
Abstract
Of the hitherto over 60 taxonomically identified species in the genus of Pseudo-nitzschia, 26 have been confirmed to be toxigenic. Nevertheless, the acquisition and evolution of the toxin biosynthesis (dab) genes by this extensive group of Pseudo-nitzschia species remains unclear. Through constructing chromosome-level genomes of three Pseudo-nitzschia species and draft genomes of ten additional Pseudo-nitzschia species, putative genomic integration sites for the dab genes in Pseudo-nitzschia species were explored. A putative breakpoint was observed in syntenic regions in the dab gene cluster-lacking Pseudo-nitzschia species, suggesting potential independent losses of dab genes. The breakpoints between this pair of conserved genes were also identified in some dab genes-possessing Pseudo-nitzschia species, suggesting that the dab gene clusters transposed to other loci after the initial integration. A “single acquisition, multiple independent losses (SAMIL)” model is proposed to explain the acquisition and evolution of the dab gene cluster in Pseudo-nitzschia species.
Similar content being viewed by others


Introduction
Species of the diatom genus Pseudo-nitzschia have attracted worldwide attention since the first reported Pseudo-nitzschia poisoning incident in 1987 at Prince Edward Island, Canada, which resulted in at least three deaths and 107 cases of illness, and the identification of the neurotoxic glutamate receptor agonist domoic acid (DA) as the toxin synthesized by P. multiseries1,2. DA synthesized by Pseudo-nitzschia species has been found to be transferred in the marine food web during bloom events, which may cause illness or death for marine organisms and humans, leading to what is called amnesic shellfish poisoning (ASP)2. An unprecedented bloom of another toxigenic P. australis in the spring of 2015 resulted in the largest recorded outbreak of DA along the North American west coast, ranging from the Aleutian Islands of Alaska, USA, to the Baja peninsula, Mexico, and resulted in prolonged closures of razor clam, rock crab, and Dungeness crab fisheries3. Similar neurotoxic symptoms have also been observed in animals, including birds and marine mammals4. In addition, numerous harmful algal bloom (HAB) events caused by Pseudo-nitzschia species have continued to erupt globally, exerting severe impacts on fisheries and the aquaculture industry5,6,7.
Pseudo-nitzschia blooms have motivated intensive research on the identification and geographical distribution of new Pseudo-nitzschia species, and on the elucidation of mechanisms of DA biosynthesis in Pseudo-nitzschia species, which resulted in the identification of key genes of the DA biosynthetic pathway in P. multiseries through comparative gene expression analysis8. This study successfully identified a gene cluster, dab, with four protein-coding genes: dabA (terpene cyclase), dabB (hypothetical protein), dabC (ɑ-dependent dioxygenase), and dabD (CYP450), which encode key enzymes catalyzing reactions extending from the substrate glutamate and geranyl pyrophosphate (GPP) to isodomoic acid A in the genome of P. multiseries8. Following this success, dab genes were readily identified in P. multistriata through genomic analysis and the search for homologous genes8, and in P. australis and P. seriata through transcriptomic analyses8,9,10. The characterization of the dab gene clusters has also facilitated research on the impact of ocean warming and acidification on DA biosynthesis11.
Pseudo-nitzschia is a genus with over 60 cosmopolitan species, up to 50% of which are capable of synthesizing DA8,12,13. This may explain the wide distribution of DA from the Pacific subarctic (58°N) to the Southern Ocean (66°S)14. Notably, Pseudo-nitzschia species that are capable of producing DA are not necessarily phylogenetically closer. Furthermore, of the 26 DA-producing Pseudo-nitzschia species, 24 were found to produce DA in some studies, but not in others2, suggesting that DA biosynthesis is not only species-specific but also strain-specific. Thus, whether the dab gene cluster was acquired independently in different Pseudo-nitzschia species, or it was acquired in the common ancestor of all Pseudo-nitzschia species remains undetermined. Ascertaining the acquisition and evolution of the dab gene cluster in Pseudo-nitzschia species will help explain why certain Pseudo-nitzschia species are toxic, while others are not, and why some species are reported to exhibit both toxic and non-toxic traits2,12,13.
In this project, we constructed the genomes of 13 Pseudo-nitzschia species, including high quality chromosome-level genomes of three species (P. delicatissima, P. multiseries, and P. pungens) and draft-quality genomes of ten species (P. americana, P. brasiliana, P. cuspidata, P. galaxiae, P. hainanensis, P. micropora, P. multistriata, P. sabit, Pseudo-nitzschia sp. (CNS00097), and Pseudo-nitzschia sp. (CNS01031)), and identified the dab gene clusters. Of the three Pseudo-nitzschia species selected for constructing chromosome-level genomes, P. multiseries has been proven to possess the dab gene cluster and is toxic8, while P. pungens was shown to be both toxic and non-toxic, depending on the strains2, P. delicatissima, on the other hand, was found to be toxic, weakly toxic, or non-toxic15. Comparative genomic analysis of syntenic regions harboring dab gene clusters, coupled with phylogenetic analysis of dab genes and structural modeling of the proteins encoded by these dab genes, enabled us to identify the putative integration sites of the dab gene cluster in the common ancestor of Pseudo-nitzschia species. Our findings suggested a “single acquisition, multiple independent losses (SAMIL)” model for the evolution of the dab gene cluster in Pseudo-nitzschia species.
Results
Construction and comparative analysis of chromosome-level genomes of three Pseudo-nitzschia species
To obtain high-quality reference genomes for the three Pseudo-nitzschia species, we generated 15.06 Gb (~442×), 20.40 Gb (~261×), and 31.87 Gb (~119×) of PacBio data (Supplementary Table 1). These data were assembled to produce the initial versions of the whole genome assemblies, which were 34.06 Mb, 67.09 Mb, and 252.10 Mb in sizes for P. delicatissima, P. pungens, and P. multiseries, respectively. These assembly results were generally consistent with the estimated sizes obtained from genome survey analysis (Supplementary Fig. 1; Supplementary Table 2).
We further carried out Hi-C (High-through chromosome conformation capture) analysis (Supplementary Table 3) to construct chromosome-level assemblies, which were 34.06 Mb, 67.11 Mb, and 252.35 Mb for P. delicatissima, P. pungens, and P. multiseries, respectively (Fig. 1; Table 1). The genome assemblies of P. delicatissima, P. pungens and P. multiseries contained 11, 12, and 11 chromosomes (Supplementary Table 4) and were highly contiguous, with 96.48%, 99.14%, and 94.90% of genome contigs anchored to chromosomes, respectively. The assemblies also had high contig N50 values of 2.90 Mb, 1.66 Mb, and 0.80 Mb for P. delicatissima, P. pungens, and P. multiseries, respectively (Table 1). BUSCO assessment indicated that the genome assemblies were 73.93%, 80.53%, and 81.52% complete for P. delicatissima, P. pungens, and P. multiseries, respectively (Supplementary Table 5).

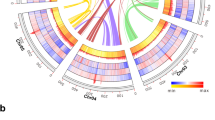
A Genome of P. delicatissima (CNS00130) consists of 11 chromosomes (outmost circle). B Genome of P. pungens (CNS00055) consists of 12 chromosomes. C Genome of P. multiseries (CNS00140) consists of 11 chromosomes. Hi-C intra-chromosomal contact maps of the genome assemblies in P. delicatissima (D), P. pungens (E), and P. multiseries (F). G Comparison between P. delicatissima and P. pungens. H Comparison between P. delicatissima and P.multiseries. I Comparison between P. pungens and P. multiseries. J Syntenic relationships among P. delicatissima, P. multiseries, and P. pungens. The sites linked by the red line indicate the putative original dab gene cluster integration site in each genome.
We explored why the genome sizes of three Pseudo-nitzschia species were so different, ranging from 34.06 Mb (P. delicatissima) to 252.10 Mb (P. multiseries). Annotation of the assembled genomes revealed that P. multiseries harbors a greater proportion of repetitive elements (68.67%) compared to P. delicatissima (1.73%) and P. pungens (34.91%). Among all types of repetitive elements, long terminal repeats (LTRs) showed the highest proportions, accounting for 0.84%, 22.94%, and 58.45% of the genomes of P. delicatissima, P. pungens, and P. multiseries, respectively (Table 2). This suggested that differential levels of LTRs were primarily responsible for the size differences among these three Pseudo-nitzschia species. Despite the differences in genome sizes, the numbers of protein-coding genes (PCGs) in these three species were similar, being 14,375, 15,472, and 18,649 in P. delicatissima, P. pungens, and P. multiseries, respectively (Table 1; Supplementary Table 6). Notably, the number of PCGs predicted in P. multiseries in this study (18,649) was similar to that annotated in a different P. multiseries strain (19,703). Most PCGs of the P. delicatissima, P. pungens, and P. multiseries genomes (93.83%, 91.44%, and 88.49%, respectively) were functionally annotated (Supplementary Table 7).
Although these three species belong to the same genus, their genomes showed extensive differences with varying numbers of chromosomes, indicating many intra-chromosomal fusion or divisions events occurred during speciation. While 11 chromosomes were constructed for P. delicatissima and P. multiseries, 12 chromosomes were constructed for P. pungens (Fig. 1). Pairwise comparative analyses revealed only small-scale syntenic blocks between the genomes of P. delicatissima and P. pungens (Fig. 1G) and between the genomes of P. delicatissima and P. multiseries (Fig. 1H), suggesting complex evolutionary relationships with numerous chromosomal exchanges. Nevertheless, relatively large syntenic block sizes were found between genomes of P. pungens and P. multiseries (Fig. 1I), consistent with their close positions in the phylogenetic tree (Supplementary Fig. 2). In particular, chromosome Pmu6 of P. multiseries and chromosome Ppu8 of P. pungens showed nearly perfect collinearity (Fig. 1I-J). A total of 10,755 collinear gene pairs were found between P. pungens and P. multiseries, 9076 collinear gene pairs between P. delicatissima and P. pungens, and 9272 collinear gene pairs between P. delicatissima and P. multiseries (Fig. 1G–I).
To explore gene family dynamic changes in evolution, PCGs of P. delicatissima, P. pungens, and P. multiseries were compared with those of nine other phytoplankton species, including Aureococcus anophagefferens, Thalassiosira oceanica, Thalassiosira pseudonana, Skeletonema marinoi, Seminavis robusta, Phaeodactylum tricornutum, Fragilariopsis cylindrus, P. multistriata, and P. multiseries (Supplementary Fig. 3A). Phylogenetic analysis using 381 single-copy orthologous genes from these 12 species showed that Pseudo-nitzschia species were tightly clustered with F. cylindrus, as expected. The divergence time of Pseudo-nitzschia species from their nearest node was estimated to be approximately 53.2 million years ago (MYA) (Supplementary Fig. 3A). Comparative analysis of the genes of P. delicatissima, P. pungens, P. multiseries, and P. multistriata revealed 7136 shared gene families (Supplementary Fig. 3B).
Comparative analysis of Pfam domains contained in the PCGs of the 12 species revealed that many gene families encoded by the Pseudo-nitzschia genomes were different from those in other species (Supplementary Fig. 3C). Notably, the family of genes encoding gametolysin peptidase M11 was substantially expanded in Pseudo-nitzschia genomes, with 17, 50, and 33 members in P. delicatissima, P. pungens, and P. multiseries, respectively (Supplementary Fig. 3C, Supplementary Fig. 3D). These gametolysin peptidase M11 domain-containing proteins, which have their origin in the ancestor of green plants and chromists, have been demonstrated to degrade cell wall in Chlamydomonas16.
Annotation of dab gene clusters and phylogenetic analysis of dab genes
From the three chromosome-level Pseudo-nitzschia genomes constructed in this study, the dab gene cluster was identified only in P. multiseries (Fig. 2A; Supplementary Table 8), which is the first Pseudo-nitzschia species known to possess the dab gene cluster8. The dab gene cluster in P. multiseries consisted of four genes: dabA, dabB, dabC, and dabD, as reported previously8. However, the dab gene cluster was not identified in either P. delicatissima or P. pungens (Fig. 2A; Supplementary Table 8), despite previous studies showing the presence of both toxic and non-toxic strains in these two Pseudo-nitzschia species2,8,15. Searches for individual genes with similarity to those in the dab gene cluster did not yield meaningful hits in the assemblies of P. delicatissima and P. pungens. The dab gene cluster was identified in the draft genomes of P. multistriata strains in this study; this species has previously been shown to possess the dab gene cluster8. The cluster was also detected in the assembly of P. cuspidata (Fig. 2A; Supplementary Table 8), a species previously reported as DA-producing2,17. No dab gene clusters were found in the draft genomes of other Pseudo-nitzschia species investigated in this study, despite some being reported as DA producers18. Interestingly, the Pseudo-nitzschia species found to possess the dab gene cluster in this study (P. multiseries, P. multistriata, and P. cuspidata), as well as two other species (P. australis and P. seriata) identified through transcriptomic analysis, were distributed throughout the phylogenetic tree of Pseudo-nitzschia species (Supplementary Fig. 2), suggesting that the acquisition and evolution of the dab gene cluster are intricate.

A Phylogenetic analysis of Pseudo-nitzschia species based on cpDNAs illustrating the positioning of the Pseudo-nitzschia species (shown in red) that possess the dab gene clusters (which are displayed to the right of the species names). B Phylogenetic tree displaying Pseudo-nitzschia DabA proteins and KabA proteins. C Phylogenetic tree displaying Pseudo-nitzschia DabB proteins and hypothetical proteins that showed the highest similarities. D Phylogenetic tree displaying Pseudo-nitzschia DabC proteins and proteins that showed the highest similarities. E Phylogenetic tree displaying Pseudo-nitzschia DabD proteins and Cytochrome CYP450 proteins that showed the highest similarities. F AlphaFold2-predicted 3D structures of DabA, DabB, DabC and DabD proteins identified in Pseudo-nitzschia species and related species. G. Comparative synteny analysis between the putative “original” integration site of the dab gene cluster in Pseudo-nitzschia species. Only the P. cuspidata genome contains a dab gene cluster between the gene pairs. H. Dot-plot illustration of the breakpoints of the “original” integration site of the dab gene cluster.
Chemical analysis performed on 13 strains of nine Pseudo-nitzschia species (P. multiseries, P. multistriata, P. delicatissima, P. americana, P. micropora, P. pungens, P. galaxiae, P. brasiliana, and Pseudo-nitzschia sp. (CNS00097)) revealed that DA was detected only in the strain of P. multiseries. No DA was detected in the P. multistriata strains, even though five P. multistriata strains were analyzed (CNS00142, CNS00781, CNS00965, CNS01237, CNS01424). DA analysis was not performed for strains of P. cuspidata, P. hainanensis, P. sabit, and Pseudo-nitzschia sp. CNS01031 due to their unavailability, as these strains unfortunately died off during the study period (Supplementary Table 8).
Acquisition and evolution of dab gene cluster
To determine how dab gene clusters were acquired by Pseudo-nitzschia species, we hypothesized that the dab gene cluster was acquired by a common ancestor of Pseudo-nitzschia species and was subsequently lost independently in different species. Based on the phylogenetic positions of Pseudo-nitzschia species that possess the dab gene cluster (Supplementary Fig. 2), we predicted that the phylogenetic relationships of the dab genes would align with the phylogenetic relationships of these species. To test this hypothesis, we conducted phylogenetic analyses of chloroplast genomes (cpDNAs) to infer the phylogenetic relationships (Fig. 2A), as rDNA molecular markers were relatively short and some species exhibited cryptic lineages with ambiguous relationships19. Separate phylogenetic analysis of the dab genes was also performed. As expected, the phylogenetic inferences for DabA (Fig. 2B), DabB (Fig. 2C), DabC (Fig. 2D), and DabD (Fig. 2E) were highly coherent with the phylogenetic relationships of the Pseudo-nitzschia species harboring the dab gene cluster (Fig. 2A; Supplementary Fig. 3). These results suggested that the dab gene cluster in these three Pseudo-nitzschia species most likely originated from a common ancestor that gained the dab genes via a horizontal gene transfer (HGT) event. In fact, the proteins encoded by dabB exhibited high similarity to proteins found in prokaryotic organisms (Fig. 2C), supporting the hypothesis of HGT. Furthermore, modeling of the 3D structures of the proteins encoded by dabB and dabC revealed high structural similarity to proteins found in prokaryotic organisms (Fig. 2F), further supporting the gene recruitment from prokaryotic donors via HGT20. Proteins encoded by dabA identified in P. multistriata (CNS00142), P. multiseries (CNS00149), and P. cuspidata (CNS00150) in this study showed high structural similarity to DabA of P. multiseries reported previously20, and proteins encoded by dabD identified in this study showed high structural similarity to Cytochrome P450 of Fragilariopsis cylindrus (Fig. 2F), indicating a high conservation of this gene in diatoms.
The hypothesis further predicted that the dab gene clusters would be present in homologous integration sites (i.e., sharing breakpoints) in the genomes of all Pseudo-nitzschia species that possess the dab gene clusters, and that deletion events (i.e., dab gene cluster losses) would be evident in the genomes of species that do not possess the dab gene clusters. Our comparative analysis of the genomes of P. multiseries, P. multistriata, and P. cuspidata, however, indicated that the genomic segments harboring the dab gene clusters were not homologous (Supplementary Fig. 4). We therefore anticipated that the dab gene clusters might have moved away from their “original” integration sites due to genome rearrangements, as all Pseudo-nitzschia species have undergone extensive shuffling and transposition (Fig. 1J).
We then searched for the putative “original” integration sites of the dab gene cluster in Pseudo-nitzschia genomes, including those with and without the dab gene clusters. To trace the loss of the dab gene cluster, we aligned the genome scaffold sequences containing the dab gene from P. multiseries, P. multistriata, and P. cuspidata with the genomic sequences of other Pseudo-nitzschia using BLAST. Our results showed that the dab gene cluster in P. cuspidata was flanked by two genes, Pde10317 (a CoA-binding protein) and Pde10588 (a protein kinase) (Fig. 2G, H). The upstream gene, Pde10317, encodes a CoA-binding protein. BLAST analysis indicated that homologous genes of Pde10317 can be found in all Pseudo-nitzschia species and many closely related species, including genera such as Nitzschia, Fragilariopsis, Phaeodactylum, and Cylindrotheca. Conversely, Pde10588 encodes a protein kinase, with homologs present in essentially all eukaryotes.
Through comparison with the SWISS-PROT database, it was found that the CoA-binding domain-containing gene exhibits homology with the Escherichia coli yccU (Percent Identity = 57.1%, Alignment Length = 137)21,22. Interestingly, this gene pair was successfully identified in all 13 Pseudo-nitzschia genomes analyzed in this project as well as in many other genomes (Fig. 2), suggesting that the putative original integration sites of the dab gene clusters remained intact in these genomes. Pairwise comparison of the genomic regions harboring the original integration sites revealed that the genomic regions between the gene pair were rather different (Supplementary Fig. 5), suggesting that the losses of the dab gene cluster were species-specific and varied among the genomes.
Discussion
HABs not only impact marine ecosystems by generating overwhelming biomass23,24, but also pose risk to humans and other life forms by producing deadly toxins25. The discovery of key genes in the DA biosynthetic pathway in P. multiseries in 20188 represented a milestone in understanding the mechanisms underlying the impact of ocean warming and acidification on DA production, and also paved the way for identifying DA biosynthesis genes in other algal species26,27. For example, kainic acid biosynthesis (kab) gene clusters were successfully identified in the genomes of two known kainic acid producers, Digenea simplex and Palmaria palmata, through homology-based searches. This was based on the observation that kainic acid and domoic acid share structural similarities (both compounds are commonly referred to as kainoids)28. Similarly, the dab biosynthesis gene cluster was found in the red alga Chondria armata, the seaweed from which DA was first characterized. Phylogenetic analysis suggested that the core DA biosynthesis genes were acquired through horizontal gene transfer29.
To facilitate the identification of dab genes across various Pseudo-nitzschia species, we assembled chromosome-level genomes for three species (P. multiseries, P. delicatissima, and P. pungens) and draft genomes for ten additional species (including two undescribed species), encompassing both toxigenic and non-toxic Pseudo-nitzschia species2. Insights gained from these genomes through comparative genomics offer valuable understanding of the evolutionary processes underlying the divergence in toxin production capabilities. Additionally, this information sheds light on how genomic context affects gene expression strength and its potential link to disruptions in genes or regulatory elements. Furthermore, these findings provide a point of reference for future transcriptomic studies and enhance our ability to investigate the genetic basis of toxin production in Pseudo-nitzschia species.
The “single acquisition, multiple independent losses (SAMIL)” model of dab gene cluster evolution in Pseudo-nitzschia species
Through mining of these genomes, we identified the DA biosynthesis dab gene cluster in only three Pseudo-nitzschia species. We proposed a putative original integration site and breakpoints for the dab gene cluster in species that lack DA metabolism. Surprisingly, the dab gene cluster-containing Pseudo-nitzschia species was low, accounting for only 20% (3 out of 13) of the species analyzed in this study. This is unexpected given that up to 50% of Pseudo-nitzschia species are known to synthesize DA8,12,13. Nevertheless, numerous studies have reported both toxic and non-toxic strains w Pseudo-nitzschia species2. It is possible that some species might have been erroneously annotated in earlier studies due to their morphological resemblance to toxigenic Pseudo-nitzschia species, particularly those identified solely through light microscopy2. The challenge of accurate species identification, especially in earlier morphology-based studies, has been recognized as a significant issue in accurately identifying Pseudo-nitzschia species30,31.
Based on the results of this study, we propose a “single acquisition, multiple independent losses (SAMIL)” model to explain the evolution of the dab gene cluster in Pseudo-nitzschia. The model suggests that the common ancestor of all Pseudo-nitzschia species acquired the dab gene cluster either via HGT from prokaryotes or through endosymbiosis with red algae (Fig. 3). Notably, since the sequence of CYP450 encoded by dabD is closest to other diatom P450s (Fig. 2E), we propose that the ancestral gene cluster gained via HGT contained only dabA, dabB, and dabC, with dabD being laterally acquired or “hijacked” for DA biosynthesis29. After this acquisition, the dab gene cluster underwent independent evolutionary changes in different Pseudo-nitzschia lineages. Under neutral or negative selection, the dab gene cluster may have been lost from the genomes of some non-DA-producing Pseudo-nitzschia lineages (e.g., P. sabit, P. americana). In addition, intraspecific variation in DA metabolism within “toxic” lineages may result from recent evolutionary events (such as pseudogenization and gene flow), likely driven by adaptations to environmental stresses (geographical or physiological adaptations10). These may have led to the loss of the dab gene cluster in certain strains/populations, ultimately affecting their DA metabolism (P. delicatissima, P. pungens) study.

The SAMIL (Single acquisition, multiple independent losses) model of the dab gene cluster evolution in Pseudo-nitzschia lineages.
The location of the dab gene cluster integration in P. cuspidata may represent a putative alternative insertion site. Our comparative analysis of the genomic locations of dab gene cluster-containing Pseudo-nitzschia species and those lacking the dab gene cluster identified a potential “original” dab gene cluster insertion site in P. cuspidata. This original insertion site is flanked by two highly conserved genes: a CoA-binding protein and a protein kinase (Fig. 3). Notably, dabD is also flanked by these two genes. Our analysis further suggested that the dab gene cluster in Pseudo-nitzschia species might have undergone genome rearrangements (e.g., segmental or dispersal duplications), as evidenced by the different genomic locations of the dab gene clusters in P. multiseries, P. multistriata, and P. cuspidata (Fig. 3). Nevertheless, it cannot be ruled out that the original dab gene cluster integration site might be located at other genomic regions, such as the shared region between P. multiseries and P. multistriata (in blue, Supplementary Fig. 4). Additional data from more dab gene cluster-containing Pseudo-nitzschia species are needed to accurately pinpoint the bona fide original dab gene cluster integration site. Figure 3 was created with BioRender.com.
The dab gene cluster and differential DA biosynthesis in Pseudo-nitzschia species
Research has shown that varying toxic characteristics can exist among different strains of the same Pseudo-nitzschia species, with some species comprised both toxic and non-toxic strains. This variability may be due to the inherent genetic diversity within Pseudo-nitzschia species32. Intraspecific variation in toxicity can be attributed to differences in the presence or absence of key biosynthetic genes within the dab gene clusters, or the presence or absence of entire dab gene clusters, as well as variations in regulatory elements controlling toxin production. In this study, we observed variability in the presence or absence of the entire dab gene cluster among different Pseudo-nitzschia species. However, no genomic differences were observed regarding the presence or absence of dab genes or the entire dab gene cluster among different strains of the same species. Of the 13 Pseudo-nitzschia species analyzed, seven (P. brasiliana, P. cuspidata, P. delicatissima, P. galaxiae, P. multiseries, P. multistriata, and P. pungens) were reported as toxigenic2,18. Despite this, the dab gene cluster was only identified in strains of three Pseudo-nitzschia species (P. cuspidata, P. multiseries, and P. multistriata). It is possible that strains of P. brasiliana, P. delicatissima, P. galaxiae, and P. pungens analyzed in this study lack the dab gene cluster, but it may exist in strains of these species from other geographical regions. Therefore, the presence of the dab gene cluster may be both species- and strain-specific.
Strains of Pseudo-nitzschia that carry the dab gene cluster may not necessarily produce DA. The toxicity of Pseudo-nitzschia species is influenced by a variety of ecological factors, both biotic and abiotic, which affect transcriptional and translational regulations, as suggested previously11,33,34. For instance, grazing by copepods and Calanus copepodite has been reported to induce toxin production in various Pseudo-nitzschia species35. Abiotic factors such as temperature, salinity, light exposure, pH, and concentrations of macronutrients and trace elements also significantly affect the toxin production capabilities of Pseudo-nitzschia species36. Even if all strains of a particular Pseudo-nitzschia species carry the dab gene cluster, different strains may still exhibit varying levels of DA production due to the differential regulation of the dab gene cluster at the transcriptional or translational levels. In fact, under similar culture conditions in this study, while strains of P. multiseries were found to produce DA, P. multistriata strains did not, despite possessing the dab gene cluster. This highlights the complex interplay between genetic diversity and environmental factors in shaping the toxicity of Pseudo-nitzschia species. Further research is needed to understand the molecular mechanisms governing toxin production at both the species and strain levels. Such insights are crucial for predicting and managing the potential impacts of harmful algal blooms on marine ecosystems and public health.
Conclusion
Our genomic analyses have enabled the identification of the dab gene cluster in P. cuspidata. The dab gene cluster identified in this study and previous studies8 suggests a possible shared evolutionary origin in Pseudo-nitzschia. The complex evolutionary trajectory of the dab gene clusters in Pseudo-nitzschia can be succinctly summarized using the SAMIL model, which illustrates the dynamic gains and losses of DA biosynthetic capabilities throughout evolutionary history.
Methods
Pseudo-nitzschia strain isolation and identification
Strains of Pseudo-nitzschia analyzed in this study were isolated from various coastal regions in China, including Qinhuangdao, Hebei Province; Jiaozhou Bay, Shandong Province; Nan’ao Island and Qinzhou Bay, Guangdong Province (Supplementary Table 8). Cells were isolated using micropipette and subsequently cultured in L1 medium (1‰ volume fraction Na2SiO3⋅9H2O was added). Cultures were maintained at 19 °C, under cool white fluorescent illumination with an intensity of 30 μmol·m−2·s−1 and a 12:12 photoperiod. Strains were identified based on their morphological features and the molecular sequences of the nuclear-encoded ITS (ITS1-5.8S-ITS2) region of the ribosomal RNA gene. The identity of some strains were described previously, including CNS00043, CNS00055, CNS00089, CNS00110, CNS00141, CNS00153, CNS00154, CNS00155, and CNS0015637, CNS0013338, CNS0013539, and CNS00090, CNS00097, CNS00130, CNS00138, CNS00142, CNS00159 and CNS0015040. This study identified eleven Pseudo-nitzschia species, including P. americana (CNS00108, CNS00138 and CNS00151), P. brasiliana (CNS01029), P. cuspidata (CNS00150), P. delicatissima (CNS00130 and CNS00135), P. galaxiae (CNS01037 and CNS01103), P. hainanensis (CNS00090), P. micropora (CNS00133 and CNS01024), P. multiseries (CNS00149, CNS00159 and CNS00771), P. multistriata (CNS00107, CNS00142, CNS00781, CNS00965, CNS01237 and CNS01424), P. pungens (CNS00043, CNS00055, CNS00089, CNS00110, CNS00141, CNS00153, CNS00154, CNS00155, CNS00156, CNS00973, CNS01028, CNS01042 and CNS01238) and P. sabit (CNS00609), plus two previously undescribed species Pseudo-nitzschia sp. strains CNS00097 and CNS01031 (Supplementary Table 9).
DNA extraction, sequencing, and draft genome assemblies
During the exponential growth phase, healthy algal cells were harvested for DNA extraction by centrifugation at 8000 rpm for 5 min. Total DNA was extracted using the DNAsecure Plant Kit (Tiangen Biotech, Beijing, China) following the manufacturers’ instructions. DNA concentration was quantified with the Qubit® DNA Assay Kit using a Qubit® 3.0 Flurometer (Invitrogen, USA). Genomic DNA from each sample (0.2 µg) was fragmented to approximately 350 bp by sonication (Covaris S220, Covaris, USA). The DNA fragments were then harvested, end-polished, A-tailed, and ligated with adapters for Illumina sequencing, followed by PCR amplification. PCR products were purified with the AMPure XP system (Beckman Coulter, Beverly, USA). Qualified libraries were sequenced on the NovaSeq 6000 PE150 platform (Illumina, San Diego, CA, USA) at Novogene (Beijing, China). Draft genomes were assembled using SPAdes v3.14.041 and Platanus-allee v2.2.242, with default parameters. Sequencing data and draft genome assembly statistics are provided in Supplementary Table 10.
DNA/RNA extraction and sequencing for chromosome-level genomes
The methods for DNA/RNA extraction and sequencing for the three chromosome-level genomes followed procedures described in our previous study43. Algal cells were processed by first powdering them in liquid nitrogen. The powdered cells were then mixed with lysis buffer and RNase A, followed by incubation. After centrifugation, the supernatants were subjected to magnetic-bead-based DNA purification and washing. High-quality DNA was obtained and sequenced using both PacBio long-reads and MGI short-reads technologies. Libraries were prepared with specific kits and sequenced on the MGISEQ-2000-PE150 and PacBio Sequel SMRT Cell platforms.
For Hi-C analysis, algal samples were cross-linked, fragmented, and labeled with biotin. DNA fragments were captured using magnetic beads, processed and sequenced on the MGISEQ-2000-PE150 platform to analyze chromatin loci proximity.
Total RNA was extracted using CTAB method. Samples were ground, mixed with CTAB lysis buffer, and subjected to several centrifugation steps. Total RNA with high quality was extracted through cetyltrimethylammonium bromide (CTAB) methods for transcriptome sequencing through MGI and PacBio platform. The mRNA sequencing library (DNBSEQ) was constructed and detected by DNF-471 Standard Sensitivity RNA Analysis Kit (AATI), BGI Optimal two-module mRNA library kit (BGI), BGI Plug-In Adapter Kit (BGI) and Qubit® ssDNA Assay Kit (Invitrogen). The sequencing library (DNBSEQ) was constructed and detected by MGIEasy Universal DNA Library Prep Set (MGI), Qubit™ dsDNA BR Assay Kit (Invitrogen) and Qubit® ssDNA Assay Kit (Invitrogen), and the sequencing strategy was MGISEQ-2000-PE150. The PacBio continuous long reads (CLR) sequencing library was constructed and detected by the SMRTbell Express Template Prep Kit 2.0 (PacBio), Qubit dsDNA HS Assay Kit 2.0 (Invitrogen) and HS Large Fragment 50 KB Analysis Kit (Agilent Technologies). The sequencing strategy was PacBio Sequel SMRT Cell 1 M.
Genome size estimation and genome assembly
To estimate the sizes of the genomes of the Pseudo-nitzschia strains, K-mer analysis was performed using Illumina DNA sequencing data and Jellyfish (v 2.1.4)44. For each dataset, k-mers were counted and aggregated (jellyfish count option), and histograms were generated with the “-histo” command. The resulting histograms were then used to estimate genome length and heterozygosity using GenomeScope v 2.045. The genome sizes, repetitive contents, and heterozygosity levels of three Pseudo-nitzschia species P. delicatissima, P. pungens, and P. multiseries were estimated by carrying out genome survey analysis using Illumina DNA sequencing results of 27.85 Gb, 44.24 Gb, and 27.98 Gb for P. delicatissima, P. pungens, and P. multiseries, respectively (Supplementary Table 11). The genome sizes of 35 Mb, 78 Mb, and 267 Mb were estimated for P. delicatissima, P. pungens, and P. multiseries, respectively (Supplementary Fig. 1; Supplementary Table 2); the heterozygosity levels of P. delicatissima, P. pungens and P. multiseries were estimated to be 1.37%, 0.82% and 0.87%, respectively; the repetitive contents were estimated to be 11.00%, 42.10% and 71.00%, respectively (Supplementary Table 2). Thus, the genome sizes of the three Pseudo-nitzschia species varied substantially.
For whole genome assemblies, long reads generated from PacBio Sequel platform were assembled using Mecat246 to generate initial versions of the genome assemblies, which were subsequently polished using Pilon47. The contigs were assembled into chromosomes through Hi-C analysis using Juicer48 and 3D-DNA49 by default parameters. These genome assemblies were further visualized, and error corrected using JucieBox50. The completeness of the assembled genomes was evaluated using BUSCO v3 (eukaryota_odb9)51.
Genome annotation
Repeat sequences in the genome of Pseudo-nitzschia species were annotated using a comprehensive analysis combining homology-based and de novo prediction methods. The homology-based approach utilized the RepBase v21.12 library52, employing RepeatMasker v4.0.7 (http://www.repeatmasker.org/). This strategy enabled the identification of sequences sharing similarity with known repetitive elements. For de novo prediction, the RepeatScout tool53, Piler54, and LTR_FINDER v1.0755 were employed to construct a de novo repeat sequence library. Subsequently, RepeatMasker was applied to predict de novo repeat elements within the genome using the constructed library. To identify tandem repeat sequences, Tandem Repeats Finder v4.0926 was used.
The genome sequences were used for homology-based, de novo, and transcriptome-based gene predictions. First, the homologous proteins from seven species including Arabidopsis thaliana, Fragilariopsis cylindrus, Phaeodactylum tricornutum, Seminavis robusta, Thalassiosira pseudonana, P. multistriata, and Skeletonema marinoi, were used to identify proteins in the repeat masked Pseudo-nitzschia species genome reference sequence with MAKER software (v.2.31.8)27. From the homology predictions, a subset of 2000 well-supported genes were selected as a training set. De novo gene prediction software, Augustus56 and SNAP57, were then trained using this set to improve accuracy in predicting gene structures. To enhance annotation accuracy, RNA-seq and Iso-Seq transcriptomic data were incorporated. RNA-seq data were aligned to the genome using HISAT2 (v2.1.0)58, and transcripts were assembled using StringTie (v1.3.4d)59. PASA pipeline (https://github.com/PASApipeline/PASApipeline) was subsequently employed for correction and refinement of the transcript information, resulting in a high confidence set of transcripts. The homology-based, de novo, and transcriptome-based were integrated using Maker for a second round of gene structure prediction and consolidation.
Protein-Protein BLAST v2.2.31 was then used to assess putative protein functions in each Pseudo-nitzschia species by comparing the protein sequences given by MAKER to the protein sequences from the annotated genomes. The predicted protein coding genes were functionally annotated based on several publicly available databases including Swissprot60, TrEMBL (http://www.uniprot.org/), InterPro61, GO62, KEGG63, and NR (http://www.ncbi.nlm.nih.gov/protein/) databases.
The tRNA sequences in the genome were identified utilizing the tRNAscan-SE 1.3.1 software64. To annotate rRNA sequences, we performed a BLASTN search against rRNA sequences of a set of closely related species. The alignment results were used to identify and annotate rRNA sequences within the genome. The miRNA and snRNA was predicted using Rfam (v1.0.4)65.
Collinearity analysis
To investigate the differences in chromosome numbers among the three species P. delicatissima, P. pungens, and P. multiseries, MCscanX in JCVI (python -m jcvi.compara.catalog ortholog)66 was utilized for pairwise comparisons between the species. To reveal the collinearity relationship between P. delicatissima, P. pungens, and P. multiseries, genome-wide synteny analysis was performed using the MCScanX pipeline within JCVI utility libraries (python -m jcvi.graphics.karyotype)66.
Gene family and evolutionary analysis
Gene families were identified between three Pseudo-nitzschia species and other nine species (A. anophagefferens, T. oceanica, T. pseudonana, S. marinoi, S. robusta, P. tricornutum, F. cylindrus, P. multistriata, and P. multiseries) using Orthofinder267. Each of the gene sets from the 12 species was filtered using a condition where, if there were multiple alternatively spliced transcripts in a gene, only the longest transcript was retained. The similarity of protein sequences was assessed by all-versus-all BLASTP with an E-value 1e-6.
A total of 381 single-copy orthologous genes were identified. Protein alignments for individual orthogroup were done by MAFFT68. The alignments were processed to remove sites with over 50% gaps and remove sequences shorter than 50% of the alignment length. To infer the species tree, we used both concatenation and multispecies coalescent approach. The concatenated dataset included all the 381 loci and was analyzed using IQ-Tree69 with ModelFinder model selection. To assess branch supports, we carried out ultrafast bootstrap and SH-aLRT analyses (both with 1000 replicates).
To estimate the divergence time of different species, the mcmctree program from PAML (v.4.9)70 was used. Calibration points for the divergence analysis were obtained from the TimeTree database (http://www.timetree.org/). The resulting phylogenetic tree was presented with 95% highest posterior density (HPD) interval.
Gene family contraction and expansion analysis were performed using the CAFE571 software based on gene family clustering data. A stochastic birth and death model were proposed in CAFE to estimate the λ value.
Pfam domains for the 12 species were identified using InterProscan72 and visualized using R packages pheatmap and ggplot2. Phylogenetic trees of genes, including PF05548 domains, were reconstructed using IQ-TREE69 with 1000 bootstrap replications.
Annotation of DA biosynthesis gene clusters
For the identification of dab genes in the chromosome-level genome assemblies of P. delicatissima, P. pungens and P. multiseries, protein sequences of DabA, DabB, DabC, and DabD from P. multiseries8 were used as queries to search for the candidate genes, dabA, dabB, dabC, and dabD.
For other Pseudo-nitzschia species, the draft genomes were assembled using SPAdes v3.14.041 and Platanus-allee v2.2.242, with default parameters. BLAST was utilized to probe for dab genes with queries mentioned above. Gene annotation was performed using Genewise73,74 and ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/), with the amino acid sequences of dab genes from P. multiseries serving as a reference. To ensure that no dab genes were missed due to incomplete assembly results, we also used the gene sequences or transcript sequences of dabA, dabB, dabC, and dabD as queries, using the BWA v0.7.1775 MEM algorithm to search for potential genomic sequences containing dab genes in assembled genomes.
Phylogenetic analysis of DA biosynthesis genes
In the phylogenetic analysis of dab genes, other than obtaining the published dab gene sequences from different Pseudo-nitzschia species8,9,10, we also incorporated radA, radC, kabA, and kabC from the red algae28,29. Protein sequences of the assembled transcripts were obtained by using the TransDecoder program (https://github.com/TransDecoder/TransDecoder). Furthermore, sequences were also acquired by querying the NCBI NR database using dab genes as references. Chloroplast genomes (cpDNAs) and ITS (ITS1-5.8S-ITS2) sequences of Pseudo-nitzschia were obtained from NCBI database and our previous study40. Sequence alignments were performed using MUSCLE76, followed by gap removal with trimAl77. Maximum Likelihood (ML) trees were constructed with IQ-Tree69, incorporating ModelFinder for optimal model selection. Branch supports were evaluated through ultrafast bootstrap, SH-aLRT (both with 1000 replicates), and an Approximate Bayes test. Protein structures were predicted using ColabFold v1.5.278 and visualized with Pymol79. Collinearity analysis was performed using Mauve80 and Dotter81.
Toxin analysis
The cultivation parameters for Pseudo-nitzschia strains followed the conditions described above. Algal cells were harvested during the late-exponential growth phase, approximately 21 days post-inoculation. For DA quantification, cell counts were performed by subsampling 5 mL of algal cells and preserved in Lugol’s iodine solution, samples were kept at 4 °C for subsequent microscopic counts. The collection, preparation, and analytical conditions of different samples refer to the previous study82. The DA detection was conducted at the Institute of Oceanology, Chinese Academy of Sciences, using ultra-performance liquid chromatography-electron spray ionization-quadrupole-time of flight-mass spectrometry (UPLC-ESI-Q-TOF-MS) (Bruker, Germany). The DA standard was sourced from the National Marine Environmental Monitoring Center (Standard Material Number: GBW(E)100782). A total of 13 Pseudo-nitzschia strains were tested for DA production (Supplementary Table 8), including P. multiseries (CNS00159), P. multistriata (CNS00142, CNS00781, CNS00965, CNS01237, CNS01424), P. delicatissima (CNS00130), P. americana (CNS00138), P. micropora (CNS00133), P. pungens (CNS00141), P. galaxiae (CNS01103), P. brasiliana (CNS01029), and Pseudo-nitzschia sp. (CNS01031).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing results (raw data) have been submitted to NCBI, and the BioProject number is PRJNA1054348. The assembled genome has been deposited in the NCBI assembly with the accession number GCA_037355735.1, GCA_037355745.1 and GCA_037355755.1. The genome annotation information has been uploaded to Figshare: 10.6084/m9.figshare.27254001.
References
Bates, S. et al. Pennate diatom Nitzschia pungens as the primary source of domoic acid, a toxin in shellfish from eastern Prince Edward Island, Canada. Can. J. Fish. Aquat. Sci. 46, 1203–1215 (1989).
Bates, S. S., Hubbard, K. A., Lundholm, N., Montresor, M. & Leaw, C. P. Pseudo-nitzschia, Nitzschia, and domoic acid: New research since 2011. Harmful Algae 79, 3–43 (2018).
McCabe, R. M. et al. An unprecedented coastwide toxic algal bloom linked to anomalous ocean conditions. Geophys. Res. Lett. 43, 10,366–310,376 (2016).
Cook, B. I., Ault, T. R. & Smerdon, J. E. Unprecedented 21st century drought risk in the American Southwest and Central Plains. Sci. Adv. 1, e1400082 (2015).
Bresnan, E. et al. Diversity and regional distribution of harmful algal events along the Atlantic margin of Europe. Harmful Algae 102, 101976 (2021).
Clark, S. et al. Pseudo-nitzschia bloom dynamics in the Gulf of Maine: 2012–2016. Harmful Algae 88, 101656 (2019).
Husson, B., Hernández-Fariñas, T., Le Gendre, R., Schapira, M. & Chapelle, A. Two decades of Pseudo-nitzschia spp. blooms and king scallop (Pecten maximus) contamination by domoic acid along the French Atlantic and English Channel coasts: Seasonal dynamics, spatial heterogeneity and interannual variability. Harmful Algae 51, 26–39 (2016).
Brunson, J. K. et al. Biosynthesis of the neurotoxin domoic acid in a bloom-forming diatom. Science 361, 1356–135 (2018).
Hardardóttir, S. et al. Transcriptomic responses to grazing reveal the metabolic pathway leading to the biosynthesis of domoic acid and highlight different defense strategies in diatoms. Bmc Mol. Biol. 20, https://doi.org/10.1186/s12867-019-0124-0 (2019).
Lema, K. A. et al. Inter- and Intra-Specific Transcriptional and Phenotypic Responses of Pseudo-nitzschia under Different Nutrient Conditions. Genome Biol. Evol. 11, 731–747 (2019).
Xu, D. et al. Plastic responses lead to increased neurotoxin production in the diatom Pseudo-nitzschia under ocean warming and acidification. ISME J. 17, 525–536 (2023).
Lelong, A., Hégaret, H., Soudant, P. & Bates, S. S. Pseudo-nitzschia (Bacillariophyceae) species, domoic acid and amnesic shellfish poisoning: revisiting previous paradigms. Phycologia 51, 168–216 (2012).
Trainer, V. L. et al. physiological ecology, phylogeny, toxicity, monitoring and impacts on ecosystem health. Harmful Algae 14, 271–300 (2012).
Silver, M. W. et al. Toxic diatoms and domoic acid in natural and iron enriched waters of the oceanic Pacific. Proc. Natl Acad. Sci. 107, 20762–20767 (2010).
Zhu, Z. et al. Understanding the blob bloom: Warming increases toxicity and abundance of the harmful bloom diatom Pseudo-nitzschia in California coastal waters. Harmful Algae 67, 36–43 (2017).
Kinoshita, T., Fukuzawa, H., Shimada, T., Saito, T. & Matsuda, Y. Primary structure and expression of a gamete lytic enzyme in Chlamydomonas reinhardtii: similarity of functional domains to matrix metalloproteases. Proc. Natl Acad. Sci. USA 89, 4693–4697 (1992).
Teng, S. T. et al. Toxic bloom of Pseudo-nitzschia cuspidata (Bacillariophyceae) and domoic acid contamination of bivalve molluscs in Malaysia Borneo. Toxicon 202, 132–141 (2021).
Bates, S. S., Lundholm, N., Hubbard, K. A., Montresor, M. & Leaw, C. P. Toxic and harmful marine diatoms. in Diatoms: fundamentals and applications (eds. Seckbach, J. & Gordon, R.) 389–434 https://doi.org/10.1002/9781119370741.ch17 (Wiley, 2019).
Niu, B.-B. et al. Morphology, molecular phylogeny and biogeography revealed two new Pseudo-nitzschia (Bacillariophyceae) species in Chinese waters. J. Syst. Evol. https://doi.org/10.1111/jse.13016 (2023).
Chekan, O. R., McKinnie, S. M. K., Noel, J. P. & Moore, B. S. Algal neurotoxin biosynthesis repurposes the terpene cyclase structural fold into an N-prenyltransferase. Proc. Natl Acad. Sci. USA 117, 12799–12805 (2020).
Butland, G. et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433, 531–537 (2005).
Hiyama, T. B. et al. Structural basis of CoA recognition by the Pyrococcus single-domain CoA-binding proteins. J. Struct. Funct. Genomics 7, 119–129 (2006).
Allen, J. I., Smyth, T. J., Siddorn, J. R. & Holt, M. How well can we forecast high biomass algal bloom events in a eutrophic coastal sea? Harmful Algae 8, 70–76 (2008).
Anderson, D. M. Approaches to monitoring, control and management of harmful algal blooms (HABs). Ocean Coast. Manag. 52, 342–347 (2009).
Van Dolah, F. M. Marine algal toxins: Origins, health effects, and their increased occurrence. Environ. Health Perspect. 108, 133–141 (2000).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinforma. 12, 491 (2011).
Chekan, J. R. et al. Scalable Biosynthesis of the Seaweed Neurochemical, Kainic Acid. Angew. Chem. Int Ed. Engl. 58, 8454–8457 (2019).
Steele, T. S. et al. Domoic acid biosynthesis in the red alga Chondria armata suggests a complex evolutionary history for toxin production. Proc. Natl Acad. Sci. USA 119, https://doi.org/10.1073/pnas.2117407119 (2022).
Li, Y. et al. Pseudo-nitzschia simulans sp. nov. (Bacillariophyceae), the first domoic acid producer from Chinese waters. Harmful Algae 67, 119–130 (2017).
Lim, H. C. et al. Phylogeny and species delineation in the marine diatom Pseudo-nitzschia (Bacillariophyta) using cox1, LSU, and ITS2 rRNA genes: A perspective in character evolution. J. Phycol. 54, 234–248 (2018).
Lelong, A., Hégaret, H., Soudant, P. & Bates, S. S. Pseudo-nitzschia(Bacillariophyceae) species, domoic acid and amnesic shellfish poisoning: revisiting previous paradigms. Phycologia 51, 168–216 (2019).
Sun, J. et al. Effects of changing pCO2 and phosphate availability on domoic acid production and physiology of the marine harmful bloom diatom Pseudo-nitzschia multiseries. Limnol. Oceanogr. 56, 829–840 (2011).
Kelly, K. J. et al. Simulated upwelling and marine heatwave events promote similar growth rates but differential domoic acid toxicity in Pseudo-nitzschia australis. Harmful Algae 127, 102467 (2023).
Harethardottir, S. et al. Dangerous Relations in the Arctic Marine Food Web: Interactions between Toxin Producing Pseudo-nitzschia Diatoms and Calanus Copepodites. Mar. Drugs 13, 3809–3835 (2015).
Sobrinho, B. F. et al. Growth, Toxin Production and Allelopathic Effects of Pseudo-nitzschia multiseries under Iron-Enriched Conditions. Mar. Drugs 15, https://doi.org/10.3390/md15100331 (2017).
Chen, Y., Wang, Y., Liu, K., Liu, F. & Chen, N. Development of a high-resolution molecular marker for tracking Pseudo-nitzschia pungens genetic diversity through comparative analysis of mitochondrial genomes. J. Appl. Phycol. 33, 2283–2298 (2021).
Chen, Y., Cui, Z., Liu, F. & Chen, N. Mitochondrial genome and phylogenomic analysis of Pseudo-nitzschia micropora (Bacillariophyceae, Bacillariophyta). Mitochondrial DNA Part B 6, 2035–2037 (2021).
He, Z. et al. Complete mitochondrial genome of the harmful algal bloom species Pseudo-nitzschia delicatissima (Bacillariophyceae, Bacillariophyta). Mitochondrial DNA Part B 6, 2541–2543 (2021).
He, Z. et al. Comparative Analysis of Pseudo-nitzschia Chloroplast Genomes Revealed Extensive Inverted Region Variation and Pseudo-nitzschia Speciation. Front. Marine Sci. 9, https://doi.org/10.3389/fmars.2022.784579 (2022).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Kajitani, R. et al. Platanus-allee is a de novo haplotype assembler enabling a comprehensive access to divergent heterozygous regions. Nat. Commun. 10, 1702 (2019).
Liu, S., Xu, Q. & Chen, N. Expansion of photoreception-related gene families may drive ecological adaptation of the dominant diatom species Skeletonema marinoi. Sci. Total Environ. 897, 165384 (2023).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Xiao, C. L. et al. MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 14, 1072–1074 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101 (2016).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 1, e323 (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Johnson, A. D. et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24, 2938–2939 (2008).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Apweiler, R. et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29, 37–40 (2001).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2021).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120 (2005).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
DeLano, W. L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 40, 82–92 (2002).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147 (2010).
Sonnhammer, E. L. & Durbin, R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167, GC1–GC10 (1995).
Dong, H. C. et al. Occurrence of Pseudo-nitzschia species and associated domoic acid production along the Guangdong coast, South China Sea. Harmful Algae 98, 101899 (2020).
Basu, S. et al. Finding a partner in the ocean: molecular and evolutionary bases of the response to sexual cues in a planktonic diatom. N. Phytologist 215, 140–156 (2017).
Acknowledgements
This research was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (XDB42000000), the Chinese Academy of Sciences Pioneer Hundred Talents Program (to Nansheng Chen), the Taishan Scholar Project Special Fund (to Nansheng Chen), the Qingdao Innovation and Creation Plan (Talent Development Program - 5th Annual Pioneer and Innovator Leadership Award to Nansheng Chen, 19-3-2-16-zhc), and the Natural Sciences and Engineering Research Council of Canada (NSERC). We are grateful to colleagues from the Jiaozhou Bay Marine Ecosystem Research Station for the opportunity to participate in the investigation expeditions. Data analysis was supported by Oceanographic Data Center, IOCAS.
Author information
Authors and Affiliations
Contributions
N.C. designed the project and wrote the manuscript with contributions from all coauthors; Z.H. and Q.X. carried out analyses and prepared figures and tables; Y.C., S.L., H.S., and H.W. identified, maintained, and characterized Pseudo-nitzschia strains used in this study; C.P.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Wendy Mok, Luke Grinham and Johannes Stortz.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, Z., Xu, Q., Chen, Y. et al. Acquisition and evolution of the neurotoxin domoic acid biosynthesis gene cluster in Pseudo-nitzschia species. Commun Biol 7, 1378 (2024). https://doi.org/10.1038/s42003-024-07068-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07068-7
This article is cited by
-
Conserved genetic markers reveal widespread diatom sexual reproduction in the global ocean
Nature Communications (2025)
