
Abstract
Cryogenic electron microscopy (cryo-EM) has revolutionized structural biology, enabling efficient determination of structures at near-atomic resolutions. However, a common challenge arises from the severe imbalance among various conformations of vitrified particles, leading to low-resolution reconstructions in rare conformations due to a lack of particle images in these quasi-stable states. We introduce CryoTRANS, a method that predicts high-resolution maps of rare conformations by constructing a self-supervised pseudo-trajectory between density maps of varying resolutions. This trajectory is represented by an ordinary differential equation parameterized by a deep neural network, ensuring retention of detailed structures from high-resolution density maps. By leveraging a single high-resolution density map, CryoTRANS significantly improves the reconstruction of rare conformations and has been validated on four real-world datasets: alpha-2-macroglobulin, actin-binding protein complexes, SARS-CoV-2 spike glycoprotein, and the 70S ribosome. CryoTRANS can also predict high-resolution structures in cryogenic electron tomography maps using a high-resolution cryo-EM map.
Similar content being viewed by others
Introduction
The elucidation of biological macromolecules’ structures is fundamental to understanding life’s complexities and biochemical mechanisms. Among various techniques, cryogenic electron microscopy (cryo-EM) has proven to be a powerful tool1,2,3,4,5, enabling structural analysis at atomic or near-atomic resolutions. Remarkably, it achieves this by rapidly vitrifying biological specimens within milliseconds6,7—a speed that notably surpasses the transition durations between conformational states8,9,10.
The functions of biological macromolecules are driven by their structural transitions. According to the dynamic equilibrium law, there is a significant disparity in the prevalence of different conformational states in vitrified particles. High-occupancy states, also known as common conformations, are markedly more likely to accumulate a substantial number of particles, thereby enabling high-resolution reconstructions in comparison to less common states. Conversely, the law also suggests that high-energy quasi-stable states are sparsely populated under equilibrium conditions11. Such states, named as rare conformations, hold biologists’ interest due to their capacity to illuminate crucial biological processes because of their functional significance7,12. However, owing to the limited availability of single-particle images, rare conformations often yield unsatisfactory resolutions in cryo-EM when processed using the conventional discrete clustering approach13,14,15.
Time-resolved cryo-EM7,10,16,17 offers a pioneering method to enhance the representation of particle images associated with rare conformations. This is achieved through meticulous control of the biochemical reaction prior to the freezing process. The innovative aspect of this technique lies in its ability to vitrify the sample precisely when the high-energy quasi-stable conformation is predominant. Successful application of time-resolved cryo-EM demands a thorough understanding of the underlying biochemical reactions, adeptness in micro-fluid mixing18, proficiency with grid preparation devices18, and seasoned expertise. An alternative strategy to address the limited occupancy of rare conformation particles is liquid cell electron microscopy (LCEM)19, which maintains the non-equilibrium conditions within the liquid cell, rather than in frozen samples. Thus, a limitation of LCEM is that, without the protective shield of cryogenic freezing, the captured images of biomolecules are of restricted resolution. Given the inherent challenges and limitations associated with these methods, there is a compelling need to craft image-processing algorithms tailored to mitigate the pronounced imbalance between various conformations observed in traditional cryo-EM.
In recent years, continuous heterogeneity reconstruction methods13,20,21,22,23,24,25 have emerged as alternatives that may enhance the resolution of these rare conformations. These techniques extrapolate deformations directly from two-dimensional particle images. However, they suffer from an imbalance in particle distribution, which is likely to impair the effectiveness of the continuous heterogeneity reconstruction methods. An alternative approach to improving the resolution of rare conformations leverages deep learning-based post-processing techniques26,27. These methods utilize neural networks trained on large datasets of cryo-EM maps to predict and refine features in low-resolution input maps. However, it is important to note that the performance of such methods heavily depends on the pre-existing structural information in the training datasets, which may introduce model bias. This bias can result in incorrect modeling for structures that are underrepresented in the training data27, thereby affecting the reliability of the enhanced maps.
In this paper, we introduce a method called CryoTRANS (TRansformations by Artificial NetworkS). Our method accurately predicts high-resolution density maps of rare conformations by identifying a pseudo-trajectory through different states in a self-supervised manner. CryoTRANS employs a deep neural network-parameterized ordinary differential equation (ODE), termed neural ODE, to model the continuum of non-rigid deformations from common to rare conformations. By incorporating a Wasserstein distance and imposing a rigidity constraint on the velocity field, we propose an optimization model for training the neural network in a self-supervised manner, eliminating the need for supervised samples or pre-training. Upon training completion, the deformed density map, governed by the ODE, yields a high-resolution approximation of the rare conformations. We demonstrate the efficacy of CryoTRANS through four real datasets, validating its capability to predict high-resolution density maps of rare conformations with high accuracy. We further demonstrate the advantages of CryoTRANS over the existing deep-learning-based post-processing methods, including DeepEMhancer26 and EMReady27. In addition, we compare the pseudo-trajectories generated by CryoTRANS with MorphOT28 and CryoDRGN22, showing that our trajectory does not suffer from the random appearance and disappearance of densities during the deformation process. In particular, the pseudo-trajectory generated by CryoTRANS exhibits consistency with molecular dynamics simulations, further validating the robustness and accuracy of our approach.
Results
Overview of CryoTRANS
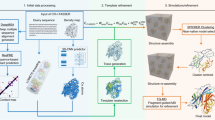
CryoTRANS enables morphing between two cryo-EM density maps using an ODE-governed deformation field parameterized by a deep neural network (DNN) (Fig. 1a). In this process, the initial state f0 corresponds to a high-resolution density map, while the terminal state f1 corresponds to a low-resolution target map. The deformation process, represented by a velocity field parameterized by the network \(\vec{V}\), is captured by varying the pseudo-time in the interval [0, 1]. The discretization of the ODE model in time naturally results in a recurrent weight-sharing network capable of mapping any point in the initial density map to a corresponding point in \({{\mathbb{R}}}^{3}\). Given t ∈ [0, 1], we define \({\hat{f}}_{t}\) as the intermediate map at time t (Fig. 1c). Specifically, the initial map \({\hat{f}}_{0}\) is equal to f0, and the terminal map \({\hat{f}}_{1}\) is called as the generated map (Fig. 1a).

a CryoTRANS leverages a high-resolution initial map denoted by f0 and a lower-resolution target map denoted by f1 as inputs. It then utilizes a neural network-parameterized velocity field \(\vec{V}\) to construct a pseudo-trajectory within the 3D coordinate space of the initial map. This pseudo-trajectory is achieved by discretizing a governing ODE into ten pseudo-time points. During training, the model minimizes a loss function that incorporates the Wasserstein distance between the final generated map \(\hat{{f}_{1}}\) and the target map f1. The gradient of this loss function is backpropagated through the neural network to optimize its parameters. b Diagram of the Wasserstein Distance: quantifies the minimum effort required to transform the densities in the generated map \({\hat{f}}_{1}\) to match those in the target map f1. c The intermediate density maps at different pseudo-time points generated by CryoTRANS using the trained velocity field \(\vec{V}\). This pseudo-trajectory, visualized as an orange cube, represents the path of a specific voxel within the density map. These example maps are simulations derived from atomic models of Mm-cpn34,35 in two conformations: the closed state (PDB 3J03) and the open state (PDB 3IYF).
To train the DNN, the squared Wasserstein distance is employed as the loss function between the generated map \({\hat{f}}_{1}\) and the target map f1, opting for it over the conventional mean squared error. The Wasserstein distance, which is the minimal transportation cost between two probability distributions, effectively captures the geometric information of the underlying space. Specifically, let \({\vec{x}}_{i}\) be location of the ith voxel in \({\hat{f}}_{1}\) and \({\vec{x}}_{j}\) be location of the jth voxel in f1, \({\parallel {\vec{x}}_{i}-{\vec{x}}_{j}\parallel }^{2}\) (squared Euclidean norm) and Pi,j represent the cost and the probability density moving from \({\vec{x}}_{i}\) to \({\vec{x}}_{j}\), respectively. Then \({P}_{i,j}{\parallel {\vec{x}}_{i}-{\vec{x}}_{j}\parallel }^{2}\) is the transportation cost that relocates from \({\vec{x}}_{i}\) in \({\hat{f}}_{1}\) to \({\vec{x}}_{j}\) in f1 (Fig. 1b). The squared Wasserstein distance corresponds to the minimal total transportation cost, which is the summation of transportation costs for all voxels, achieved by finding a suitable transport plan P. Recent studies have shown the advantages of Wasserstein distance over classical Euclidean distances with theoretical analysis29, which validates the mathematical soundness of CryoTRANS. Additionally, a rigidity constraint is placed on the velocity field \(\vec{V}\) to preserve the mass of the density map in each layer and prevent the dispersion of density. The network is optimized using the adaptive moment estimation (ADAM) algorithm30, and we provide a more detailed exposition of CryoTRANS in the “Methods” section.
Within the trained velocity field \(\vec{V}\), it is observed that the high-resolution details of the initial map are maintained throughout the deformation process (Fig. 1c). Thus, the generated map can be seen as a high-resolution prediction of the target map, effectively capturing the structural features.
Validation of the CryoTRANS-generated maps
To evaluate the quality and accuracy of the CryoTRANS-generated maps, we conducted experiments on simulated datasets of four biological macromolecules. For each macromolecule, two distinct conformations have been resolved in previous studies using experimental approaches, and corresponding atomic models have been constructed. Specifically, the four are the heterodimeric ABC exporter TmrAB31 in ATP-bound outward-facing open conformation (PDB 6RAH) and ATP-bound outward-facing occluded conformation (PDB 6RAI), another ABC exporter PCAT132 in the inward-facing wide conformation (PDB 7T55) and the inward-facing narrow conformation (PDB 7T57), human prestin33 in complex with chloride (PDB 7V73) and salicylate (PDB 7V75), and Mm-cpn34,35 in its open state (PDB 3IYF) and closed state (PDB 3J03). To mimic target maps with different resolutions, we low-pass filtered the atomic model of one conformation state to 3, 5, and 7 Å. We compared the generated maps to the atomic models and calculated the model-to-map Fourier shell correlation (FSC) (Fig. 2). Additional quantitative metrics, including Q-score36, TM-score37, and chain comparison score of Phenix38, can be found in Supplementary Note I.

The four biological macromolecules tested by CryoTRANS are shown from top to bottom. Each macromolecule exists in two distinct conformations, with atomic models of these conformations resolved using experimental methods. The left column shows the initial maps for CryoTRANS, generated from one conformation of each protein at a resolution of 3 Å. The middle columns display the target maps (upper row) and the corresponding generated maps by CryoTRANS (lower row). The target maps were generated at resolutions of 3 Å (green), 5 Å (yellow), and 7 Å (orange). The right column presents the model-to-map FSCs between the ground truth atomic model and the generated maps.
If both the initial and target density maps are at 3 Å resolution, the generated maps have a resolution higher than 3.2 Å (Fig. 2). Additionally, when the target maps are at 5 or 7 Å resolution, the generated maps preserve information up to an approximate resolution of 4 Å (Fig. 2). These results demonstrate that the density maps generated by CryoTRANS can preserve the correct structural details using the learned velocity field.
Furthermore, we explored the performance of CryoTRANS when simultaneously morphing multiple different target conformations based on the simulated data of TmrAB (Supplementary Note II). The results indicated that incorporating constraints from an intermediate conformation can further improve the quality of the CryoTRANS-generated density for the target map.
CryoTRANS predicts high-resolution maps for rare conformations
We validated the effectiveness of CryoTRANS by applying it to three real-world cryo-EM datasets: the alpha-2-macroglobulin (A2M)39 (Fig. 3a), a component of the actin-binding protein complexes (Arp2/3)40 (Fig. 3f) and the 70S ribosome41 (Supplementary Note III). In these cases, the original atomic models, constructed from low-resolution target maps, inherently contained uncertainties and inaccuracies. To address these uncertainties and validate the quality of our generated map, we refined the target model using our generated map through real space refinement in Phenix38. The root mean square deviation (RMSD) was kept below 3 Å, and the maximum deviation (calculated as the infinity norm of the averaged error vector between all atoms) was restricted to <0.5 Å (Fig. 3d, g). Subsequently, the model-to-map FSC of the generated map with the refined atomic model was computed (Fig. 3e, h). These experiments demonstrate that CryoTRANS produces a density map that accurately reflects the high-resolution structures in the target conformation.

a CryoTRANS analysis of A2M from the induced state (EMDB-32052, blue) to the native state (EMDB-32051, yellow). b Side chains of the generated map (green) and the target map (yellow) for A2M. c Density of an A2M subunit obtained by CryoTRANS, EMReady, and DeepEMhancer. d The target model (PDB 7VON) of one subunit of A2M before and after structural refinement is superimposed for depiction, labeled with various metrics of similarity. e The model-to-map FSC curves for the initial map (with the induced state atomic model), generated maps via various methods, and the target map (with the refined native state atomic model) of A2M. f Similar to a, but the data analysis involves two conformations of Arp2/3, with a home-collected initial map and EMDB-10960 as the target map. g Similar to d, but the atomic model (PDB 6YW7) corresponds to the target map of Arp2/3. h Similar to e, but for the analysis of Arp2/3. i Atomic models of Arp2/3 are fitted to the target map and the generated map, respectively, and compared with maps generated by EMReady and DeepEMhancer.
A2M, a protease inhibitor comprised of four identical subunits, exhibits both an induced state (EMDB-32052) and a native state (EMDB-32051), reported to have half-map resolutions of 3.8 and 5.3 Å, respectively39. The 5.3 Å resolution of native A2M poses challenges in atomic model building. Utilizing the induced state as the starting map (model-to-map FSC of 4.32 Å) and the native state as the target map (model-to-map FSC of 8.02 Å), CryoTRANS generated a high-quality pseudo-trajectory (Fig. 3a). The refined target model’s model-to-map FSC (PDB 7VON) with the generated map achieved a resolution of 4.84 Å. Comparative visualization of the side chains of the generated (in green) and target (in yellow) maps is shown in Fig. 3b, with the atomic model integrated into these density maps. This demonstrates that the generated map maintains the initial map’s high-resolution features and effectively approximates the native state.
We applied CryoTRANS to Arp2/3, a component of the actin-binding protein complexes. Due to its flexibility, the target conformation (EMDB-10960) exhibits a section of missing density. By applying the CryoTRANS-generated deformation to the initial map (Fig. 3d), the missing density in the target conformation is seamlessly supplemented, revealing many high-resolution details. The model-to-map FSC between the generated map and the refined atomic model (PDB 6YW7) maintains a resolution of 3.79 Å, comparable to the initial map’s resolution of 3.72 Å and significantly higher than the target map’s 9.82 Å resolution (Fig. 3e). These results validate our model’s ability to generate high-resolution approximations for rare conformations, even in cases where the target exhibits missing density sections.
To further validate the effectiveness of CryoTRANS, we compared it with post-processing methods based on deep learning, including DeepEMhancer26 and EMReady 27, which directly enhance the resolution of target conformations (Fig. 3c, i). For a fair comparison, we applied generated densities from DeepEMhancer and EMReady to refine the target model, respectively, and calculated the model-to-map FSC relative to the refined model (Fig. 3e, h). The results show that CryoTRANS significantly outperforms these methods in terms of the resolution of the generated maps. It is worth noting that CryoTRANS is a self-supervised method, which does not require pretraining on large datasets and does not suffer from model bias risks.
CryoTRANS predicts high-resolution details for cryo-ET map
CryoTRANS demonstrates the potential in bridging cryo-EM single-particle analysis (SPA) and cryogenic electron tomography (cryo-ET). By utilizing a high-resolution SPA map as the starting point, CryoTRANS can effectively predict high-resolution structures from low-resolution cryo-ET maps. In the case of the SARS-CoV-2 spike glycoprotein (Fig. 4a), the initial map presents three receptor binding domains (RBDs) in a closed state at 3.57 Å (model-to-map FSC) from cryo-EM SPA (EMDB-22001), and the target map depicts one RBD in an open state at 13.2 Å (model-to-map FSC) from subtomogram averaging of cryo-ET (EMDB-16697, PDB 6ZGG). CryoTRANS successfully generates a rotation of the RBD domain while maintaining a high-resolution structure (Fig. 4b). The model-to-map FSC resolution (with the refined model) of the generated density map by CryoTRANS reaches 3.78 Å (Fig. 4c), surpassing the resolutions achieved by deep learning post-processing strategies EMReady and DeepEMhancer, which are 9.62 and 9.34 Å, respectively. Furthermore, to validate the accuracy of the CryoTRANS-generated map, we compared it with the SPA map (EMDB-21999) using the corresponding model (PDB 6X2A) as the reference. The computed model-to-map FSC resolution of CryoTRANS-generated density and the SPA structure was both 3.78 Å (Fig. 4d). This validation underscores CryoTRANS’s ability to effectively leverage the structure obtained in SPA to enhance cryo-ET density resolution, offering valuable insights for cross-modal fusion of different imaging modalities.

a The initial map is the SARS-CoV-2 spike protein with three RBDs down obtained from cryo-EM SPA (EMDB-22001, model-to-map FSC resolution is 3.57 Å) and the target map is chosen as the SARS-CoV-2 spike protein one RBD up (EMDB-16697, model-to-map FSC resolution is 13.2 Å) obtained from cryo-ET subtomogram averaging. b The CryoTRANS pseudo-trajectory between the SPA map and cryo-ET map is depicted by selecting five intermediate density maps along the trajectory. c The model-to-map FSC curves for the initial map (with respect to the corresponding model PDB 6X2C), generated map (with respect to PDB 6ZGG after structure refinement), and target map of SARS-CoV-2 are compared, along with the generated results from EMReady and DeepEMhancer (with respect to PDB 6ZGG after structure refinement). d The panel shows the density maps from CryoTRANS and cryo-EM SPA (left), and their corresponding model-to-map FSC curves (right), comparing the maps with the model (PDB 6X2A). The SPA map is of the SARS-CoV-2 spike protein with one RBD up (EMDB-21999, model-to-map FSC resolution is 3.78 Å).
The comparison between pseudo-trajectories generated from CryoTRANS and other methods
We first compared CryoTRANS with MorphOT28, another morphing method for two cryo-EM density maps, using two selected cases for this comparison. The first is the simulated data derived from Mm-cpn34,35 in its open state (PDB 3IYF) and closed state (PDB 3J03). The second example involves two distant conformations of the SARS-CoV-2 spike glycoprotein (EMDB-22001, EMDB-21999)42. In the Mm-cpn simulated dataset, the inherent constraints of MorphOT’s regularization process in solving optimal transport result in indistinct interpolations at coarser resolutions. Such a limitation hinders the preservation of intricate structural nuances throughout interpolation. Conversely, our methodology adeptly avoids this issue, permitting an elevated degree of accuracy and consistent retention of structural details (Supplementary Fig. 1a). The commendable efficacy of our approach accentuates its utility as a potent instrument for morphing cryo-EM density maps. When considering the real-world scenario involving the SARS-CoV-2 spike glycoprotein, the performance disparity between MorphOT and CryoTRANS trajectories echoes the patterns identified in the Mm-cpn case. Besides, there is a noticeable distortion in the intermediary density maps from MorphOT (Supplementary Fig. 1b).
We next compared CryoTRANS with two representative continuous heterogeneity reconstruction methods, CryoDRGN22 and 3DFlex20. CryoDRGN predicts a density-map trajectory by traversing a linear path within the latent space. The training of CryoDRGN involves two-dimensional particle images, while CryoTRANS relies solely on the initial and target maps. We compared the trajectories generated from two methods using a pre-catalytic spliceosome dataset (EMPIAR-10180). Specifically, we first selected two density maps from the K-means clusters22 generated in the CryoDRGN latent space as the initial and target maps (Fig. 5a). Using these two density maps, we generated a linear path in the latent space as the trajectory from CryoDRGN and applied CryoTRANS to align these two maps for comparison (comparison with other trajectories can be found in Supplementary Note IV). The intermediate states of the two trajectories did not match. The trajectory generated by CryoDRGN was unrealistic, with densities emerging and disappearing sporadically, deviating from basic physical constraints. In contrast, CryoTRANS effectively captured the relative motion of the SF3b component (Fig. 5b). Furthermore, our analysis with the simulated TmrAB dataset indicated that CryoDRGN tends to struggle with the imbalance of particle distribution across conformations; notably, it fails to observe the rare conformations whereas CryoTRANS effectively overcomes this issue (Supplementary Note V).

a The UMAP visualization of latent encodings (middle) after training a 10-dimensional latent variable with CryoDRGN on the spliceosome dataset (EMPIAR-10180). Five positions are pinpointed along a linear path (the trajectory of CryoDRGN) within the latent space, each representing a density map encoded by the VAE's decoder within CryoDRGN. The initial and target maps are defined as the density maps at the starting and ending points of CryoDRGN's trajectory, respectively. b The trajectories of CryoDRGN and CryoTRANS are juxtaposed for comparison, considering both front and side perspectives. For a clearer visualization of the movements, the contour of the density map is also displayed. A local magnification of the side view is showcased through a highlighted frame.
3DFlex is another deep learning-based algorithm that predicts protein motion from the learned latent space. We conducted a comparative experiment with 3DFlex, also using simulated particles of TmrAB (Supplementary Note V). In this experiment, the high-resolution initial map and the low-resolution target map for training CryoTRANS were reconstructed from a simulated dataset with an imbalanced distribution of particles corresponding to each conformation. The trajectory produced by 3DFlex, trained on all simulated particles, was then compared with CryoTRANS (Supplementary Note V). The results revealed that the 3DFlex software’s default trajectory predominantly produced local perturbations of the initial map. We manually chose a trajectory that yielded a target map close to the occluded conformation. However, this density map is still inferior to the one obtained from CryoTRANS, as measured by the Q-score and residue distances.
To further validate the accuracy of the CryoTRANS pseudo-trajectory, we compared it with the trajectory generated by molecular dynamics (MD) for SARS-CoV-243 (Supplementary Note VI). The MD trajectory investigates the conformational changes of SARS-CoV-2 in the receptor-binding domain (RBD) at spaced time intervals from 0 to 180 nanoseconds (ns). The first frame of the MD trajectory (t = 0 ns) was used as the initial map, and the last frame (t = 180 ns) was used as the target map. We then generated the trajectory between these two maps using CryoTRANS. The results showed that while there are some discrepancies in the temporal distribution between the two methods, the overall trend of deformation remains consistent. After adjusting the true timeline (0 to 180 ns) in MD to the pseudo-timeline ([0, 1]) of CryoTRANS, the CryoTRANS pseudo-trajectory closely matches the MD trajectory. The error (3–5 Å) may largely be due to the Brownian motions in MD simulations, which introduce thermal variations that CryoTRANS cannot accurately capture. Previous studies have shown that the Brownian motion of proteins in water is on the order of Angstroms44. It is important to note that Brownian motion is inherent to the simulation and is accounted for in the dynamics of the system, which can vary depending on the specific conditions of the simulation. Furthermore, it is worth mentioning that these findings are specific to the studied case, and further investigation is needed to make broader generalizations.
Discussion
In this study, we introduced a novel method named CryoTRANS to predict high resolutions for rare conformations in cryo-EM studies by generating a pseudo-trajectory across various conformations. CryoTRANS leverages an ODE, parameterized by a deep neural network, to model a continuous, non-rigid deformation from common high-resolution conformations to rare ones. The method also features the novel incorporation of a Wasserstein distance and the application of a rigidity constraint on the velocity field, together forming a variational model. Mathematically, the Wasserstein distance provides a metric in the probability space, whereas the conventional Euclidean distance does not. Theoretical advantages of the Wasserstein distance over the Euclidean distance have been studied previously29,45, demonstrating success in machine learning46 and seismic data processing47. The rigidity constraint helps in preserving the detailed structure of the high-resolution density map. This design obviates the need for supervised samples or pre-training during the neural network training process. After training, the deformed density map, governed by the ODE, offers a high-resolution counterpart of the rare conformations. The potential and effectiveness of CryoTRANS have been demonstrated through its application in various case studies, where it succeeded in predicting high-resolution features of rare conformations and cryo-ET maps.
The exploration of rare conformations in structural biology holds significant value, as these rare states often yield crucial insights into the function and behavior of biological macromolecules. However, the infrequent occurrence of these rare conformations presents a challenge in their analysis. This is where deformation models prove to be instrumental. The utilization of deformation models to generate intricate, high-resolution trajectories of macromolecules, especially from multi-modal data, holds promise in other applications as well. For instance, particle data obtained from liquid-phase electron microscopy48 contain valuable temporal information. However, reconstructing high-resolution movies from liquid-phase EM is still an unsolved challenge, primarily due to constraints in the number of available particles and the high level of noise. Our future endeavors will focus on exploring CryoTRANS as an adaptable solution to address and overcome this ongoing issue. In conclusion, CryoTRANS serves as a tool for exploring rare conformations in structural biology by utilizing neural network deformation models. This approach may open up promising avenues for gaining deeper insights into the function and behavior of biological macromolecules.
CryoTRANS is a predictive approach, although it is self-supervised and does not require labeled data or pretraining. While CryoTRANS has shown successful results in various cases, the generated maps and pseudo-trajectories may be influenced by the choice of loss function and need further validation from both experimental and computational perspectives. In the future, more interactions between image registration-based methods and molecular dynamics simulations could provide cross-validation in computations. Additionally, using the pseudo-trajectory generated by CryoTRANS to identify particles belonging to rare conformations—typically overlooked by conventional classification methods due to imbalance clustering issues—deserves future work.
Methods
Generation of of simulated datasets
Simulated density maps with varying resolutions were generated, and morphing from the initial map to the simulated density maps was performed using CryoTRANS. Four biological macromolecules were selected for generating simulated datasets, each containing two conformations resolved by experimental approaches. These macromolecules include the heterodimeric ABC exporter TmrAB (PDB 6RAH and PDB 6RAI), the peptidase-containing ABC transporter 1 (PDB 7T55 and PDB 7T57), thermostabilized human prestin (PDB 7V73 and PDB 7V75), and Mm-cpn (PDB 3IYF and PDB 3J03). The corresponding atomic model of the simulated dataset was converted to the density map using the molmap function of Chimera. The density map of one conformation was low-passed to 3 Å, serving as the initial high-quality map for morphing using CryoTRANS. The density map of the other conformation was treated as the target density map and was low-passed to different resolutions of 3, 5, and 7 Å, respectively.
Mathematical principles of CryoTRANS
Basic formulation of CryoTRANS self-supervised training
CryoTRANS trains a flow map, \(\vec{\Phi }\left(\vec{x},t\right)\in {{\mathbb{R}}}^{3},\ t\in [0,1]\), in a self-supervised manner between the high-resolution density map f0 and the low-resolution density map f1. The high-resolution prediction of f1 is given by \({f}_{0}({\vec{\Phi }}^{-1}(\cdot ,1))\). This flow map, \(\vec{\Phi }(\vec{x},t)\), is determined by an ODE model:
The velocity field \(\vec{V}\) is formulated as a multi-layer perceptron (MLP) with two hidden layers:
where σ is the activation function, and \({W}_{1}\in {{\mathbb{R}}}^{n\times 3}\), \({b}_{1}\in {{\mathbb{R}}}^{n}\), \({W}_{2}\in {{\mathbb{R}}}^{n\times n}\), \({b}_{2}\in {{\mathbb{R}}}^{n}\), \({W}_{3}\in {{\mathbb{R}}}^{3\times n}\), and \({b}_{3}\in {{\mathbb{R}}}^{3}\) are learnable parameters. Here, θ denotes all the learnable parameters. In this study, the width of the hidden layer, n, is set to 100 by default, and σ is chosen as the leaky ReLU function with a slope of 0.01.
To learn the velocity field, the loss is defined as the squared Wasserstein distance between \({f}_{0}({\vec{\Phi }}^{-1}(\cdot ,1))\) and f1. The high-resolution density map is obtained by solving the following optimization model:
Here, \({{{{\mathcal{W}}}}}_{2}\) is the squared Wasserstein distance49. After solving this optimization problem, the velocity field \(\vec{V}\) and the corresponding flow \(\vec{\Phi }\left(\vec{x},t\right)\) can be obtained by solving Eq. (1).
Discretization of the neural ODE
The optimization of the neural ODE, as described in Eq. (1), requires discretization before it can be computed. Without loss of generality, the computational domain is chosen as [0, 1]3 and is discretized using uniform grids:
The ODE (1) is discretized using the forward Euler method:
where \({t}_{m}=\frac{m}{M},\ m\in \left\{0,1,\cdots \,,M-1\right\}\) and \(\tau =\frac{1}{M}\). A typical choice for M is 10. Then \({\hat{f}}_{1}\) can be obtained as follows:
In the computation, the target density map f1 is evaluated on the uniform grids \({\vec{x}}_{ijk}\). To compute \({{{{\mathcal{W}}}}}_{2}({\hat{f}}_{1},{f}_{1})\), \({\hat{f}}_{1}\) also needs to be evaluated over \({\vec{x}}_{ijk}\). This is done using bilinear interpolation (see Supplementary Note VII).
Implementation of CryoTRANS training
The Wasserstein distance \({{{{\mathcal{W}}}}}_{2}({\hat{f}}_{1},{f}_{1})\) is computed using the Sinkhorn algorithm50, which is an efficient, GPU-friendly algorithm that employs entropic regularization and allows for efficient gradient backpropagation (detailed in Supplementary Note VIII). To alleviate computational overhead, the first 5 iterations of the Sinkhorn algorithm are performed in each round, and the penalty coefficient of the entropic regularization is set to 0.0001.
The gradient is computed based on automatic gradient propagation in PyTorch (see Supplementary Note VII). The weights of the neural network are initialized following a normal distribution with a mean of zero and a variance of 0.01. The model is trained using the ADAM optimizer with a learning rate of 0.001. A multi-scale algorithm is also introduced during the training process, achieving acceleration by a factor of 5 to 10 across different datasets (Supplementary Note IX).
Pseudo-trajectory inference after CryoTRANS training
Once a neural velocity field is trained with optimal parameters θ*, (5) is further iterated with θ* to obtain an optimal moving trajectory for each voxel \({\vec{x}}_{ijk}\). All the trajectories obtained in this process are gathered and bilinear interpolation is utilized to generate the final trajectory of the density map:
The density map \({\hat{f}}_{1}\) corresponds to the generated density map at the end of the trajectory.
Density maps partition strategy with rigidity constraints during CryoTRANS training
The procedure described above is utilized for training simulated data. However, when training CryoTRANS with real data (A2M, Arp2/3, SARS-CoV-2), where different domains follow different velocity fields, a modified approach is employed. This approach utilizes two velocity fields with rigidity constraints based on a manual partition of the density map, enhancing the model’s representation capability.
Specifically, the whole volume is manually divided into two disjoint parts,
and f0 is consequently divided into \({f}_{0}^{a}\) and \({f}_{0}^{b}\), where
Subsequently, two separate neural velocity fields, characterized by different parameters θ a and θ b, are employed to generate trajectories on these individual partitions. The trajectory for each partition can be described as follows:
The generated maps for each partition, \({\hat{f}}_{1}^{a}\) and \({\hat{f}}_{1}^{b}\), are obtained as follows:
For employing rigidity constraints, the loss function is modified to consist of two components: the squared Wasserstein distance between the combined deformed map \({\hat{f}}_{1}^{a}+{\hat{f}}_{1}^{b}\) and the target density map f1, and the local rigidity constraint Lrigid( ⋅ ) on the two velocity fields20 (Supplementary Note X). Consequently, the total loss of the model is
Here, λa and λb are hyperparameters of the model that control the rigidity degree of the velocity field. The selection of λa and λb, as well as the partitioning process, are described in detail in Supplementary Note XI. The model is trained using ADAM optimization with the same parameters as before. After obtaining the optimal parameters θa* and θb*, the final optimal trajectory can be generated as follows:
where
Metrics for density map quality
The model-to-map FSC is used to measure the similarity between the CryoTRANS-generated density and the atomic model. The unmasked FSC curves are depicted in this study. This is because CryoTRANS requires density maps to be represented as probability distributions, necessitating that each voxel’s value be positive. During the preprocessing stage, any negative values in the density map are reset to zero. This process effectively acts as a mask applied to the density map itself.
The Q-score of the CryoTRANS-generated density map is compared with the Q-score of the target map. To further evaluate the accuracy of CryoTRANS, map-to-model in Phenix is used to build new atomic models from both the generated map and the target map, respectively. Subsequently, the TM-score between the reconstructed model from Phenix and the corresponding reference model from the PDB bank is calculated, serving as an additional measure of structure accuracy. Chain-comparison in Phenix is also used to compute the backbone coverage, providing further insights into the accuracy of the generated structures.
Statistics and reproducibility
The model is described in detail through mathematical formulation, and both the Python code and raw data are uploaded. The experiments mentioned in the article can be reproduced using the provided code and data. The neural network of CryoTRANS employs random initialization, so the results of experiments will exhibit some randomness, which may cause fluctuations in the model-to-map FSC curves of the generated densities. The displayed results are based on the best outcomes from three repeated experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All source data underlying the graphs and charts in the main figures are available in Supplementary Data 1. The datasets analyzed in this study were downloaded from the EMPIAR repository. Atomic models are obtainable using the Protein Data Bank (PDB) ID mentioned in the text. Experimental results, including trajectories, of the datasets incorporated in this study have been deposited at https://github.com/mxhulab/cryotrans-demos.
Code availability
CryoTRANS is open source and released on GitHub with its homepage available at https://github.com/mxhulab/cryotrans.
References
Cao, E., Liao, M., Cheng, Y. & Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 (2013).
Kühlbrandt, W. The resolution revolution. Science 343, 1443–1444 (2014).
Bai, X.-c, McMullan, G. & Scheres, S. H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 40, 49–57 (2015).
Cheng, Y. Single-particle cryo-EM—how did it get here and where will it go. Science 361, 876–880 (2018).
Zhu, J. et al. A minority of final stacks yields superior amplitude in single-particle cryo-em. Nat. Commun. 14, 7822 (2023).
Kasas, S., Dumas, G., Dietler, G., Catsicas, S. & Adrian, M. Vitrification of cryoelectron microscopy specimens revealed by high-speed photographic imaging. J. Microsc. 211, 48–53 (2003).
Mäeots, M.-E. & Enchev, R. I. Structural dynamics: review of time-resolved cryo-EM. Acta Crystallogr. Sect. D: Struct. Biol. 78, 927–935 (2022).
White, H., Walker, M. & Trinick, J. A computer-controlled spraying-freezing apparatus for millisecond time-resolution electron cryomicroscopy. J. Struct. Biol. 121, 306–313 (1998).
Frauenfelder, H. The Physics of Proteins: an Introduction to Biological Physics and Molecular Biophysics (Springer Science & Business Media, USA, 2010).
Frank, J. Time-resolved cryo-electron microscopy: recent progress. J. Struct. Biol. 200, 303–306 (2017).
Henzler-Wildman, K. & Kern, D. Dynamic personalities of proteins. Nature 450, 964–972 (2007).
Ourmazd, A. Cryo-EM, XFELs and the structure conundrum in structural biology. Nat. Methods 16, 941–944 (2019).
Toader, B., Sigworth, F. J. & Lederman, R. R. Methods for Cryo-EM single particle reconstruction of macromolecules having continuous heterogeneity. J. Mol. Biol. 435, 168020 (2023).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Dandey, V. P. et al. Time-resolved cryo-EM using Spotiton. Nat. Methods 17, 897–900 (2020).
Mäeots, M.-E. et al. Modular microfluidics enables kinetic insight from time-resolved cryo-EM. Nat. Commun. 11, 3465 (2020).
Lu, Z. et al. Monolithic microfluidic mixing-spraying devices for time-resolved cryo-electron microscopy. J. Struct. Biol. 168, 388–395 (2009).
Ross, F. M. Liquid Cell Electron Microscopy (Cambridge University Press, Cambridge, UK, 2017).
Punjani, A. & Fleet, D. J. 3DFlex: determining structure and motion of flexible proteins from cryo-EM. Nat. Methods 1–11 https://www.nature.com/articles/s41592-023-01853-8 (2023).
Herreros, D. et al. Estimating conformational landscapes from Cryo-EM particles by 3D Zernike polynomials. Nat. Commun. 14, 154 (2023).
Zhong, E. D., Bepler, T., Berger, B. & Davis, J. H. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 18, 176–185 (2021).
Levy, A., Wetzstein, G., Martel, J. N. P., Poitevin, F. & Zhong, E. Amortized inference for heterogeneous reconstruction in Cryo-EM. Adv. Neural Inf. Process. Syst. 35, 13038–13049 (2022).
Wu, Z., Chen, E., Zhang, S., Ma, Y. & Mao, Y. Visualizing conformational space of functional biomolecular complexes by deep manifold learning. Int. J. Mol. Sci. 23, 8872 (2022).
Schwab, J., Kimanius, D., Burt, A., Dendooven, T. & Scheres, S. H. DynaMight: estimating molecular motions with improved reconstruction from cryo-EM images. Nat. Methods, 1–8 https://doi.org/10.1038/s41592-024-02377-5 (2024).
Sanchez-Garcia, R. et al. Deepemhancer: a deep learning solution for cryo-em volume post-processing. Commun. Biol. 4, 874 (2021).
He, J., Li, T. & Huang, S.-Y. Improvement of cryo-em maps by simultaneous local and non-local deep learning. Nat. Commun. 14, 3217 (2023).
Ecoffet, A., Poitevin, F. & Dao Duc, K. MorphOT: transport-based interpolation between EM maps with UCSF ChimeraX. Bioinformatics 36, 5528–5529 (2020).
Singer, A. & Yang, R. Alignment of density maps in wasserstein distance. Biological Imaging 4, 5 (2024)
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. arXiv preprint arXiv:1412.6980 (2014).
Hofmann, S. et al. Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571, 580–583 (2019).
Kieuvongngam, V. & Chen, J. Structures of the peptidase-containing ABC transporter PCAT1 under equilibrium and nonequilibrium conditions. Proc. Natl Acad. Sci. USA 119, e2120534119 (2022).
Futamata, H. et al. Cryo-EM structures of thermostabilized prestin provide mechanistic insights underlying outer hair cell electromotility. Nat. Commun. 13, 6208 (2022).
Zhang, J. et al. Cryo-EM structure of a Group II chaperonin in the prehydrolysis ATP-bound state leading to lid closure. Structure 19, 633–639 (2011).
Zhang, J. et al. Mechanism of folding chamber closure in a group II chaperonin. Nature 463, 379–383 (2010).
Pintilie, G. et al. Measurement of atom resolvability in cryo-em maps with q-scores. Nat. Methods 17, 328–334 (2020).
Zhang, Y. & Skolnick, J. Tm-align: a protein structure alignment algorithm based on the tm-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Adams, P. D. et al. Phenix: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 66, 213–221 (2010).
Huang, X. et al. Cryo-EM structures reveal the dynamic transformation of human alpha-2-macroglobulin working as a protease inhibitor. Sci. China Life Sci. 65, 2491–2504 (2022).
von Loeffelholz, O. et al. Cryo-em of human arp2/3 complexes provides structural insights into actin nucleation modulation by arpc5 isoforms. Biol. Open 9, bio054304 (2020).
Xue, L. et al. Visualizing translation dynamics at atomic detail inside a bacterial cell. Nature 610, 205–211 (2022).
Henderson, R. et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 27, 925–933 (2020).
Fallon, L. et al. Free energy landscapes from sars-cov-2 spike glycoprotein simulations suggest that rbd opening can be modulated via interactions in an allosteric pocket. J. Am. Chem. Soc. 143, 11349–11360 (2021).
Erban, R. From molecular dynamics to brownian dynamics. Proc. R. Soc. A 470, 20140036 (2014).
Yang, Y., Engquist, B., Sun, J. & Hamfeldt, B. F. Application of optimal transport and the quadratic Wasserstein metric to full-waveform inversion. Geophysics 83, R43–R62 (2018).
Arjovsky, M., Chintala, S. & Bottou, L. Wasserstein generative adversarial networks. In International Conference on Machine Learning (eds Precup, D. & Teh, Y. W.) 214–223 (PMLR, 2017).
Engquist, B. & Yang, Y. Optimal transport based seismic inversion: beyond cycle skipping. Commun. Pure Appl. Math. 75, 2201–2244 (2022).
Wu, H., Friedrich, H., Patterson, J. P., Sommerdijk, N. A. & De Jonge, N. Liquid-phase electron microscopy for soft matter science and biology. Adv. Mater. 32, 2001582 (2020).
Peyré, G. & Cuturi, M. et al. Computational optimal transport: With applications to data science. Found. Trends® Mach. Learn. 11, 355–607 (2019).
Cuturi, M. Sinkhorn distances: lightspeed computation of optimal transport. In Advances in Neural Information Processing Systems, Vol. 26 (eds Burges, C. J. Bottou, L., Welling, M., Ghahramani, Z. & Weinberger, K. Q.) (Curran Associates, Inc., 2013).
Acknowledgements
This work was supported by the National Key R&D Program of China (No.2021YFA1001300) (to C.B.), the National Natural Science Foundation of China (No.12271291) (to C.B.), Beijing Frontier Research Center of Biological Structure (to M.H.), Shenzhen Medical Academy of Research and Translation (to M.H.), and the National Natural Science Foundation of China (No.12071244) (to Z.S.). Lili Ju’s research was partially supported by U.S. Department of Energy under grant number DE-SC0022254. We are grateful to Dr. Xing Zhang and Prof. Hongwei Wang for providing the Arp2/3 dataset and for their fruitful discussions.
Author information
Authors and Affiliations
Contributions
C.B., M.H., and Z.S. initiated the project. C.B., M.H., Z.S., X.F., Q.Z., and L.J. clarified the mathematical principles. X.F., Q.Z., M.H., and C.B. developed CryoTRANS and carried out testing. H.Z. provided support in analyzing the A2M dataset. X.F., Q.Z., M.H., and C.B. analyzed the data. M.H., C.B., Z.S., X.F., and J.Z. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Ruben Sanchez-Garcia, Genki Terashi and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editors: Manidipa Banerjee and Manuel Breuer. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, X., Zhang, Q., Zhang, H. et al. CryoTRANS: predicting high-resolution maps of rare conformations from self-supervised trajectories in cryo-EM. Commun Biol 7, 1058 (2024). https://doi.org/10.1038/s42003-024-06739-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06739-9
This article is cited by
-
Artificial intelligence in cryo-EM: emerging deep neural network methods from sample preparation, particle picking, map reconstruction, modelling to enhanced resolution
BMC Artificial Intelligence (2026)
-
Fine-tune AlphaFold with limited cryo-EM observations
Communications Chemistry (2026)
