase of other non-specific fungi. Thus, if termites weed out Pseudoxylaria by 48 h, then the comb can still perhaps be used to grow Termitomyces, but beyond 72 h, Termitomyces is all but wiped out by the proliferation of other non-specific fungi (Fig. 3 and Supplementary Fig. S8).
Pseudoxylaria, with its faster growth and constitutive fungicidal activity (Fig. 4, Supplementary Fig. S13, and Table S8), can be a more efficient inhibitor of non-specific fungi than Termitomyces. This hints towards its potential beneficial role in this symbiosis, but such a contention remains controversial. Pseudoxylaria has been considered to be an opportunistic fungus which can take over the crop monoculture when conditions are suitable50. As termites cannot feed on any other fungi51, the invasion of Pseudoxylaria is considered to be indicative of the collapse of this symbiosis52. However, the weedy or parasitic nature of Pseudoxylaria also raises some questions. Both in vitro and in vivo studies show that Pseudoxylaria can easily take over Termitomyces53,54,55, then why “sit-and-wait” if it can easily take over Termitomyces? Which microbe or what forces keep it under check in a healthy comb? How does Pseudoxylaria remain viable for the duration of its “sit-and-wait” phase and not be displaced by other microbes? Therefore, a more parsimonious explanation than “sit-and-wait” is that Pseudoxylaria keeps on flourishing but is prevented from taking over a comb by the termite workers and bacterial symbiotes as several such bacterial strains have been identified from these same mounds33. For, only upon the removal of termites do the proliferation of Pseudoxylaria begins. Therefore, if the anti-fungal capabilities of Pseudoxylaria and Termitomyces, as seen in culture assays (Fig. 4), are a reliable indicator of their capabilities within a fungus comb, then it is clear that Termitomyces is a less efficient inhibitor of non-specific fungi than Pseudoxylaria. This efficiency of Pseudoxylaria can point towards the acquisition of trait that provides benefits to compete and survive in the pool of other microbes or acts as a selection pressure for the accommodation of Pseudoxylaria into this symbiosis. Some evidence towards the symbiotic role of Pseudoxylaria is provided by its presence in the core-mycobiota (Fig. 2 and Supplementary Data 1) and also in the alates of O. obesus (Fig. 2) which hints at successful transmission across generations. The extensive taxonomic review of this group56,57 puts Pseudoxylaria to be exclusively associated with Macrotermitinae termites. Therefore, Pseudoxylaria is either an active member of this symbiosis or is an extremely specialized weed or parasite. Second, Fricke et al.53 reported that the genome architecture of Pseudoxylaria is consistent with a symbiotic lifestyle as they reveal significantly reduced genome sizes with a loss of lignin metabolizing capabilities. Therefore, this could be indicative of a mutualistic role for Pseudoxylaria, involved in preventing other fungi, while simultaneously being dependent on Termitomyces for nutrition through lignocellulose digestion. Third, a mutualistic role of Pseudoxylaria predicts its presence in appreciable amounts even in a fresh comb. As Fig. 3 indicates, the OTU data barely supports this as only 51 reads identified as Pseudoxylaria from the fresh fungus comb. However, qPCR estimates, indicate the presence of 6.0 × 105 copies (95% confidence intervals; Supplementary Table S6). When taken together, these two estimates prove the presence of Pseudoxylaria in fresh combs and are contrary to previous estimates24. However, whether these numbers represent actively growing Pseudoxylaria or not remains to be seen. Fourth, Visser et al.50 reported that both Pseudoxylaria and Termitomyces can use the same Carbon sources to grow, making them strong competitors with the potential of one eliminating the other within a fungus comb. However, that does not seem to be true in O. obesus, as Pseudoxylaria is widely present in this symbiosis (Fig. 2 and Supplementary Data 1). Moreover, upon the removal of termites Pseudoxylaria starts proliferating and very quickly displaces Termitomyces (Fig. 3).
We propose a complex and interdependent relationship between Termitomyces, Pseudoxylaria, and the symbiotic bacteria in maintaining this nutritional symbiosis. The broad outlines of these arguments are detailed in Fig. 6 as a verbal model. This model posits that Termitomyces monocultures face a constant threat from incoming non-specific fungi, possibly brought by foragers into the mound. Since termites construct an ideal growth chamber to grow Termitomyces, the presence of these non-specific fungi can be detrimental as they can easily compete upon the same resources. However, these fungi are prevented by Termitomyces (Fig. 4 and Supplementary Fig. S12), Pseudoxylaria (Fig. 4 and Supplementary Fig. S13), as well as by some bacterial symbiotes (Fig. 5 and Supplementary Figs. S15–S22) from proliferating. The second prediction of this model assumes the presence of bacterial symbiotes in the fresh comb, possibly for the prevention of both Pseudoxylaria and other non-specific fungi. Supplementary Data 2 indicates the presence of 991 OTU’s from the four genera of bacterial symbiotes in fresh comb samples33. As a representative of these mutualists, we undertook qPCR estimation of Pseudomonas and found 3.0 × 105 copies (95% confidence intervals; Supplementary Table S6) in the same fresh comb samples. Therefore, the presence of these bacteria indicates that the control of Pseudoxylaria can perhaps be enforced by secondary symbiotic bacteria in these colonies with additional control by termites through weeding23. However, how such precise control of these various microbes is achieved remains to be investigated.

It suggests a possible mutualistic role of the weed, Pseudoxylaria, as it can control the growth of many non-specific fungi brought in by the termites. It also proposes that Pseudoxylaria, which can overgrow Termitomyces, is kept in control by bacterial symbiotes in fresh fungus combs. The non-specific fungi that compete for resources can also be inhibited by the Termitomyces and bacterial symbiotes. These interactions suggest a complex network of microbial management, and is important for understanding the role of Pseudoxylaria within these termite colonies.
The hypothesis that Pseudoxylaria is an additional symbiont still needs to be established as the culture-dependent studies do not replicate the full complexity of these interactions as they occur in nature. Theory suggests that two or more mutualistic partners cooperate and benefit each other without harming the other partner58,59. The instances where one partner attempts to exploit the relationship by not reciprocating the benefits leads to the extinction of the mutualism. In this case, by inhibiting the sole crop of termites, Pseudoxylaria does not meet the criteria of a symbiont in this system. The presence of Pseudoxylaria in fresh fungus combs might indicate their less harmful role, initially, for entry into the colony. This can change with the age of the colony, where it can overtake the termite garden and behave as a typical parasite or weed of these fungus gardens. As our study used fresh and mature combs, additional work is needed to definitively demonstrate a role for Pseudoxylaria in the early stages of the comb formation, and a particularly important question to resolve in this regard is how a growing culture of Termitomyces might be immune from the generic anti-fungal properties of this weed. Moreover, as our study also viewed the microbial dynamics after removing the termites, it is not entirely clear at this point if bacterial symbiotes play a role in defending Termitomyces against Pseudoxylaria. However, for Pseudoxylaria to be a mutualist, it must be present in a fresh fungus comb in appreciable numbers. We have shown that this is true for the two mounds of O. obesus used in this study. However, if such further evidence is not obtained from other mounds, then this evidence can be considered as incidental and an artifact of the two mounds used. Several studies have discussed the coevolution of Pseudoxylaria with the fungus-farming termites, yet there still remains some confusion on the ecological role of Pseudoxylaria in this symbiosis52,53. While some reports propose its role in biomass degradation39,53, others suggest an antagonistic relationship based on culture-dependent studies54. Several aspects of Pseudoxylaria functioning remains unclear, like its role during the formation of a comb as it necessitates the demonstration of the anti-fungal effects of Pseudoxylaria54 even in these early stages. Some of the experiments detailed here are relatively difficult to do because they involve sampling substantial amounts of fungus combs and consequently, only large and mature colonies can be used, which are few and far between. Therefore, these studies need to be confirmed with additional sampling from different fungus-growing termites. Thus, the role of Pseudoxylaria in fungus-growing termites is mostly considered that of a weed and a threat to the termite colony unless concrete evidence emerges to the contrary. Therefore, our verbal model awaits empirical validation as future studies with more mounds and different species of termites are needed to establish whether this model is validated or the role of Pseudoxylaria remains that of a weed and/or parasite.
Methods
Collection of termites, fungus combs, and their DNA extraction
Different castes (major workers, minor workers, male and female alates and nymphs) and fungus combs of O. obesus were collected from two different mounds within the IISER Mohali campus (Fig. 1). These are the same mounds from which the core-microbiota was obtained in a previous study33. Male and female alates were collected from these mounds during swarming, which began after the first rains in June 2018, while other castes (major workers, minor workers, and nymphs) were collected along with the fungus combs (Fig. 1). The details of the collection and DNA extraction are detailed in Supplementary Notes 1 and 2.
Fresh fungus combs were collected across three months (August–October 2018) from the same two mounds as mentioned above and were set up to decay for 120 h. The collected combs were immediately removed from their resident termites, kept in sterile plastic containers, then crushed with autoclaved pestles, divided into roughly 0.5 g portions, and incubated at 30 °C inside sterilized glass containers which were then screwed shut to prevent any possible contamination (Supplementary Fig. S1). For each incubation assay, three such containers were set up from each mound with 12 such incubation assays in all. DNA was extracted from these combs consecutively for six days, i.e., Fresh comb (0 h) to 120 h. For fresh comb, DNA was extracted from each portion separately within 3 hours of the collection. DNA samples were amplified in replicates and then the PCR products from the same day were pooled in equimolar concentrations (20-25 ng µl−1) into a single sample. Six such pooled PCR products, one from each day (Fresh comb (0 h)– 120 h), were prepared for microbiota and mycobiota analysis on the Nanopore platform (Supplementary Table S3).
In total, we have sampled 12 pieces of fresh fungus comb, 30 termites across different castes and 36 samples of the decaying comb across various stages for Nanopore sequencing to have a far better understanding of how the microbes within a termite mound functions.
Isolation and identification of fungal strains
Termitomyces was directly isolated from fresh nodules growing on the fungus comb, successively washed with 50% ethanol and sterilized water, inoculated on plates containing Potato Dextrose Agar (PDA) with Yeast Malt Extract (PYME)22 and incubated at 30 °C. Pseudoxylaria hyphae were obtained from a 48–72 h old post-collection fungus comb and directly cultured on Potato Dextrose Agar (PDA) plates. To isolate other fungi, comb fragments and termite castes were washed with sterilized water, homogenized in 500 µl of sterile 1× PBS buffer (pH −7.4) and a dilution series (10×–10−6×) of these homogenates were plated on PDA plates containing 250 μg/mL chloramphenicol (Himedia), and incubated at 30 °C for 4–7 days. Fungal cultures obtained were initially screened visually for unique growth characteristics and identified by sequencing the Internal Transcribed Spacer (ITS) region, which was amplified with the primer set ITS4/ITS560 (Supplementary Note 3). Any fungal cultures with more than one Single Nucleotide Polymorphism (SNP) were considered unique and used in the study (Supplementary Table S1). The sequences of these fungi have been submitted to GenBank accession number (MN913749 –MN913772). Since multiple strains within some fungal genera were obtained, the nomenclature of these strains was modified to read as “Genus-NCBI Accession Number” (Supplementary Table S1).
Running the nanopore platform and obtaining fungal and bacterial operational taxonomic units (OTU’s)
The mycobiota was obtained by amplifying the ITS fragment with the primer set ITS5/ITS460, while the V3-V4 region of the 16 S rRNA gene was amplified to characterize the microbiota using the primers 341F/806R61. DNA from different castes and six different comb samples of varying degrees of incubation were used to obtain the pan-mycobiota (Supplementary Tables S2 and S3). These six comb samples were also amplified to characterize their microbiota (Supplementary Table S3).
The sequencing library was run on FLO-MIN106 flowcell for 48 h using MinKNOW software with the protocol NC_48 h_sequencing_FLO-MIN106_SQK-LSK108_plus_Basecaller (Supplementary Note 4). All the cleaned sequences were deposited to NCBI Sequence Read Archive (SRA). The five mycobiota were deposited under the BioProject PRJNA665655 (SAMN16262892–SAMN16262897), the microbiota of decaying comb samples were deposited under BioProject PRJNA758817 (SAMN21036816–SAMN21036821) and the mycobiota of decaying comb as PRJNA758827 (SAMN21037110–SAMN21037115), respectively.
Taxonomic identification of OTU’s
The fungal OTU’s were identified by comparing them against the fungus repository from the NCBI FTP site with an additional modification. Initial annotation revealed several “Pseudoxylaria-like” sequences, which were not identified till the genus level. Therefore, to refine the annotation we used the previous study by Hsieh et al.57 to phylogenetically characterize these sequences from NCBI to see whether these belong to Pseudoxylaria or not. This was done by amplifying the α-actin gene using the primer set ACT-512F/ACT-783R57 and comparing it with the other members of the Xylariaceae family. The α-actin sequences accession number (OQ503187 and OQ503188) from the Pseudoxylaria isolated from the two mounds under study were initially aligned with Clustal Omega62 and manually edited in Bioedit v 7.0.5.363. Phylogenetic analysis of these sequences was performed in MrBayes v 3.2.764. After confirming that the two sequences belonged to the Pseudoxylaria clade, we aligned them with sequences from NCBI annotated as Xylaria, Xylariaceae, and Xylariales and performed another phylogenetic analysis. Sequences that formed a monophyletic clade with these two previously identified Pseudoxylaria sequences were renamed as Pseudoxylaria (Supplementary Figs. S2 and S3 and Supplementary Data 1) and updated in the reference database used for annotation. Annotation was done using LAST v 973, with the following parameters: match score of 1, gap opening penalty of 1, and gap extension penalty of 165. The identified reads were sorted from the phylum to the genus level and their relative abundances were calculated. To estimate the sequencing depth, rarefaction curves were generated using the Vegan package v 2.5-4 in R v 3.6.2. The estimated community richness (Chao1) and diversity indices (Evenness, Shannon, Simpson, and Inv Simpson) were also calculated in R v 3.6.2. To test how representative these communities are across the different fungus-growing termites, a comparison was done with the data obtained in this study with previously published data from refs. 20 and 24.
The bacterial sequences generated from the six comb samples (Fresh comb (0 h)–120 h) were identified by comparing them against the customized microbial repository made by combining 16 S rRNA gene fragments from NCBI FTP site and the DictDb v 3.0 database66. The OTU’s for the potential four bacterial symbiotes were filtered, combined and compared with the absolute abundances of Termitomyces and Pseudoxylaria.
Quantitative PCR for Termitomyces, Pseudoxylaria, and Pseudomonas in fungus combs
The decaying comb samples (0–120 h) were also used to quantify Termitomyces, Pseudoxylaria, and Pseudomonas copy numbers. For qPCR analysis, 72 samples of degrading comb (12 for each stage) were used to quantify the absolute abundances of Termitomyces, Pseudoxylaria and Pseudomonas (details in Supplementary Table S6). Moreover, each of these samples was run as a triplicate for the qPCR assays to increase the accuracy of the analysis. The specificity of the designed primers was checked with Primer-BLAST (last performed in July 2021). The sequence of these primers and their respective annealing temperatures are given in Supplementary Table S4.
qPCR was performed in a CFX96TM Real-Time System (Bio-Rad) with SYBR Green I assay. The amplification reaction was prepared to a final volume of 10 µl containing 2.7 µl of sterilized distilled water, 5 µl of iTaq Universal SYBR® Green Supermix (BIO-RAD), 0.05 µl (10 µM) of each primer, 0.2 µl of Bovine Serum Albumin (BSA) and 20 ng of template DNA. The qPCR amplification began with an initial incubation for 3 minutes at 95 °C, followed by 40 cycles of 95 °C for 10 s and a 30 s incubation for primer annealing (Supplementary Table S4). All the reactions were set up in triplicates and a mixture of DNA from other non-target fungi was used as the negative control, while autoclaved distilled water was used as the non-template control. DNA of pure cultures of Termitomyces, Pseudoxylaria and Pseudomonas were amplified using their specific primer sets (Supplementary Table S4) in a serial dilution of 10−1× to 10−5 to obtain the standard curve. Target copy numbers for each reaction were calculated from the standard curves67 using the formula:
Number of copies/µl = ((amount used in ng) * (6.022 * 1023 molecules/mole))/(length of amplicon) * (650 g/mole).
The specificity of the reactions was assessed by the analysis of the melting curve. The copy numbers obtained were then used to generate the number of amplicons in the target samples with the following formula68:
Xosample = EAMPsample [babs *log (EAMPsample EAMPabs)-Cqsample]
Xosample = number of copies
EAMPsample= Exponential amplification of the sample
EAMPabs= Exponential amplification of the standards
Cqsample= Quantification cycle for the sample
babs= Standard curve of the intercept.
The statistical significance of these copy number variations was done with a two-tailed t test at P < 0.05.
In vitro anti-fungal assays with Pseudoxylaria and Termitomyces
In vitro interaction experiments were done to identify whether Termitomyces and Pseudoxylaria can prevent the growth of the non-specific fungi (Supplementary Note 5). The magnitude of inhibition by test fungi was evaluated by using the following formula from ref. 69:
% Inhibition = ((total area of test fungus in control in mm2) – (total area of test fungus in the interaction in mm2)) * 100/total area of test fungus in control in mm2.
For easy comprehension, the interactions were categorized into the following three categories: more than 50% inhibition, less than 50% inhibition, and no inhibition. To explore the effect of Termitomyces, the spread method was used33 where a liquid suspension of Termitomyces nodules/mycelia was prepared in 1× PBS and spread on PYME22 plates (Supplementary Fig. S12). These plates were first incubated for 36 h at 30 °C before placing a plug of the test fungus at the center. The pre-incubation time of 36 h was selected on the basis of the pilot experiments where Termitomyces was grown for both 24 h and 36 h before the introduction of the competing fungus. We found no difference in the results after 7 days of interaction (Supplementary Fig. S10). In nature fresh fungus combs are dominated by Termitomyces. Therefore, we wanted to mimic this feature of the fresh fungus comb in our in vitro plates, as far as possible, and tried covering the entire plate with Termitomyces culture, before the introduction of the test fungus. Going beyond the 36 h of incubation time may not yield accurate results as any inhibition seen can be attributed to the depletion of nutrients from the media or Termitomyces dying than to the anti-fungal activity of Termitomyces. For interaction with Pseudoxylaria, a dual plug assay was used (Supplementary Fig. S13) in 90 mm diameter Petri dishes containing PDA and 250 µg/mL chloramphenicol. Pseudoxylaria being a fast-growing fungus can fill an entire plate within 5 days, therefore plugs of Pseudoxylaria were used (Supplementary Fig. S11). Plates were inoculated separately with a 1-cm2 disc of actively growing Pseudoxylaria, 10 mm away from the edge of the plates. In all, 1-cm2 disc of the test fungus was placed separately in the same plate but 60 mm away from the Pseudoxylaria plug (Supplementary Fig. S13). The control growth plates were set up for the test fungi and Pseudoxylaria. These were then incubated in the dark at 30 °C in HerathermTM Compact Microbiological Incubator (Thermo-Fisher Scientific). All the interaction plates (with both Pseudoxylaria and Termitomyces) were evaluated for growth inhibition by taking photographs using the Panasonic Lumix G2 camera. Photographs from the 7th day were taken as the final data and were analyzed in Adobe Photoshop CS6 by measuring the area of test fungi growth in their respective interactions (both test and control plates) in pixels and converting it into millimeters (100 pixels = 1 mm). All interaction assays were repeated with sample sizes ranging from 3 to 5. Representative figures for all the interaction assays are given in Supplementary Figs. S12 and S13.
In vitro anti-fungal assays with symbiotic bacteria
The non-specific fungi which were inhibited to less than 50% by both the Termitomyces and Pseudoxylaria were further selected for the bacterial-fungal assays. Paper disc diffusion assays were performed to identify whether the previously identified eight bacterial symbiotes from the same mounds33 prevented the growth of these non-specific fungi. We tested four strains of Bacillus (MN908297, MN908298, MN908304 and MN908305), single strain of Burkholderia (MN908310), two strains of Pseudomonas (MN908321 and MN908322), and a single strain of Streptomyces (MN908327) against five non-specific fungi. The interactions were categorized into four broad categories of clear zone of inhibition, reduced growth of fungus near the bacteria, contact inhibition, and negligible inhibition33. The magnitude of inhibition of the fungal growth by these bacteria was evaluated using the formula mentioned above from ref. 69. The cultures of Phoma and Fusarium grow much slower than the other fungi tested. To avoid any discrepancy in the results due to differences in growth, the spread method was used for these two fungi and four bacterial discs were placed compared to a single disc. The culture of Paraconiothyrium-MN913759 was lost during this study and therefore, bacterial interactions against this fungus could not be done.
Statistics and reproducibility
Fresh fungus combs were collected over three months (August–October 2018) from two mounds. For each day, three containers were set up from each mound. Three separate DNA extractions, per mound, were pooled in equimolar concentrations, and these pools were further combined with DNA from the other mound, creating a total of six replicates for each decaying time point. For termite castes, three replicates per mound were used and pooled, resulting in a total of six replicates per caste. For alates, collections were done from one mound, where three individual DNA extractions from males and females were pooled. For Nanopore sequencing, no data were excluded from the analyses and no statistical method was used to predetermine sample size. Diversity indices were calculated using the phyloseq package in Rstudio v3.6.2. The heatmap for the myco- and microbiota was generated using the pheatmap package in Rstudio v3.6.2. Details of the phylogenetic analysis are provided in the Methods section. All the statistical analysis for the qPCR data can also be found in Methods section, with data values available in the supplementary data files. The interaction assays were conducted with a minimum of three replicates and graphs for these assays were prepared in Rstudio v3.6.2. The data values for all graphs can be found in the supplementary data files.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mycobiome dataset generated is available as Supplementary Data 1 and 2 files of this article. ITS gene fragments obtained have been submitted to GenBank accession number (MN913749–MN913772) and Nanopore sequencing generated have been submitted to NCBI Sequence Read Archive (SRA) under the BioProjects PRJNA665655 (SAMN16262892–SAMN16262897), PRJNA758817 (SAMN21036816–SAMN21036821) and PRJNA758827 (SAMN21037110–SAMN21037115). Source data for graphs presented in the main figures can be found in Supplementary Data 3–5. All other data are available from the corresponding author upon reasonable request.
References
Martínez, Á. T., Ruiz-Dueñas, F. J., Martínez, M. J., Del Río, J. C. & Gutiérrez, A. Enzymatic delignification of plant cell wall: from nature to mill. Curr. Opin. Biotechnol. 20, 348–357 (2009).
Raychoudhury, R. et al. Comparative metatranscriptomic signatures of wood and paper feeding in the gut of the termite Reticulitermes flavipes (Isoptera: Rhinotermitidae). Insect Mol. Biol. 22, 155–171 (2013).
Breznak, J. A. & Brune, A. Role of microorganisms in the digestion of lignocellulose by termites. Annu. Rev. Entomol. 39, 453–487 (1994).
Bignell, D. E. & Eggleton, P. In Termites: Evolution, Sociality, Symbioses, Ecology (eds Abe, T. et al.) 287–306 (Kluwer Academic Publishers, 2000).
Mueller, U. G. & Gerardo, N. Fungus-farming insects: multiple origins and diverse evolutionary histories. Proc. Natl. Acad. Sci. USA 99, 15247–15249 (2002).
Li, H. et al. Lignocellulose pretreatment in a fungus-cultivating termite. Proc. Natl. Acad. Sci. USA 114, 4709–4714 (2017).
Chiu, C. I., Ou, J. H., Chen, C. Y. & Li, H. F. Fungal nutrition allocation enhances mutualism with fungus-growing termite. Fungal Ecol. 41, 92–100 (2019).
Aanen, D. K. et al. High symbiont relatedness stabilizes mutualistic cooperation in fungus-growing termites. Science 326, 1103–1106 (2009).
Aanen, D. K. & Eggleton, P. Fungus-growing termites originated in African rain forest. Curr. Biol. 15, 851–855 (2005).
Mueller, U. G., Gerardo, N. M., Aanen, D. K., Six, D. L. & Schultz, T. R. The evolution of agriculture in insects. Annu. Rev. Ecol. Evol. Syst. 36, 563–595 (2005).
Eggleton, P. An introduction to termites: biology, taxonomy and functional morphology. In Biology of Termites: A Modern Synthesis (eds Bignell, D. E. et al.) 1–26 (Springer, 2011).
Nobre, T., Eggleton, P. & Aanen, D. Vertical transmission as the key to the colonization of Madagascar by fungus-growing termites. Proc. R. Soc. B: Biol. Sci. 277, 359–365 (2010).
Sieber, R. Establishment of fungus comb in Laboratory colonies of Macrotermes michaelseni and Odontotermes montanus (Isoptera, Macrotermitinae). Insects Soc. 30, 204–209 (1983).
Aanen, D. K. As you reap, so shall you sow: coupling of harvesting and inoculating stabilizes the mutualism between termites and fungi. Biol. Lett. 2, 209–212 (2006).
Batra, L. R. & Batra, S. W. Fungus-growing termites of tropical India and associated fungi. J. Kans. Entomol. Soc. 39, 725–738 (1966).
Korb, J. Thermoregulation and ventilation of termite mounds. Naturwissenschaften 90, 212–219 (2003).
Korb, J. & Linsenmair, K. Thermoregulation of termite mounds: what role does ambient temperature and metabolism of the colony play?. Insect Soc. 47, 357–363 (2000).
Boddy, L. Fungal community ecology and wood decomposition processes in angiosperms: from standing tree to complete decay of coarse woody debris. Ecol. Bull. 49, 43–56 (2001).
Gams, W. The analysis of communities of saprophytic microfungi with special reference to soil fungi. In Fungi in Vegetation Science (ed. W. Winterhoff) 183–223 (Kluwer Academic Publishers, 1992).
Bos, N. et al. You don’t have the guts: a diverse set of fungi survive passage through Macrotermes bellicosus termite guts. BMC Evol. Biol. 20, 1–11 (2020).
Nagam, V. et al. Diversity of fungal isolates from fungus‐growing termite Macrotermes barneyi and characterization of bioactive compound from Xylaria escharoidea. Insect Sci. 28, 392–402 (2021).
Thomas, R. Selective medium for isolation of Termitomyces from termite nests. Trans. Br. Mycol. Soc. 84, 519–526 (1985).
Katariya, L. et al. Fungus-farming termites selectively bury weedy fungi that smell different from crop fungi. J. Chem. Ecol. 43, 986–995 (2017).
Otani, S. et al. Disease-free monoculture farming by fungus-growing termites. Sci. Rep. 9, 1–10 (2019).
Li, H. & Greening, C. Termite-engineered microbial communities of termite nest structures: a new dimension to the extended phenotype. FEMS Microbiol. Rev. 46, fuac034 (2022).
Mathew, G. M., Ju, Y. M., Lai, C. Y., Mathew, D. C. & Huang, C. C. Microbial community analysis in the termite gut and fungus comb of Odontotermes formosanus: the implication of Bacillus as mutualists. FEMS Microbiol. Ecol. 79, 504–517 (2012).
Schmidt, S., Kildgaard, S., Guo, H., Beemelmanns, C. & Poulsen, M. The chemical ecology of the fungus-farming termite symbiosis. Nat. Prod. Rep. 39, 231–248 (2022).
Yin, C., Jin, L., Li, S., Xu, X. & Zhang, Y. Diversity and antagonistic potential of Actinobacteria from the fungus-growing termite Odontotermes formosanus. 3 Biotech 9, 1–7 (2019).
Aylward, F. O. et al. Convergent bacterial microbiotas in the fungal agricultural systems of insects. mBio 5, e02077–02014 (2014).
Barcoto, M. O. et al. Fungus-growing insects host a distinctive microbiota apparently adapted to the fungiculture environment. Sci. Rep. 10, 1–13 (2020).
Fukuda, T. T. et al. Insights into the ecological role of Pseudomonas spp. In an ant-plant symbiosis. Front. Microbiol. 12, 621274 (2021).
Saati-Santamaría, Z., Rivas, R., Kolařik, M. & García-Fraile, P. A new perspective of Pseudomonas—host interactions: distribution and potential ecological functions of the genus Pseudomonas within the bark beetle Holobiont. Biology 10, 164 (2021).
Agarwal, R., Gupta, M., Antony, A., Sen, R. & Raychoudhury, R. In vitro studies reveal that Pseudomonas, from Odontotermes obesus colonies, can function as a defensive mutualist as it prevents the weedy fungus while keeping the crop fungus unaffected. Microb. Ecol. 84, 1–13 (2021).
Vega, F. E. & Biedermann, P. H. On interactions, associations, mycetangia, mutualists and symbiotes in insect-fungus symbioses. Fungal Ecol. 44, 100909 (2020).
Beemelmanns, C. et al. Macrotermycins A–D, glycosylated macrolactams from a termite-associated Amycolatopsis sp. M39. Org. Lett. 19, 1000–1003 (2017).
Benndorf, R. et al. Natural products from Actinobacteria associated with fungus-growing termites. Antibiotics 7, 83 (2018).
Um, S., Fraimout, A., Sapountzis, P., Oh, D. C. & Poulsen, M. The fungus-growing termite Macrotermes natalensis harbors bacillaene-producing Bacillus sp. that inhibit potentially antagonistic fungi. Sci. Rep. 3, 1–7 (2013).
Carr, G. et al. Microtermolides A and B from termite-associated Streptomyces sp. and structural revision of vinylamycin. Org. Lett. 14, 2822–2825 (2012).
Visser, A. A., Nobre, T., Currie, C. R., Aanen, D. K. & Poulsen, M. Exploring the potential for actinobacteria as defensive symbionts in fungus-growing termites. Microb. Ecol. 63, 975–985 (2012).
Thomas, R. J. Factors affecting the distribution and activity of fungi in the nests of Macrotermitinae (Isoptera). Soil Biol. Biochem. 19, 343–349 (1987).
Liang, S. et al. Exploring the effect of plant substrates on bacterial community structure in termite fungus-combs. PLoS ONE 15, e0232329 (2020).
Otani, S., Hansen, L. H., Sørensen, S. J. & Poulsen, M. Bacterial communities in termite fungus combs are comprised of consistent gut deposits and contributions from the environment. Microb. Ecol. 71, 207–220 (2016).
Moriya, S. et al. Fungal community analysis of fungus gardens in termite nests. Microbes Environ. 20, 243–252 (2005).
Thomas, R. J. Distribution of Termitomyces and other fungi in the nests and major workers of several Nigerian Macrotermitinae. Soil Biol. Biochem. 19, 335–341 (1987).
Wood, T. G. & Thomas, R. J. The mutualistic association between Macrotermitinae and Termitomyces. In Insect–Fungus Interactions (eds Wilding, Collins, N. M., Hammond, P. M. & Webber, J. F.) 69–92 (Academic Press, 1989).
Zhang, T. & Fang, H. Applications of real-time polymerase chain reaction for quantification of microorganisms in environmental samples. Appl. Microbiol. Biotechnol. 70, 281–289 (2006).
Diouf, M. et al. Succession of the microbiota in the gut of reproductives of Macrotermes subhyalinus (Termitidae) at colony foundation gives insights into symbionts transmission. Front. Ecol. Evol. 10, 1055382 (2022).
de Fine Licht, H. H., Boomsma, J. & Aanen, D. K. Presumptive horizontal symbiont transmission in the fungus‐growing termite Macrotermes natalensis. Mol. Ecol. 15, 3131–3138 (2006).
Katariya, L., Ramesh, P., Sharma, A. & Borges, R. M. Local hypoxia generated by live burial is effective in weed control within termite fungus farms. Insectes Soc. 65, 561–569 (2018).
Visser, A. A., Kooij, P. W., Debets, A. J., Kuyper, T. W. & Aanen, D. K. Pseudoxylaria as stowaway of the fungus-growing termite nest: Interaction asymmetry between Pseudoxylaria, Termitomyces and free-living relatives. Fungal Ecol. 4, 322–332 (2011).
Aanen, D. K. et al. The evolution of fungus-growing termites and their mutualistic fungal symbionts. Proc. Natl. Acad. Sci. USA 99, 14887–14892 (2002).
Visser, A. et al. Levels of specificity of Xylaria species associated with fungus‐growing termites: a phylogenetic approach. Mol. Ecol. 18, 553–567 (2009).
Fricke, J. et al. Adaptations of Pseudoxylaria towards a comb-associated lifestyle in fungus-farming termite colonies. ISME J. 17, 733–747 (2023).
Guo, H. et al. Pseudoxylallemycins A–F, cyclic tetrapeptides with rare allenyl modifications isolated from Pseudoxylaria sp. X802: a competitor of fungus-growing termite cultivars. Org. Lett. 18, 3338–3341 (2016).
Rogers, J. D., Ju, Y.-M. & Lehmann, J. Some Xylaria species on termite nests. Mycologia 97, 914–923 (2005).
Boedijn, K. On a new family of the Sphaeriales. Pers. Mol. Phylogeny Evol. 1, 15–19 (1959).
Hsieh, H. M. et al. Phylogenetic status of Xylaria subgenus Pseudoxylaria among taxa of the subfamily Xylarioideae (Xylariaceae) and phylogeny of the taxa involved in the subfamily. Mol. Phylogenet. Evol. 54, 957–969 (2010).
Connor, R. C. J. B. R. The benefits of mutualism: a conceptual framework. Biol. Rev. 70, 427–457 (1995).
Leigh, E. G. Jr The evolution of mutualism. J. Evol. Biol. 23, 2507–2528 (2010).
White, J. S. et al. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods Applications (eds Innis, M. A. et al.) 315–322 (Academic Press, 1990).
Hansen, C. et al. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia 55, 2285–2294 (2012).
Sievers, F. & Higgins, D. G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079, 105–116 (2014).
Hall, T., Biosciences, I. & Carlsbad, C. BioEdit: an important software for molecular biology. GERF Bull. Biosci. 2, 60–61 (2011).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Shin, J. et al. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Sci. Rep. 6, 29681 (2016).
Mikaelyan, A. et al. Diet is the primary determinant of bacterial community structure in the guts of higher termites. Mol. Ecol. 24, 5284–5295 (2015).
Gallup, J. M. & Ackermann, M. R. The ‘PREXCEL-Q method’for qPCR. Int. J. Biomed. Sci. 4, 273 (2008).
Gallup, J. M. qPCR inhibition and amplification of difficult templates. In PCR Troubleshooting and Optimization: The Essential Guide (eds Kennedy, S. & Oswald, N.) 23–65 (Horizon Scientific Press, 2011).
Royse, D. & Ries, S. The influence of fungi isolated from peach twigs on the pathogenicity of Cytospora cincta. Phytopathology 68, 603–607 (1978).
Acknowledgements
The authors thank Abin Antony and Raunak Dhar for their help with the fieldwork, Dr. Shashi Bhushan Pandit and Paras Verma for their technical help with the data analysis. The authors acknowledge all the EVOGEN lab members for their valuable comments on the manuscript. The authors thank IISER Mohali for providing funds to conduct this study. R.R. and R.S. were also supported by the SERB-CRG grant CRG/2021/007010. R.A. was supported by PhD fellowship from IISER Mohali. M.G. was supported by the University Grant Commission (UGC) Junior Research Fellowship #325974. A.P. was supported by University Grant Commission (UGC) Junior Research Fellowship #623.
Author information
Authors and Affiliations
Contributions
R.R. and R.A. devised the study. R.A. performed field collections, sequencing, microbial culturing, data collection and analysis. A.P. participated in data collection. N.E.S. helped in the fieldwork, culturing of fungi and pilot experiments. Nanopore sequencing was done by R.A. and M.G. and its analysis was done by R.A. The statistical analysis of interaction experiments was done by R.A. and R.S. R.A., R.S., and R.R. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Suzanne Schmidt and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: David Favero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Agarwal, R., Gupta, M., Sen, R. et al. Investigation into how Odontotermes obesus maintains a predominantly Termitomyces monoculture in their fungus combs suggests a potential partnership with both fungi and bacteria. Commun Biol 7, 1010 (2024). https://doi.org/10.1038/s42003-024-06708-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06708-2
